

Abstract
Protein tyrosine phosphatase (PTP) 4A3 is frequently overexpressed in human solid tumors and hematologic malignancies and is associated with tumor cell invasion, metastasis, and a poor patient prognosis. Several potent, selective, and allosteric small molecule inhibitors of PTP4A3 were recently identified. A lead compound in the series, JMS-053 (7-imino-2-phenylthieno[3,2-c]pyridine-4,6(5H,7H)-dione), has a long plasma half-life (∼ 24 hours) in mice, suggesting possible binding to serum components. We confirmed by isothermal titration calorimetry that JMS-053 binds to human serum albumin. A single JMS-053 binding site was identified by X-ray crystallography in human serum albumin at drug site 3, which is also known as subdomain IB. The binding of JMS-053 to human serum albumin, however, did not markedly alter the overall albumin structure. In the presence of serum albumin, the potency of JMS-053 as an in vitro inhibitor of PTP4A3 and human A2780 ovarian cancer cell growth was reduced. The reversible binding of JMS-053 to serum albumin may serve to increase JMS-053’s plasma half-life and thus extend the delivery of the compound to tumors.
SIGNIFICANCE STATEMENT
X-ray crystallography revealed that a potent, reversible, first-in-class small molecule inhibitor of the oncogenic phosphatase protein tyrosine phosphatase 4A3 binds to at least one site on human serum albumin, which is likely to extend the compound’s plasma half-life and thus assist in drug delivery into tumors.
Introduction
Albumin’s concentration in serum ranges from 35 to 50 mg/ml (530–750 μM), comprising more than half of the total amount of all human serum proteins. Its surface contains multiple hydrophobic binding pockets, and the protein functions as a natural transporter of diverse ligands, such as fatty acids, hormones, bile acids, amino acids, heme, metal ions, lipophilic xenobiotics, and drugs (Sudlow et al., 1976; Larsen et al., 2016). Many commonly used drugs bind preferentially to albumin at specific sites (Czub et al., 2020). Human serum albumin (HSA) has a long circulatory half-life of ∼19 days (Peters, 1995). HSA is not, however, just a transport protein; it protects bound drugs from rapid metabolic destruction, decreases renal clearance, and extends drug half-life. The presence of albumin in the lymphatic system also provides an endogenous delivery vehicle for deep drug tissue penetration (Larsen et al., 2016).
Tumor tissues have very high metabolic activities, and they exploit the endogenous transporting properties of albumin to assist in the delivery of nutrients and macromolecules. To meet the demands of high energy consumption, tumor cells and their associated stromal and endothelial cells frequently have elevated levels of albumin-binding proteins, such as secreted protein, acidic, cysteine rich (SPARC; also known as osteonectin); megalin; and calreticulin (Zhao et al., 2018). Clinically important oncology drugs are known to complex with HSA (Wang et al., 2013), which has been shown to enhance the tumor delivery of anticancer drugs. Albumin-drug binding has even been used as a drug delivery system. For example, paclitaxel has been formulated as an albumin-drug nanoparticle and is clinically approved and marketed as Abraxane (Merlot et al., 2014; Zhao et al., 2018).
The PTP4A phosphatases comprise a unique group of three highly homologous, membrane-associated intracellular enzymes: PTP4A1, PTP4A2, and PTP4A3 (also known as Protein of Regenerating Liver-1 (PRL-1, PRL-2, and PRL-3). All three PTP4A family members, which are considered dual specificity phosphatases with the ability to dephosphorylate tyrosine, threonine, and serine side chains, promote the hallmarks of malignancy—namely, cell migration, invasion, and anchorage-independent growth (Tasker et al., 2019b). PTP4A3 and to a lesser extent PTP4A1 and PTP4A2 have been reported to be overexpressed in many human solid tumors, including colorectal, ovarian, liver, lung, breast, brain, stomach, bladder, pancreatic, thyroid, and prostate cancers (Tasker et al., 2019b; Wei et al., 2020). For example, high PTP4A3 expression has been observed in ∼80% of 151 tumor tissues examined from 11 different tumor types (Thura et al., 2019). PTP4A3 is also prominently expressed at both the mRNA and protein levels in many leukemias and lymphomas (Hjort et al., 2017, 2018; Wei et al., 2020). It is notable that PTP4A3 is barely detectable in most nonmalignant tissues, consistent with an acquisitional role for PTP4A3 in the transformation process. The pro-oncogenic involvement of the PTP4A family members is thought to be due at least in part to their direct interactions with Cyclin M CNNM magnesium transport regulators, which enable enhanced energy metabolism in cancer cells (Hardy et al., 2019). Elevated PTP4A expression is associated with poor patient prognosis (Bai et al., 2016; den Hollander et al., 2016), which reinforces the attractiveness of this potential therapeutic target.
The emergence of potent small molecules that target PTP4A phosphatases (Stadlbauer et al., 2015; Salamoun et al., 2016; Yu and Zhang, 2018; Lazo et al., 2019; Tasker et al., 2019a,b) highlights the unique opportunity to exploit the high expression levels of this oncogenic family in human cancers for therapeutic applications. Members of the thienopyridone and thienopyridinedione class of PTP4A inhibitors reduce the growth and survival of cells derived from solid tumors (Daouti et al., 2008; Salamoun et al., 2016; McQueeney et al., 2017, 2018; Lazo et al., 2019; Tasker et al., 2019a,b) and T-cell acute lymphoblastic leukemia (Wei et al., 2020). The novel iminopyridinedione JMS-053 has been shown to be a reversible and allosteric PTP4A3 phosphatase inhibitor (McQueeney et al., 2017; Lazo et al., 2019). Furthermore, although PTPs are generally susceptible to reactive oxygen species–mediated deactivation because of the presence of a catalytically essential cysteine residue at the active site, JMS-053 lacks the ability to generate detectable reactive oxygen species levels in vitro or in cancer cells (McQueeney et al., 2017; Lazo et al., 2019). In the current studies, we determine the structural interaction of this unique, first-in-class PTP4A3 inhibitor with HSA. We further provide insights into the pharmacological properties of JMS-053, which could help guide its further preclinical development.
Materials and Methods
Compounds and Reagents.
JMS-053 (7-imino-2-phenylthieno[3,2-c]pyridine-4,6(5H,7H)-dione) was synthesized and subjected to the rigorous quality control procedures we described previously (Salamoun et al., 2016; Tasker et al., 2019a). Specifically, JMS-053 was subjected to chromatographic purification, 1H and 13C NMR, high-resolution mass spectrometry, and elemental analysis and was >98% pure, as previously described (Salamoun et al., 2016; Tasker et al., 2019a). JMS-053 was always solubilized in DMSO immediately prior to usage. All other reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Human Ovarian Cancer Cell Growth Inhibition Assays.
Authenticated human A2780 cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were monitored at least every 6 months and found negative for mycoplasma contamination using the Universal Mycoplasma Detection Kit (#20–10121; American Type Culture Collection) and never carried beyond 20 passages. The CellTiter-Glo (Promega, Madison, WI) 3D assay system was used to test compound effects on cell viability. Cells were seeded (250 cells/20 µl) in complete cell culture medium into 384-well ultralow attachment U-bottom microplates (Corning, Corning, NY). Plates were incubated at 37°C and 5% CO2 to allow for spheroid formation. DMSO (0.5% final concentration) vehicle control or ten different concentrations of JMS-053 (2.5 μl), ranging from 0.1 to 50 μM, were added to the plates 24 hours after cell addition along with 0.5%, 1%, 5%, or 10% FBS. Spheroids were cultured for an additional 48 hours, at which time viability was measured with CellTiter-Glo 3D as previously described (McQueeney et al., 2017). The number of biologic replicates (n = 4) and the method of data analysis (one-way ANOVA) were decided before the start of the experiment. Data were analyzed and EC50 values were calculated using Prism 8.0 software (GraphPad Software, San Diego, CA).
Isothermal Titration Calorimetry Studies.
Lyophilized HSA, which was >99% globulin-free but not fatty acid–free, was obtained commercially from Sigma-Aldrich (#A8763). Lyophilized protein was dissolved in 25 mM Tris (pH 7.4) and 50 mM NaCl to a concentration of 100 mg/ml and centrifuged to remove insoluble matter. The protein solution was purified using a Sephacryl S300 resin column equilibrated with 25 mM Tris (pH 7.4) and 50 mM NaCl. Fractions corresponding to full-length, monomeric HSA, detected by SDS-PAGE, were consolidated and used for experimentation. Isothermal titration calorimetry (ITC) experiments were performed on a MicroCal VP ITC200 instrument. Experiments were conducted at 30.0°C using purified HSA (1 mM) in the syringe injected into a cell with JMS-053 (100 μM). We employed 40 injections of 7 µl each to obtain heat generation over the full spectrum of binding concentrations. Experiments were corrected for dilution enthalpy. Raw data were fit using OriginPro with a MicroCal module add-on to both a one-site or two-site binding model.
HSA Purification and Cocrystallization with JMS-053.
Lyophilized HSA (#A8763; Sigma-Aldrich) was dissolved in a buffer containing 25 mM Tris (pH 7.4) and 50 mM NaCl and purified using size-exclusion chromatography with a Superdex 200 column attached to an ÄKTA Fast Protein Liquid Chromatography Fast Proetin Liquid Chromatography (GE Healthcare, Pittsburgh, PA) to separate the dimeric and monomeric fractions of HSA. A purification buffer consisting of 25 mM Tris (pH 7.4) and 50 mM NaCl was used as the mobile phase. The absorbance (280 nm) was measured with a Nanodrop 2000 (Thermo Scientific, Waltham, MA) to estimate protein concentrations using the extinction coefficient (ε280 = 34,440 M−1 cm−1) and molecular mass (66,470 Da). The monomeric fractions of HSA were concentrated to 100 mg/ml using an Amicon Ultra Centrifugal Filter (#UFC903024; Millipore Sigma) with a molecular mass cutoff of 30 kDa. Prior to crystallization, myristic acid (dissolved in ethanol) was added to the protein to reach a final concentration of 5 mM fatty acid. The protein complexed with myristic acid was incubated at 37°C for several hours, and then crystallization was performed in 96-well hanging drop plates (Swissci 3-Well Midi) that were set using a Mosquito crystallization robot (TTP Labtech, Boston, MA). Aliquots of 200 or 300 nl of HSA solution with myristic acid were mixed with 200 or 300 nl of reservoir solution [25% polyethylene glycol (PEG) 3350, 50 mM K2HPO4 at pH 7.0] in ratios 1:1, 2:1, or 1:2. The crystallization plate was incubated at 37°C for several days and, after growth of the first HSA crystals, the plate was transferred to room temperature. JMS-053 powder (∼50 μg) was added to each crystallization drop containing crystals and then incubated for 48 hours before harvesting. Harvested crystals were flash-cooled in liquid nitrogen using 30% PEG 3350 as a cryoprotectant.
Structure Determination.
Diffraction data were collected at the Structural Biology Center - Collaborative Access Team 19-ID beamline at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). Data collection was performed for a single crystal at 100 K using a 0.979-Å wavelength. HKL-3000 (Otwinowski and Minor, 1997; Minor et al., 2006) was used to process, integrate, and scale the data. The structure was determined by molecular replacement using the structure of HSA complexed with fatty acids (PDB ID: 6HSC) as the template. Structure determination and refinement of the HSA complex with JMS-053 were performed using HKL-3000 interacted with MOLREP (Vagin and Teplyakov, 2010), Fitmunk (Porebski et al., 2016), REFMAC (Murshudov et al., 2011), Coot (Emsley et al., 2010), and auxiliary programs (Winn et al., 2011). The most recent guidelines were used during the refinement process (Shabalin et al., 2018). The length of modeled fatty acids was based only on how many atoms were visible in the electron density; the actual fatty acid chains could be longer. We employed the TLS Motion Determination Server (Painter and Merritt, 2006; Zucker et al., 2010) to determine the TLS groups used during refinement. Stereochemical restraints for the JMS-053 molecule were generated using Grade Web Server (http://grade.globalphasing.org/). A standardized placement of the model in the unit cell was achieved using The ACHESYM server (Kowiel et al., 2014). MOLPROBITY (Chen et al., 2010) and wwPDB validation servers (Gore et al., 2017) were used to validate the protein model. PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.3; Schrödinger, LLC) was used to visualize protein structures. Dali server (Holm and Laakso, 2016) was used to calculate RMSD values between the aligned Cα atoms presented in Supplemental Table 3. Diffraction images are available at the Integrated Resource for Reproducibility in Macromolecular Crystallography (Grabowski et al., 2016). Statistics for data collection, structure refinement, and structure quality are displayed in Table 1. The atomic coordinates and structure factors were deposited in the PDB with accession code ID 6WUW.
TABLE 1.
Data collection, structure refinement, and structure quality statistics
Values in parentheses are for the highest resolution shell. Ramachandran plot statistics are calculated by MolProbity.
| PDB ID | 6WUW |
|---|---|
| Diffraction images DOI | 10.18430/m3.irrmc.5549 |
| Data collection statistics | |
| Resolution (Å) | 50.00–2.20 |
| (2.24–2.20) | |
| Beamline | 19-ID |
| Wavelength (Å) | 0.979 |
| Space group | P1 |
| Unit-cell dimensions (Å) | a = 38.4 |
| b = 93.4 | |
| c = 94.9 | |
| Unit-cell angles (o) | α = 74.6 |
| β = 89.6 | |
| γ = 80.4 | |
| Protein chains in the ASU | 2 |
| Completeness (%) | 92.9 (91.0) |
| Number of unique reflections | 59,720 (2981) |
| Redundancy | 2.6 (2.5) |
| <I>/<σ(I)> | 18.5 (1.7) |
| CC 1/2 | (0.61) |
| Rmerge | 0.051 (0.678) |
| Refinement statistics | |
| Rwork/Rfree | 0.192/0.247 |
| Bond lengths RMSD (Å) | 0.003 |
| Bond angles RMSD (°) | 1.1 |
| Mean B value (Å2) | 44 |
| Mean B value for JMS-053 (Å2) | 67 |
| Number of protein atoms | 9296 |
| Mean B value for protein atoms (Å2) | 44 |
| Number of water molecules | 433 |
| Mean B value for water molecules (Å2) | 38 |
| Clashscore | 2.63 |
| MolProbity score | 1.05 |
| Rotamer outliers (%) | 0.88 |
| Ramachandran outliers (%) | 0.00 |
| Ramachandran favored (%) | 98.1 |
In Vitro PTP4A3 Phosphatase Assay.
Inhibition of PTP4A3 activity was determined as previously described (Salamoun et al., 2016) using triplicate 384-well microtiter plates, each containing four wells per sample, with recombinant His6-tagged PTP4A3, the substrate 4-methylumbelliferyl phosphate (15 μM), and concentrations of BSA ranging from 0.1% to 5% and seven different concentrations of JMS-053 ranging from 31.2 nM to 2 μM. PTP4A3 inhibition assays were conducted in triplicate as technical replicates. The number of biologic replicates (n = 4 to 5) was determined based on enzyme and compound availability. The solution was incubated at 25°C for 20 minutes in 40 mM Tris-HCl (pH 7.0), 75 mM NaCl, 2 mM EDTA, and 4 mM dithiothreitol buffer, and fluorescence was measured using a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA) at A358excitation/A455emission. The data were analyzed at the end of the study with GraphPad Prism 8.0 software using a one-way ANOVA Dunnett’s multiple comparison test. These findings should be viewed as exploratory, as recommended by Michel et al. (2020).
In Vitro JMS-053 Metabolism and Pharmacokinetics.
Incubations consisted of pooled CD1 male mouse liver microsomes (Xenotech pool of 1143; 0.5 mg/ml), compound of interest (2 μM), MgCl2 (5 mM), NADPH (1 mM, if applicable), phosphate buffer (100 mM, pH 7, 200 μl), and liquid chromatography–mass spectrometry (LC-MS)–grade water (106 μl) for a total volume of 400 μl in an Eppendorf tube (500 μl). Compounds and microsomes were preincubated (37°C, 5 minutes) in a Thermo Scientific MIDI 40 Incubator before NADPH was added (if applicable). Samples (50 μl) at t = 0 minutes and after an incubation time of 10, 15, 30, 45, and 60 minutes were quenched with ice-cooled acetonitrile (200 μl) containing quinidine (2 μM) as an internal standard and centrifuged (950g, 40 minutes) in an Eppendorf centrifuge 5702, and the supernatant (90 μl) was transferred to an LC-MS vial with LC-MS–grade water (90 μl) for analysis. Control experiments without NADPH were performed in parallel. Verapamil was used as a control in all experiments. Analyses of the supernatant were performed using a Thermo Scientific Exactive Orbitrap LC-MS (electrospray ionization source-positive ion mode) coupled to a Thermo Scientific Accela High Performance Liquid Chromatography system using a 3.5-μm Water XTerra C18 column (2.1 × 50 mm; 10-minute gradient elution with MeCN/H2O/MeOH containing 0.1% formic acid at a flow rate of 500 μl/min from 3:92:5 at 0–0.5 minutes, to 93:2:5 at 4.0, and back to 3:92:5 from 6.0 to 7.5 minutes). Injections (10 μl) were performed in duplicate. The molecular ion for each compound was monitored using Xcalibur software. The total ion chromatogram peak for each time point was integrated and calculated as a percentage of the area of the peak at time = 0 minutes, corrected for the internal standard.
Pharmacokinetic studies were conducted by Sai Life Sciences Ltd. (Hinjewadi, India) using 20% 1-methyl-2-pyrrolididone, 25% Kolliphor HS 15, and 55% 1× phosphate-buffered saline as the vehicle and a single i.p. dose of 10 mg/kg JMS-053 injection into nine male C57Bl/6 mice (8–12 weeks old) per group. The study was conducted in accordance with the guidelines provided by the Committee for the Purpose of Control and Supervision of Experiments on Animal as published in the Gazette of India (December 15, 1998) and had the prior approval of the Institutional Animal Ethics Committee. Blood samples (60 μl) were collected under light isoflurane anesthesia from retro-orbital plexus predose and at eight time points, the last being at 24 hours. Three mice were used for each time point. Immediately after the blood samples were collected, plasma was harvested by centrifugation, and the samples were stored at −70°C. At the time of analysis, proteins were precipitated using acetonitrile, and the sample was analyzed using a Waters XSelect CSH C18 (50 × 2.1 mm, 2.5 μm) column (Milford, MA) with an initial liquid chromatography (0.8 ml/min) elution mobile phase consisting in 98% of 0.1% formic acid in acetonitrile and in 2% of 0.1% formic acid in H2O, followed by elution with the secondary mobile phase of 0.1% formic acid in H2O. The composition of the eluent was monitored by mass spectroscopy.
Mouse plasma binding studies were conducted by Cyprotex, LLC (Watertown, MA), using equilibrium dialysis. Duplicate samples of JMS-053 (5 μM) were incubated with 50% mouse plasma at 37°C in vitro for 4 hours, at which time the samples were analyzed by LC-MS. The binding of JMS-053 to serum albumin was calculated using the formulas found below from Bocedi et al. (2004), in which W is the molar fraction of the drug-bound serum albumin, and Y is the molar fraction of the serum albumin–bound drug. The JMS-053 concentration was based on the experimentally determined KD of 118 μM, Tmax of 233.8 ng/ml (913 nM), and the albumin concentration used for C57BL/6 mice was 27 mg/ml (406 μM) (Zaias et al., 2009) and for human serum albumin was 45 mg/ml (700 μM) (Bocedi et al., 2004).
 |
Statistical Analysis.
All statistical analyses were performed with GraphPad Prism 8.0. Data are presented as means ± S.D. P values were calculated with one-way ANOVA as indicated. P < 0.05 was considered statistically significant. For the in vitro PTP4A3 inhibition studies, the biologic replicate sample number (n = 4 to 5) was determined based on enzyme and compound availability. The data were analyzed with GraphPad Prism 8.0 software. Concentration-response curves were fit to the data using the log(inhibitor) versus response-variable slope (four parameter) model. The top value (MAXmaximum) was the fluorescence value with 0.5% DMSO vehicle control, and the bottom value (minimum) was the value obtained with 2 mM Na2OV4. The use of a one-way ANOVA Dunnett’s multiple comparison test was decided after the data were collected. These findings should, therefore, be viewed as exploratory, as recommended by Michel et al. (2020). For the cell spheroid studies, the number of biologic replicates (n = 3) and the method of data analysis were decided before the start of the experiment. The data were analyzed with GraphPad Prism 8.0 software. Concentration-response curves were fit to the data using the log(inhibitor) versus response-variable slope (four parameter) module. The top value (maximum) was the fluorescence value with 0.5% DMSO (final concentration) vehicle control, and the bottom value (minimum) was the value obtained with 10% DMSO (final concentration).
Results
JMS-053 Is Rapidly Metabolized but Has a Long Mouse Plasma Half-Life.
We observed a time-dependent loss of JMS-053 when it was incubated with mouse liver microsomes and NADPH, which in the first 30 minutes was comparable to that seen with the control compound, verapamil (Fig. 1A). After a 60-minute incubation, 42% of JMS-053 remained. In the absence of NADPH, we also observed an approximate 40% loss of JMS-053 (Supplemental Fig. 1), which may reflect a non–NADPH-dependent metabolism of JMS-053 or some hydrolytic instability. We next examined the pharmacokinetics of JMS-053 using a dose, formulation, and route of administration that we previously showed has antitumor activity against a drug-resistant human ovarian cancer xenograft in mice (McQueeney et al., 2017). The mean plasma concentrations of JMS-053 after a single i.p. injection of 10 mg/kg of JMS-053 in mice are presented in Fig. 1B. The Tmax for JMS-053 of 233.8 ng/ml (0.913 μM) was observed at 0.5 hours, which was followed by a 30% decrease at 2 hours and then a rebound to almost the 0.5-hour concentration at 4 hours. Surprisingly, in light of the in vitro mouse liver microsome data (Fig. 1A), the area under the plasma JMS-053 concentration curve was 4.425 h*μg/ml, which indicated a sustained presence of compound. The JMS-053 plasma half-life was estimated to be ∼24 hours. We postulate that the transient decrease of plasma levels followed by the restoration of higher plasma levels might reflect an enterohepatic recirculation process (Roberts et al., 2002). We further hypothesized that the relatively high plasma area under the curve and the prolonged JMS-053 plasma half-life might reflect binding of this weakly basic compound [pKa 7.0, calculated with ChemAxon InstantJChem v. 5.9 (Boston, MA)] to plasma components. Therefore, we compared the binding of JMS-053 to two common clinically employed drugs, propranolol and warfarin, using mouse plasma protein and equilibrium dialysis (Supplemental Table 1). More than 99% of JMS-053 was found bound to mouse plasma compared with 74.1% and 82.3% of propranolol and warfarin, respectively. These data supported the hypothesis that JMS-053 bound to some components in plasma.
Fig. 1.

Mouse liver microsomal metabolism and pharmacokinetics of JMS-053. (A) JMS-053 (red circles) and verapamil (black squares) in vitro metabolism by mouse liver microsomes. Each symbol is from an independent assay. The line represents the mean value of the two experiments. (B) Mouse plasma pharmacokinetics. Each symbol represents the mean value from three independent determinations from mice treated i.p. with 10 mg/kg JMS-053. The bar indicates S.D. unless the value was smaller than the symbol, and the dotted line represents the 95% confidence interval.
JMS-053 Binds to Human Serum Albumin.
Albumin comprises more than half of all plasma proteins (Zaias et al., 2009) and is a likely candidate for compound binding. Although there are multiple X-ray crystal structures of drugs bound to albumin (Wang et al., 2013; Czub et al., 2020), there are no generally accepted guidelines that would predict drug binding proclivity or the site of drug interactions with albumin. It is notable that drugs frequently bind to the same sites on serum albumin from different species (Wang et al., 2013; Sleep, 2015) ostensibly because the albumins share considerable homology and at least 70% amino acid identity (Supplemental Table 2). Thus, we initially tested whether JMS-053 binds to HSA using isothermal titration calorimetry. Titrating HSA into a solution of JMS-053 yielded an exothermic binding curve that could be fit to a one-site model of binding with N = 1.0 ± 0.1; KA = 8.5 × 103 ± 1.5 × 103 M; Gibb’s free energy (ΔG) = 23 ± 4 kJ/mol; and a KD of 118 ± 21 µM (Fig. 2, A and B). The exothermic binding curve data also could be fit to a two-site binding model with N1 = 0.21 ± 0.02; KA1 = 1.5 × 106 ± 8.0 × 105 M; Gibb’s free energy (ΔG1) = 36 ± 19 kJ/mol; a KD1 of 0.63 ± 0.34 µM; N2 = 1.4 ± 0.04; KA2 = 2.3 × 104 ± 3.2 × 103 M; Gibb’s free energy (ΔG2) = 25 ± 4 kJ/mol; and a KD2 of 43 ± 6.0 µM. Many clinically used drugs have KD values similar to these (Kratochwil et al., 2002; Bocedi et al., 2004).
Fig. 2.

Measurement of JMS-053 binding to HSA by isothermal titration calorimetry. (A) Raw data for the titration of 100 μM JMS-053 with 1 mM HSA at pH 7.4 and 30.0°C showing the calorimetry responses as successive injections of HSA are added to JMS-053 in the sample cell. (B) Integrated heat profile of the calorimetric titration shown in (A) fitted to a one-site binding model. The red solid line represents the best nonlinear least-squares fit to a one-site binding model. (C) Integrated heat profile of the calorimetric titration shown in (A) fitted to a two-site model. The blue solid line represents the best nonlinear least-squares fit to a two-site binding model.
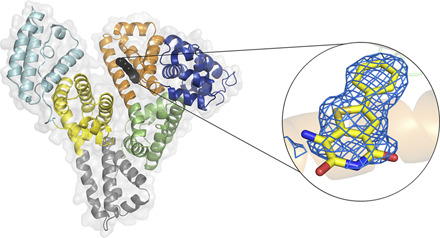
To probe further into the specific interactions between JMS-053 and HSA, we determined the structure of a complex of HSA and JMS-053 using X-ray crystallography at a resolution of 2.2 Å (Fig. 3; Table 1). HSA was cocrystallized with myristic acid, and the crystals were then soaked with JMS-053. Albumin crystals grew in the P1 space group and contained two HSA molecules in the asymmetric unit, which were nearly identical. JMS-053 bound in both copies of the protein chain to drug site 3, which is located in subdomain IB (Fig. 3A). The electron density for JMS-053 at drug site 3 was observed in several structures that were determined from data collected for different albumin crystals soaked with the compound. We selected the highest resolution data for further consideration and PDB deposition. In the structures collected for albumin crystals obtained from the same crystallization condition but not soaked with JMS-053, the analogous position in drug site 3 remains unoccupied. The phenyl and thiophene portions of JMS-053 were well ordered (Fig. 3B; Supplemental Figs. 2 and 3) and stabilized by hydrophobic interactions with the following residues: Ile142, Phe149, Leu154, Phe157, Tyr161, and aliphatic parts of Arg186 and Lys190 side chains. The pyridinedione moiety was oriented toward the solvent and appeared less ordered and more flexible. Its carbonyl groups were engaged in hydrogen bonding with the side chain nitrogen atoms of His146 and Lys190 with a distance ∼3.5 Å. Drug site 3 is known to be a fatty acid binding site. An elongated fragment of electron density observed in the vicinity of drug site 3 suggested the possible binding of a fatty acid close to the position of JMS-053. However, the molecule of myristic acid modeled at this site clashes with the JMS-053 in chain A but not in chain B. To further explore other possibilities, we decided to examine the possibility of binding of a PEG molecule at this location (25% PEG 3350 was also in the crystallization cocktail). The PEG molecule modeled at this site does not cause clashes with the JMS-053 molecule, but based on the electron density alone, we were unable to distinguish between fatty acid and PEG molecules. To avoid any misinterpretation, we decided to model a PEG molecule and label it as an unknown ligand. Most of the crystallization conditions for HSA, including ours, contain highly concentrated PEGs that may affect proper identification of bound fatty acids. Nevertheless, in the determined structure, we modeled myristic and caprylic acids in several locations previously reported to bind fatty acids, such as FA2, FA3, FA4, FA5, and FA6 (Curry et al., 1998; Simard et al., 2006).
Fig. 3.

HSA domains and JMS-053 binding site. (A) Domains are labeled with Roman numerals and subdomains with letters. Each subdomain is shown in a different color; JMS-053 at drug site 3 is shown with atoms as black spheres. (B) JMS-053 binding site with omit electron density difference map (mFo–DFc), calculated after 10 refinement cycles without a drug, RMSD: 2.5). Oxygen atoms are shown in red, nitrogen in blue, and sulfur in gold; carbon atoms of JMS-053 are shown in yellow, PEG in gray, and protein residues in orange. Gly189 is not shown. The electron density can be inspected interactively and in detail at the Molstack platform: https://molstack.bioreproducibility.org/project/view/gpt44gh1A2dDZOKWUqer/.
Drug site 3, which we are proposing as a binding site for JMS-053, is shared by other clinically used anticancer drugs, such as idarubicin and teniposide (Wang et al., 2013). Figure 4 shows the binding modes of JMS-053 and these compounds at drug site 3. Binding of JMS-053 to drug site 3 did not markedly alter the albumin structure (Fig. 5; Supplemental Fig. 4; Supplemental Table 3). We did not see any crystallographic evidence for a second JMS-053 binding site. Assuming a single binding site with a KD of 118 µM from the abovementioned isothermal titration calorimetry data, we calculated that 77.4% of the JMS-053 would be bound to albumin at the Tmax when the concentration of JMS-053 was observed to be 233.8 ng/ml (0.913 μM). This degree of albumin binding is similar to that reported for other clinically used drug molecules (Kratochwil et al., 2002; Bocedi et al., 2004).
Fig. 4.

Interactions of JMS-053 and other anticancer drugs with domain IB amino acids in HSA. (A) Binding environment of JMS-053 and teniposide. (B) Binding environment of JMS-053 and idarubicin.
Fig. 5.

(A) Superposition of crystal structures of HSA complexes with JMS-053 and with myristic acid (PDB ID: 1BJ5). Binding of JMS-053 to drug site 3 does not alter albumin’s structure. (B) Superposition of HSA complex with myristic acid (PDB ID: 1BJ5) and HSA ligand free (PDB ID: 4K2C). (C) Superposition of crystal structures of ligand free HSA (PDB ID: 4K2C) and BSA (PDB ID: 3V0C). Molecules of JMS-053, myristic acid, and PEG are shown in stick representation.
Serum Alters JMS-053 PTP4A3 Inhibition and Cytotoxicity.
We hypothesized that if JMS-053 binds to albumin, then the presence of this protein would reduce the biochemical potency of JMS-053 as an in vitro inhibitor of PTP4A3. As illustrated in Fig. 6A, increasing concentrations of BSA indeed markedly decreased the potency of JMS-053. In the absence of BSA, the in vitro JMS-053 IC50 for recombinant PTP4A3 was 28.5 ± 1.9 (S.D.) nM, consistent with our previously reported values (McQueeney et al., 2017, 2018; Tasker et al., 2019a). Inclusion of 0.1% BSA did not alter the potency of JMS-053—namely, IC50 = 25.6 ± 2.2 nM. In contrast, the JMS-053 IC50 values for samples coincubated with 1% or 5% (calculated to be ∼750 μM) BSA were 64.3 ± 2.6 and 1997 ± 5 nM, respectively, consistent with substantial JMS-053 binding to albumin (Fig. 6A). Similarly, FBS concentrations altered the EC50 cytotoxicity values for JMS-053 with human A2780 ovarian cancer cells grown as pathologically relevant spheroids (Fig. 6B). The EC50 of JMS-053 with 10% FBS was 2.6 ± 0.3 μM (95% CI: 1.9–3.4), which was different than that seen with 5% FBS (1.6 ± 0.3 μM; 95% CI: 1.4–1.8; P = 0.0002), with 1% FBS (1.1 ± 0.1 μM; 95% CI: 0.6–1.4; P = 0.0002), and with 0.5% FBS (0.98 ± 0.21 μM; 95% CI: 0.9–1.1; P = 0.0002).
Fig. 6.

Reduction in JMS-053 inhibition of PTP4A3 and ovarian cancer spheroid viability with the presence of albumin. (A) The IC50 values for JMS-053 PTP4A3 inhibition in vitro with increasing concentrations of BSA. The mean values are indicated for the four or five independent observations, which are marked as black circles. Bars indicate S.D., and P = 0.015 as determined with one-way ANOVA with Dunnett’s test for comparisons involving multiple groups. (B) Increase in A2780 ovarian cancer spheroid survival with reduction of FBS concentrations. Blue circles = 10% FBS; red squares = 5% FBS; open black squares = 1% FBS; black diamonds = 0.5% FBS. N = 3, and bars = S.D.
Discussion
HSA is a flexible protein with multiple potential drug binding sites (Sudlow et al., 1976; Wang et al., 2013; Sleep, 2015; Czub et al., 2020). HSA binding has been viewed as a liability for drug candidates because it can reduce the free drug concentration and availability in the blood, but there has been growing appreciation that HSA binding can actually be a valuable means of drug delivery (Larsen et al., 2016). HSA binding reduces drug metabolism and clearance, acts as a drug depot, and facilitates deep tissue delivery (Larsen et al., 2016). Indeed, HSA binding can be one of the major pharmacokinetic determinants for many drugs, including those used to treat cancer (Wang et al., 2013).
Albumin is an endogenous carrier of many nutrients that are required to fulfill the needs of rapidly growing cancer cells and the energy demands of the tumor microenvironment (Hoogenboezem and Duvall, 2018; Park et al., 2019). Tumors are thought to accumulate considerable quantities of albumin as a consequence of at least two mechanisms: 1) the enhanced permeability and retention effect (Hoogenboezem and Duvall, 2018) and 2) albumin-binding protein and receptor-mediated accretion (Park et al., 2019). The fundamental principle involved in the enhanced permeability and retention effect is tumor vasculature hyperpermeability, which allows albumin to extravasate and accumulate in the tumor interstitial space, which lacks fully functional lymphatic drainage. The enhanced permeability and retention effect is generally viewed as an energetically passive process. The fundamental principle involved in albumin-binding protein and receptor-mediated accretion is elevated albumin transcytosis across the tumor vascular endothelium and endocytosis into tumor cells using an assortment of albumin-binding proteins, including SPARC, megalin, calreticulin, and albondin, which also contributes to the movement of nutrients and drugs into tumors. An important role for albumin-binding proteins has been illustrated, for example, by using short hairpin RNA to reduce SPARC in human cancer cells, which attenuates HSA accumulation (Park et al., 2019). The transcytosis and endocytosis of albumin requires functional caveolae and the protein caveolin-1, which is highly expressed in many tumor types (Hoogenboezem and Duvall, 2018). The accumulation of HSA into tumors has been exploited in the creation of albumin-drug conjugates, such as the HSA-paclitaxel hybrid Abraxane, which is now clinically used (Hoogenboezem and Duvall, 2018).
We observed that JMS-053 binds preferentially to a hydrophobic groove on HSA formed by α-helices h7, h8, and h9 and an extended polypeptide domain, which is one of the most common drug binding sites on HSA (Wang et al., 2013). Albumin binding may explain the potency differences seen in vitro with PTP4A3 inhibition versus cellular migration and survival assays in which substantial FBS is present (McQueeney et al., 2017, 2018; Lazo et al., 2019). This may also explain the prolonged in vivo half-life of JMS-053 observed in mice, which is opposite to the rapid microsomal metabolism. We note that the JMS-053 binding site is located in a region of albumin that varies among different species. Nonetheless, the three residues that create a hydrophobic hot spot at this site are conserved among HSA, BSA, and MSA: Phe149, Leu154, and Tyr161. Moreover, His146, which interacts with JMS-053’s carbonyl group, is also conserved (Figs. 3 and 5; Supplemental Table 2). The four other contact residues retain the same amino acid characteristics: Ile142 is retained in BSA and modified to Val in MSA, Phe157 is replaced by Tyr in both MSA and BSA, and Arg186 is retained in BSA and exchanged for Lys in MSA (Arg186 contributes only to hydrophobic interactions with JMS-053). The exchange of Lys190 to Leu, which is observed in BSA and MSA, could potentially be an important change; however, it contributes mostly to hydrophobic interactions, like Arg186. Thus, we believe it is likely that our observations with HSA will be applicable to BSA and MSA.
As previously mentioned, our ITC results fit both a one-site and a two-site binding model with similar goodness-of-fit scores (Fig. 2). Although we only observed a single binding site by X-ray crystallography, JMS-053 could interact at two binding sites. The ITC data suggest that one of these two sites would be preferred, with a KD that is two orders of magnitude lower. It should also be noted that myristic acid was included in the HSA used for the crystallography studies because it was essential for crystal formation, but we did not include myristic acid in the ITC studies. Thus, it is possible that the lack of myristic acid could alter the binding properties of JMS-053. In addition, we cannot exclude the possibility that JMS-053 binds to other components in serum. Nevertheless, human serum albumin concentrations are normally between 530 and 750 μM, so it will be in vast excess of the characterized KD values, and a majority of JMS-053 will be bound to serum albumin. Moreover, the binding constants we observed are consistent with many clinically used drugs, which have extended plasma half-lives (Kratochwil et al., 2002; Bocedi et al., 2004).
Our structural studies define at least one major binding site of this potent inhibitor of an oncogenic protein tyrosine phosphatase inhibitor. Although there are many X-ray crystal structures of HSA bound to lipids and drugs, only a very small fraction (<2%) of the Food and Drug Administration–approved drugs have deposited cocrystal structures, and most of those are not with anticancer drugs (Czub et al., 2020). A detailed understanding of the molecular interactions between HSA and drugs, especially anticancer agents, is quite important because of the central role combination chemotherapy has in the treatment of human malignant diseases. Our work on this first-in-class tyrosine phosphatase inhibitor designed as an anticancer agent is also noteworthy for several reasons. First, computational analyses indicate JMS-053 is structurally unrelated to other known compounds (McQueeney et al., 2017). Second, our study generates hypotheses about potential drug interactions, which can be experimentally tested. Moreover, our study could help guide future synthetic advances that would fine-tune the interactions between this class of compounds and HSA. Therapeutic optimization based on HSA binding has previously been performed with anticancer agents from other chemical classes, such as the B-cell lymphoma 2 (Bcl-2) inhibitors (Oltersdorf et al., 2005). We imagine that the lead compound JMS-053 might be chemically altered to reduce HSA binding if it is formulated for intravenous or oral rather than intraperitoneal delivery, as was previously used (McQueeney et al., 2017). It is important to note, however, that even closely related chemical structures, such as etoposide and teniposide, can exhibit marked differences in the nature of their interactions with HSA (Wang et al., 2013). Thus, it will be important to determine the binding architecture of any newly synthesized analog.
Acknowledgments
We thank Changsoo Chang at Structural Biology Center for his assistance in data collection and Ivan G. Shabalin for his assistance with the crystallographic data interpretation. Results shown in this report are derived from work performed at Argonne National Laboratory (ANL), Structural Biology Center (SBC), at the Advanced Photon Source (APS) under US Department of Energy, Office of Biological and Environmental Research, contract DE-AC02-06CH11357.
Abbreviations
- ASU
asymmetric unit
- BSA
bovine serum albumin
- CI
confidence interval
- FA
fatty acid binding site
- HSA
human serum albumin
- ITC
isothermal titration calorimetry
- LC-MS
liquid chromatography-mass spectrometry
- MSA
murine serum albumin
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- PRL
protein of regenerating liver
- PTP
protein tyrosine phosphatase
- RMSD
root-mean-square-deviation
- SPARC
secreted protein, acidic, cysteine rich
- TLS
Translation/Libration/Screw
- Tmax
time required to reach maximum plasma concentration
Authorship Contributions
Participated in research design: Bushweller, Minor, Wipf, Sharlow, Lazo.
Conducted experiments: Czub, Boulton, Rastelli, Tasker, Maskrey, Blanco, McQueeney.
Contributed new reagents or analytic tools: Czub, Boulton, Rastelli, Tasker, McQueeney.
Performed data analysis: Czub, Boulton, Rastelli, Tasker, Minor, Wipf, Sharlow, Lazo.
Wrote or contributed to the writing of the manuscript: Czub, Boulton, McQueeney, Bushweller, Minor, Wipf, Sharlow, Lazo.
Footnotes
This work was supported by National Institutes of Health National Cancer Institute [Grant R21-CA191944] (to J.S.L.), [Grant R43-CA228774] (to J.S.L.), [Grant F31-CA196062] (to K.E.M.), and [Grant P30-CA044579] (to J.S.L.); the National Institute of General Medical Sciences [Grant R01-GM132595] (to W.M.); and funds from NIH National Institute of Allergy and Infectious Diseases, US Department of Health and Human Services, under contract no. [HHSN272201700060C] (to W.M.) and NIH [Shared Instrument Grant S10-OD021723] (to J.S.L.). M.P.C. was additionally supported by the Robert R. Wagner Fellowship at the University of Virginia. This work was also supported by the Department of Defense [Award W81XWH- 18-1-011, BC170507] (to P.W. and J.S.L.), the Fiske Drug Discovery Fund (to J.S.L.), the Owens Foundation (to J.S.L.), the Cure Alzheimer’s Fund (to E.R.S. and J.S.L.), and the Ivy Foundation (to J.S.L.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Bai Y, Yu ZH, Liu S, Zhang L, Zhang RY, Zeng LF, Zhang S, Zhang ZY. (2016) Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res 76:4805–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocedi A, Notaril S, Narciso P, Bolli A, Fasano M, Ascenzi P. (2004) Binding of anti-HIV drugs to human serum albumin. IUBMB Life 56:609–614. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry S, Mandelkow H, Brick P, Franks N. (1998) Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol 5:827–835. [DOI] [PubMed] [Google Scholar]
- Czub MP, Handing KB, Venkataramany BS, Cooper DR, Shabalin IG, Minor W. (2020) Albumin-based transport of nonsteroidal anti-inflammatory drugs in mammalian blood plasma. J Med Chem 63:6847–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daouti S, Li WH, Qian H, Huang KS, Holmgren J, Levin W, Reik L, McGady DL, Gillespie P, Perrotta A, et al. (2008) A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130Cas cleavage. Cancer Res 68:1162–1169. [DOI] [PubMed] [Google Scholar]
- den Hollander P, Rawls K, Tsimelzon A, Shepherd J, Mazumdar A, Hill J, Fuqua SA, Chang JC, Osborne CK, Hilsenbeck SG, et al. (2016) Phosphatase PTP4A3 promotes triple-negative breast cancer growth and predicts poor patient survival. Cancer Res 76:1942–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore S, Sanz García E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, et al. (2017) Validation of structures in the protein data bank. Structure 25:1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK, et al. (2016) A public database of macromolecular diffraction experiments. Acta Crystallogr D Struct Biol 72:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Kostantin E, Wang SJ, Hristova T, Galicia-Vázquez G, Baranov PV, Pelletier J, Tremblay ML. (2019) Magnesium-sensitive upstream ORF controls PRL phosphatase expression to mediate energy metabolism. Proc Natl Acad Sci USA 116:2925–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjort MA, Abdollahi P, Vandsemb EN, Fenstad MH, Lund B, Slørdahl TS, Børset M, Rø TB. (2017) Phosphatase of regenerating liver-3 is expressed in acute lymphoblastic leukemia and mediates leukemic cell adhesion, migration and drug resistance. Oncotarget 9:3549–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjort MA, Hov H, Abdollahi P, Vandsemb EN, Fagerli UM, Lund B, Slørdahl TS, Børset M, Rø TB. (2018) Phosphatase of regenerating liver-3 (PRL-3) is overexpressed in classical Hodgkin lymphoma and promotes survival and migration. Exp Hematol Oncol 7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Laakso LM. (2016) Dali server update. Nucleic Acids Res 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboezem EN, Duvall CL. (2018) Harnessing albumin as a carrier for cancer therapies. Adv Drug Deliv Rev 130:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowiel M, Jaskolski M, Dauter Z. (2014) ACHESYM: an algorithm and server for standardized placement of macromolecular models in the unit cell. Acta Crystallogr D Biol Crystallogr 70:3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil NA, Huber W, Müller F, Kansy M, Gerber PR. (2002) Predicting plasma protein binding of drugs: a new approach. Biochem Pharmacol 64:1355–1374. [DOI] [PubMed] [Google Scholar]
- Larsen MT, Kuhlmann M, Hvam ML, Howard KA. (2016) Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo JS, Blanco IK, Tasker NR, Rastelli EJ, Burnett JC, Garrott SR, Hart DJ, McCloud RL, Hsu KL, Wipf P, et al. (2019) Next-generation cell-active inhibitors of the undrugged oncogenic PTP4A3 phosphatase. J Pharmacol Exp Ther 371:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueeney KE, Salamoun JM, Ahn JG, Pekic P, Blanco IK, Struckman HL, Sharlow ER, Wipf P, Lazo JS. (2018) A chemical genetics approach identifies PTP4A3 as a regulator of colon cancer cell adhesion. FASEB J 32:5661–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueeney KE, Salamoun JM, Burnett JC, Barabutis N, Pekic P, Lewandowski SL, Llaneza DC, Cornelison R, Bai Y, Zhang ZY, et al. (2017) Targeting ovarian cancer and endothelium with an allosteric PTP4A3 phosphatase inhibitor. Oncotarget 9:8223–8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlot AM, Kalinowski DS, Richardson DR. (2014) Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol 5:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Murphy TJ, Motulsky HJ. (2020) New author guidelines for displaying data and reporting data analysis and statistical methods in experimental biology. Mol Pharmacol 97:49–60. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859–866. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435:677–681. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62:439–450. [DOI] [PubMed] [Google Scholar]
- Park CR, Jo JH, Song MG, Park JY, Kim YH, Youn H, Paek SH, Chung JK, Jeong JM, Lee YS, et al. (2019) Secreted protein acidic and rich in cysteine mediates active targeting of human serum albumin in U87MG xenograft mouse models. Theranostics 9:7447–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JT. (1995) All About Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, CA. [Google Scholar]
- Porebski PJ, Cymborowski M, Pasenkiewicz-Gierula M, Minor W. (2016) Fitmunk: improving protein structures by accurate, automatic modeling of side-chain conformations. Acta Crystallogr D Struct Biol 72:266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. (2002) Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet 41:751–790. [DOI] [PubMed] [Google Scholar]
- Salamoun JM, McQueeney KE, Patil K, Geib SJ, Sharlow ER, Lazo JS, Wipf P. (2016) Photooxygenation of an amino-thienopyridone yields a more potent PTP4A3 inhibitor. Org Biomol Chem 14:6398–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin IG, Porebski PJ, Minor W. (2018) Refining the macromolecular model - achieving the best agreement with the data from X-ray diffraction experiment. Crystallogr Rev 24:236–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JR, Zunszain PA, Hamilton JA, Curry S. (2006) Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J Mol Biol 361:336–351. [DOI] [PubMed] [Google Scholar]
- Sleep D. (2015) Albumin and its application in drug delivery. Expert Opin Drug Deliv 12:793–812. [DOI] [PubMed] [Google Scholar]
- Stadlbauer S, Rios P, Ohmori K, Suzuki K, Köhn M. (2015) Procyanidins negatively affect the activity of the phosphatases of regenerating liver. PLoS One 10:e0134336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow G, Birkett DJ, Wade DN. (1976) Further characterization of specific drug binding sites on human serum albumin. Mol Pharmacol 12:1052–1061. [PubMed] [Google Scholar]
- Tasker NR, Rastelli EJ, Blanco IK, Burnett JC, Sharlow ER, Lazo JS, Wipf P. (2019a) In-flow photooxygenation of aminothienopyridinones generates iminopyridinedione PTP4A3 phosphatase inhibitors. Org Biomol Chem 17:2448–2466. [DOI] [PubMed] [Google Scholar]
- Tasker NR, Rastelli EJ, Burnett JC, Sharlow ER, Lazo JS, Wipf P. (2019b) Tapping the therapeutic potential of protein tyrosine phosphatase 4A with small molecule inhibitors. Bioorg Med Chem Lett 29:2008–2015. [DOI] [PubMed] [Google Scholar]
- Thura M, Al-Aidaroos AQ, Gupta A, Chee CE, Lee SC, Hui KM, Li J, Guan YK, Yong WP, So J, et al. (2019) PRL3-zumab as an immunotherapy to inhibit tumors expressing PRL3 oncoprotein. Nat Commun 10:2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66:22–25. [DOI] [PubMed] [Google Scholar]
- Wang ZM, Ho JX, Ruble JR, Rose J, Rüker F, Ellenburg M, Murphy R, Click J, Soistman E, Wilkerson L, et al. (2013) Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim Biophys Acta 1830:5356–5374. [DOI] [PubMed] [Google Scholar]
- Wei M, Haney MG, Rivas DR, Blackburn JS. (2020) Protein tyrosine phosphatase 4A3 (PTP4A3/PRL-3) drives migration and progression of T-cell acute lymphoblastic leukemia in vitro and in vivo. Oncogenesis 9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZH, Zhang ZY. (2018) Regulatory mechanisms and novel therapeutic targeting strategies for protein tyrosine phosphatases. Chem Rev 118:1069–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaias J, Mineau M, Cray C, Yoon D, Altman NH. (2009) Reference values for serum proteins of common laboratory rodent strains. J Am Assoc Lab Anim Sci 48:387–390. [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Wang Y, Wu A, Rao Y, Huang Y. (2018) Roles of albumin-binding proteins in cancer progression and biomimetic targeted drug delivery. ChemBioChem 19:1796–1805. [DOI] [PubMed] [Google Scholar]
- Zucker F, Champ PC, Merritt EA. (2010) Validation of crystallographic models containing TLS or other descriptions of anisotropy. Acta Crystallogr D Biol Crystallogr 66:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]