

Abstract
K-ras mutations are major genetic events that drive cancer development associated with aggressive malignant phenotypes, while expression of the immune checkpoint molecule PD-L1 plays a key role in cancer evasion of the immune surveillance that also profoundly affects the patient outcome. However, the relationship between K-ras oncogenic signal and PD-L1 expressions as an important area that requires further investigation. Using both in vitro and in vivo experimental models of K-ras-driven cancer, we found that oncogenic K-ras significantly enhanced PD-L1 expression through a redox-mediated mechanism. Activation of K-rasG12V promoted ROS generation and induced FGFR1 expression, leading to a significant upregulation of PD-L1. We further showed that exogenous ROS such as hydrogen peroxide alone was sufficient to activate FGFR1 and induce PD-L1, while antioxidants could largely abrogate PD-L1 expression in K-ras mutant cells, indicating a critical role of redox regulation. Importantly, genetic knockout of FGFR1 led to a decrease in PD-L1 expression, and impaired tumor growth in vivo due to a significant increase of T cell infiltration in the tumor tissues and thus enhanced T-cell-mediated tumor suppression.
Our study has identified a novel mechanism by which K-ras promotes PD-L1 expression, and suggests that modulation of ROS or inhibition of the FGFR1 pathway could be a novel strategy to abrogate PD-L1-mediated immunosuppression and thus potentially improve the efficacy of immunotherapy in K-ras-driven cancers.
Keywords: PD-L1, K-ras, ROS, FGFR1
Highlights
-
•
Oncogenic K-Ras up-regulates PD-L1 expression in vitro and in vivo.
-
•
ROS play a major role in mediating K-Ras-induced FGFR1 activation leading to PD-L1 expression in K-Ras-driven cancers.
-
•
Antioxidants are able to modulate PD-L1 expression in K-Ras mutant cancer cells.
-
•
Suppression of FGFR1 enhances CD8+ T cell infiltration and inhibits tumor growth.
Abbreviations
- Akt (PKB)
protein kinase B
- AP-1
activator protein-1
- CAT
catalase;
- CM
conditionated medium
- DAB
3,3'-diaminobenzidine
- EGF(R)
epidermal growth factor (receptor)
- ELISA
enzyme-linked immunosorbent assay
- FGF(R)
fibroblast growth factor (receptor)
- Glox
glucose oxidase
- HEK
human embryonic kidney
- hTERT-HPNE
human pancreatic nestin expressing cells (transfected with human telomerase reverse transcriptase)
- IL
interleukin
- INF-γ
interferon gamma
- JAK
janus kinase
- K-ras
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PD-1
programmed death 1
- PDAC
pancreatic ductal adenocarcinoma
- PD-L1
programmed death ligand 1
- PI3K
phosphoinositide-3 kinase
- PTEN
phosphatase and tensin homolog
- ROS
reactive oxygen species
- STAT
signal transducer and activator of transcription
- TGF-β
transforming growth factor beta
- TNF-α
tumor necrosis factor alpha
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly malignant disease and one of the leading cause of cancer-related death around the world [1]. A better understanding of the biology of PDAC and its relationship with K-ras mutation and immune response would be important for the development of more effective therapeutic strategies. Aberrant activation of K-ras by mutation is frequently observed in human tumors, especially in pancreatic cancer where over 90% of malignant cells exhibit constitutive activation of K-ras [2]. In lung [3] and colon [4] cancer K-ras mutations are also frequent (30-40%). The presence of K-ras mutations in cancer cells is correlated with disease progression and poor outcome, due in part to activation of several downstream pathways that promote cell proliferation, cell survival,drug resistance, and invasion [5,6]. Other factors such as reactive oxygen species (ROS), which are often elevated in tumor cells [7], have also been suggested to contribute to ras-driven malignant transformation and cancer development [8]. Indeed, our previous study using a doxycycline-inducible system showed that induction of the K-rasG12V expression caused a significant increase in ROS production associated with altered metabolism and malignant cell behaviors [9]. Given the critical role of tumor immunity in cancer development and disease progression, it is important to evaluate if activation of K-ras might affect cancer immunity.
PD-L1 (programmed death-ligand 1) is expressed on the surface of many cell types including antigen-presenting cells, T cells, B cells and epithelial cells [10,11]. The interaction with its receptor PD-1 (programmed death 1) expressed on T cells is a physiologic mechanism to regulate immune function and avoid auto-immune attacks [[10], [11], [12], [13]]. Expression of PD-L1 is also detected in various types of tumors as a mechanism of immune evasion [12], and its high expression is correlated with poor clinical prognosis in cancer patients due to PD-L1/PD-1-mediated suppression of antitumor immunity [14,15]. Thus, targeting this immune checkpoint by disrupting PD-1/PD-L1 interaction is a validated antitumor strategy [16]. Antibodies against PD-1 or PD-L1 have been shown to be effective against various types of cancers [[17], [18], [19]]. Recent studies suggest that PD-L1 expression can be regulated by multiple factors including the PTEN/PI3K/Akt signaling [20], micro-RNAs [21], AP-1 [22], MAPK [23], JAK/STAT [24], and NF-κB [25]. Tumor microenvironment factors such as hypoxia [26] and cytokines such as interferon gamma (IFN-γ), transforming growth factor beta (TGF-β), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6) have also been reported to affect the expression of PD-L1 protein [21,25,[27], [28], [29]]. A study in epidermal growth factor receptor (EGFR)-driven lung cancer suggested that K-ras mutation might be associated with PD-L1 expression [30]. However, the mechanistic link between oncogenic K-ras and PD-L1 expression and its role in affecting K-ras-mediated cancer development remain unclear. The main goal of this study was to investigate the potential role of K-ras in regulation of PD-L1 expression and its underlying mechanisms.
2. Results
2.1. Activation of oncogenic K-ras induces PD-L1 expression
To test the potential role of oncogenic K-ras in regulating PD-L1 expression and its physiological relevance in vivo, we first examined the expression of PD-L1 protein in tumor tissues from K-ras-driven pancreatic cancer specimens, and observed that PD-L1 protein was highly expressed in vivo in both mouse and human pancreatic cancer tissues (Fig. 1A). We then used a doxycycline-inducible K-rasG12V expression cell system, designated as T-Rex/K-ras cells in our previous study [9], to directly test if activation of oncogenic K-rasG12V could induce PD-L1 expression. As shown in Fig. 1B, qRT-PCR assay revealed that activation of K-rasG12V caused a significant increase in PD-L1 mRNA expression in a time-dependent manner, and this increase in PD-L1 expression remained high during a long-term (2 months) K-ras induction. Consistently, there was a substantial increase in PD-L1 protein expression after K-rasG12V induction, as revealed by immunoblotting (Fig. 1C) and by flow cytometry analysis of cell surface PD-L1 (Fig. 1D). This induction was specific for PD-L1, since activation of K-rasG12V did not induce any increase in expression of its receptor PD-1 nor in the expression of another ligand PD-L2 whose mRNA levels were barely detected (Fig. 1E). Similar results were also observed in the hTERT-immortalized human pancreatic cells (HPNE), where expression of K-rasG12D did not increase the expression of PD-1 or PD-L2 (Fig. S1A). It is interesting to note that PD-1 was barely detectable by immunoblotting analysis in several pancreatic cancer cell lines (Fig. S1B), while PD-L2 protein was detected in Capan-2, SW1990 and CFPAC-1 pancreatic cancer cells (Fig. S1C).
Fig. 1.

Activation of oncogenic K-ras promotes PD-L1 expression. (A) Immunostaining of PD-L1 and H&E staining in K-ras-driven mouse (subcutaneous tumor tissues derived from KPC mice) and human pancreatic cancer cells (tissue micro-array). The scale bars represent 50 μm. (B) T-Rex/K-ras cells were incubated with 100 ng/ml doxycycline to induce K-rasG12V expression for the indicated times. PD-L1 mRNA was measured by qRT-PCR. (C) T-Rex/K-ras cells were incubated without (OFF) or with (ON) doxycycline for 72 h. K-ras and PD-L1 proteins were detected by western blotting. (D) T-Rex/K-ras cells incubated without (OFF) or with (ON) doxycycline for 72 h were stained with control antibody (IgG cont) or specific antibody against human PD-L1. Cell surface PD-L1 was analyzed by flow cytometry (representative of three separate experiments). (E) T-Rex/K-ras cells after induction by doxycycline for 72 h, and PD-1 and PD-L2 mRNA levels were quantified by qRT-PCR. Statistical analysis: Data represent means ± SEM of three separate experiments; One-way ANOVA followed by Tukey post hoc test for B; Two-tailed unpaired t-test for E. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
2.2. K-ras-induced PD-L1 expression is mediated by growth factor signaling
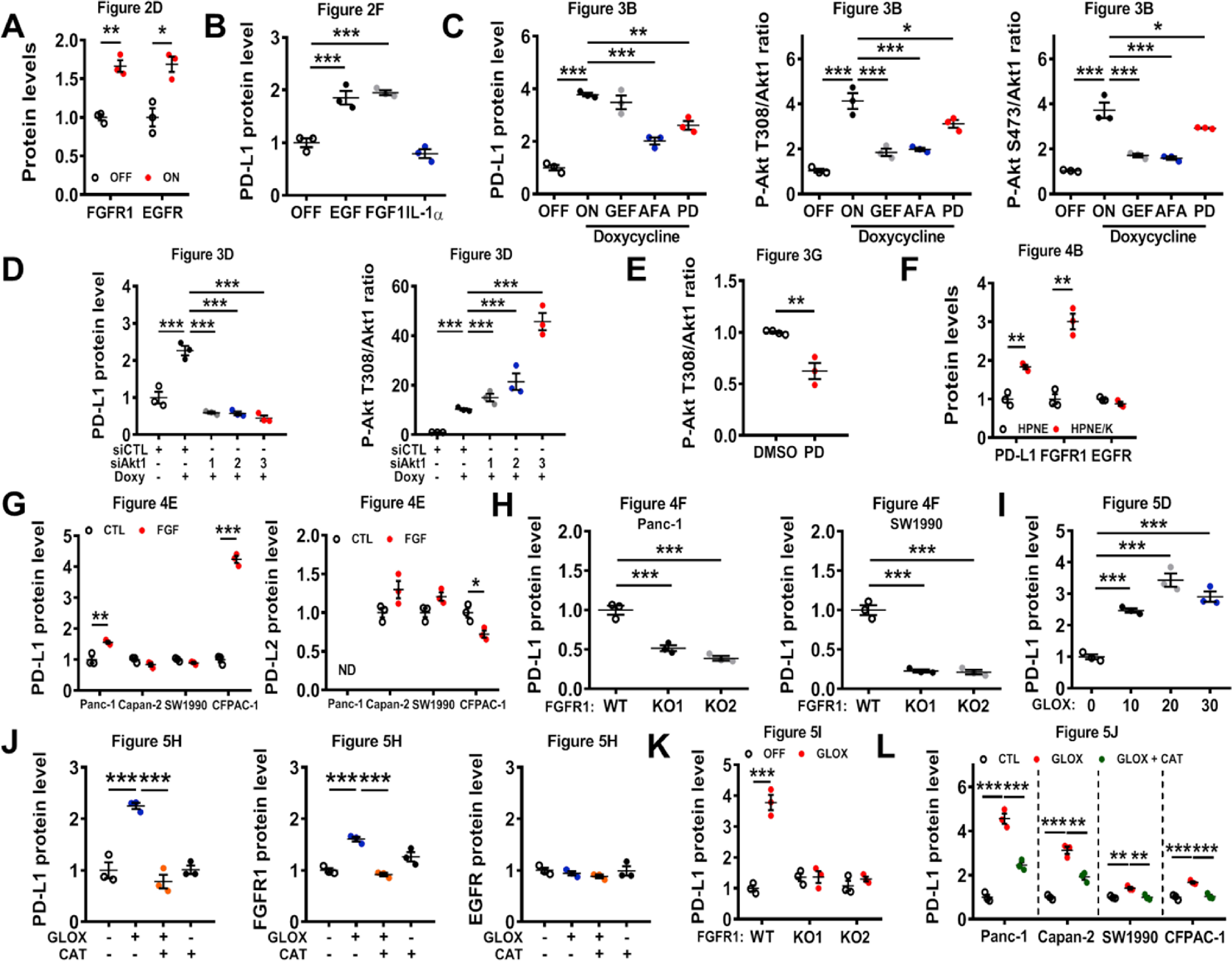
Although the role of PD-L1 in immune checkpoint is well established, the regulation of its expression by oncogenic signal remains poorly understood. Based on the recent reports that certain cytokines (IFN-γ, TGF-β, TNF-α, and IL-6) and the epidermal growth factor (EGF) in the microenvironment could stimulate PD-L1 expression [21,25,[27], [28], [29]], we postulated that some of these factors might be involved in K-ras-induced PD-L1 expression. To test this possibility, we first obtained the conditioned medium (CM) from T-Rex/K-Ras cells (which are HEK-293T cells containing the doxycycline-inducible K-ras expression vector) cultured with or without K-ras induction by doxycycline, and then added the CM to the control HEK293T cells without harboring the doxycycline-inducible expression vector. As shown in Fig. 2A, incubation of the control HEK293T cells with the CM from T-Rex/K-ras/On cells (CM/ON) led to a significant increase in PD-L1 mRNA levels, whereas the CM from T-Rex/K-ras/Off cells (CM/OFF) or doxycycline (Doxy) did not induce any increase in PD-L1 expression. These data suggest that secreted factors in the CM, regulated by oncogenic K-rasG12V, might stimulate PD-L1 expression. Indeed, qRT-PCR assay revealed that induction of K-rasG12V caused a significant increase in expression of EGF, FGF1 (fibroblast growth factor 1), IL-1α (interleukin-1α) as well as their respective receptors EGFR, FGFR1, and IL1R1A (Fig. 2B). The increased secretion of EGF, FGF1, and IL-1α into the culture medium from K-rasG12V/On cells was confirmed by ELISA assay (Fig. 2C), and the elevated expression of EGFR and FGFR1 in K-rasG12V/On cells was confirmed by Western blot analysis (Fig. 2D, Fig. S7A), further supporting an autocrine mechanism by which K-rasG12V stimulated growth factor signaling. Interestingly, when T-Rex/K-ras were incubated with each of these cytokines, EGF and FGF1 were able to induce PD-L1 expression (mRNA and protein), but IL-1α did not show any significant effect (Fig. 2E–G, Fig. S7B).
Fig. 2.

Regulation of PD-L1 expression by growth factor signaling. (A) HEK293T were incubated for 24 h with doxycycline (Doxy), or with conditioned medium (CM) from T-Rex/K-ras cells incubated with (ON) or without (OFF) doxycycline. PD-L1 mRNA was measured by qRT-PCR. (B) EGF, EGFR, FGF1, FGFR1, IL-1α and IL1R1A mRNA levels were quantified by qRT-PCR in T-Rex/K-ras cells incubated with or without doxycycline for 72 h. (C) Secretion of EGF, FGF1 and IL-1α in the culture medium of K-ras/On or Off cells, measured by ELISA. The results were normalized by protein contents of the corresponding cell samples. (D) FGFR1, EGFR and K-Ras protein levels were detected by immunoblotting in T-Rex/K-ras/On (72 h) or Off cells. Data are representative of three separate experiments. (E) T-Rex/K-ras cells were incubated with EGF (20 ng/ml), FGF1 (10 ng/ml), or IL-1α (100 ng/ml) for 24 h, and PD-L1 mRNA was quantified by qRT-PCR. (F) T-Rex/K-ras cells were incubated with EGF (20 ng/ml), FGF1 (10 ng/ml) of IL-1α (100 ng/ml) for 48 h. PD-L1 protein was detected by immunoblotting. (G) T-Rex/K-ras cells were incubated with EGF (20 ng/ml), FGF1 (10 ng/ml) or IL-1α (100 ng/ml) for 72 h, and cell surface PD-L1 was analyzed by flow cytometry. Statistical analysis: Data are means ± SEM of three separate experiments; One-way ANOVA followed by Tukey post hoc test for A, C, E, G; Two-tailed unpaired t-test for B. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
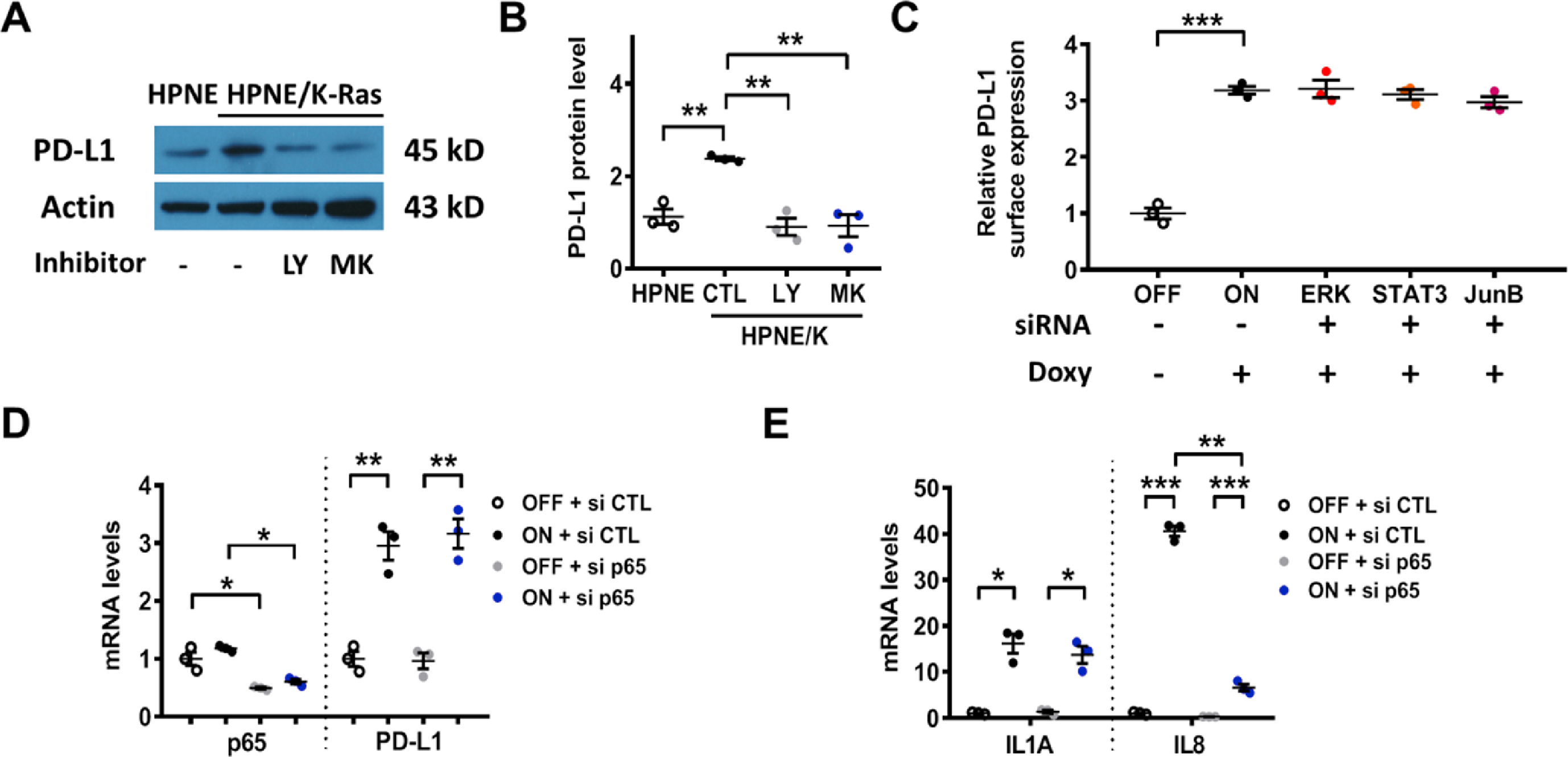
Since the ability of EGFR to affect PD-L1 expression was recently observed in lung cancer [23] but the impact of FGFR1 on PD-L1 expression has not been reported previously, specific inhibitors of EGFR and FGFR1 were used to further test their relative influence on K-rasG12V-induced PD-L1 expression. As shown in Fig. 3A, the EGFR inhibitors (Gefitinib and Afatinib) and FGFR1 inhibitor (PD173074) were able to suppress K-rasG12V-induced PD-L1 mRNA expression. Afatinib and PD173074 also decreased the K-ras-induced expression of PD-L1 protein, wheras the impact of Gefitinib on PD-L1 protein expression was not significant at 48 h (Fig. 3B–C, Fig. S7C). We also observed Akt activation by K-ras, as evidenced by an increase in phosphorylation at Ser473 and Thr308 while the total Akt protein did not increase (Fig. 3B, Fig. S7C). Consistently, EGFR or FGFR inhibitors could also suppress surface PD-L1 protein expression (Fig. 3C). This was consistent with the ability of growth factor signaling to activate Akt, which is known to promote PD-L1 expression [20]. To test the potential role of Akt in mediating K-rasG12V-induced PD-L1 expression, siRNA was used to silence the expression of Akt1, the major isoform of Akt found in HEK293T cells [31]. Silencing of Akt1 substantially suppressed PD-L1 expression in T-Rex/K-ras/On cells (Fig. 3D, Fig. S7D). Induction of K-ras by doxycycline consistently caused an increase of Akt phosphorylation (Fig. 3B, D), suggesting that K-ras activated Akt mainly through the phosphorylation of the Akt protein. The role of Akt in K-rasG12V-induced PD-L1 expression was further confirmed by the use of PI3K inhibitor LY294002 and Akt inhibitor MK-2206 (Fig. 3E–F, Figs. S2A–B). Importantly, we also found that inhibition of FGFR1 using PD173074 in mice (20 mg/kg; 48 h) could significantly reduce the levels of phosphorylated Akt and PD-L1 protein expression in vivo (Fig. 3G–H, Fig. S7E).
Fig. 3.

Effect of inhibition of EGFR and FGFR on PD-L1 expression. (A) T-Rex/K-ras cells were incubated with or without doxycycline for 48 h, then with 1 μM Gefitinib (GEF), 1 μM Afatinib (AFA), or 1 μM PD173074 (PD) as indicated for an additional 24 h. PD-L1 mRNA was measured by qRT-PCR. (B) T-Rex/K-ras cells were incubated with or without doxycycline for 48 h and then incubated with 1 μM GEF, 1 μM AFA, or 1 μM PD173074 for additional 48 h. PD-L1 protein was measured by immunoblotting. Data are representative of three separate experiments. (C) T-Rex/K-ras cells were incubated with or without doxycycline for 48 h and then incubated with 1 μM GEF, 1 μM AFA, or 1 μM PD173074 for an additional 48 h in presence of doxycycline. Cell surface PD-L1 expression was quantified by FACS analysis. (D) T-Rex/K-ras cells were first incubated with doxycycline for 48 h, then transfected with 100 nM siRNA against Akt1 for 48 h. PD-L1 protein level was measured by immunoblotting analysis. Data are representative of three separate experiments. (E-F) T-Rex/K-ras cells were incubated with 100 ng/ml doxycycline for 48 h. Then, cells were incubated with PI3K inhibitor LY294002 (5 μM) or Akt inhibitor MK2206 (50 nM) for 72 h. PD-L1 and K-ras protein levels were analyzed by immunoblotting and quantified. (G-H) C57BL/6 mice were inoculated with syngeneic pancreatic cancer cells expressing K-rasG12D (2 × 106 cells per injection). The mice were divided into 2 groups (3 mice/group). PD173074 (20 mg/kg) was administrated orally on days 7 and 8. The control mice were treated with DMSO. Tumors were harvested 6 h after the last treatment and proteins were extracted. The expression of PD-L1, p-Akt (T308) and total Akt protein was measured by immunoblotting analysis and quantified. Statistical analysis: Data are mean ± SEM of three independent experiments; One-way ANOVA followed by Tukey post hoc test for A, C and F; Two-tailed unpaired t-test for H. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Considering that other pathways including ERK (MAPK), STAT3, JunB, and NF-κB were previously suggested to affect PD-L1 expression, we tested their involvement in mediating K-rasG12V-induced PD-L1 expression. As shown in Supplementary Figs. S2C–E, siRNA silencing of these genes failed to modify PD-L1 expression in T-Rex/K-ras/On cells, suggesting that these pathways might not play a significant role in the context of K-ras-driven PD-L1 expression.
FGFR1 signaling plays a major role in K-ras-induced PD-L1 expression in pancreatic cells.
To further investigate the relative roles of EGFR and FGFR1 signaling pathways in mediating K-rasG12V-induced PD-L1 expression in human pancreatic cells, we compared the expression of PD-L1, EGFR, and FGFR1 in HPNE cells and in their K-ras-transfected counterpart (HPNE/K cells). Quantitative RT-PCR analysis showed that the expression of PD-L1, EGFR and FGFR1 mRNA increased in HPNE/K-ras cells (Fig. 4A). Unlike T-Rex/K-ras cells, the expressions of EGF and FGF1 did not change in HPNE cell lines (Fig. 4A). Western blot analysis showed that only FGFR1 protein substantially increased in HPNE/K-ras cells, whereas EGFR expression remained unchanged (Fig. 4B, Fig. S7F), suggesting that the FGFR1 signaling might be the main pathway that mediated PD-L1 expression in this cell line.
Fig. 4.

Effect of inhibition of EGFR and FGFR on PD-L1 expression in cancer cells. (A) Impact of K-ras on expression of PD-L1, EGF, EGFR, FGF1 and FGFR1 mRNA in HPNE cells, measured by qRT-PCR analysis. (B) Expression of PD-L1, FGFR1, and EGFR in HPNE cells stably transfected with K-ras (HPNE/K) in comparison with their parental HPNE cells. Protein levels were measured by immunoblotting analysis. Data are representative of three separate experiments. (C–D) Four human pancreatic cell lines were incubated with FGF1 and FGF2 (50 ng/ml). Cell surface PD-L1 and PD-L2 expressions were quantified by FACS analysis. (E) Four human pancreatic cell lines were incubated with FGF1 and FGF2 (50 ng/ml). Expressions of PD-L1 and PD-L2 were measured by immunoblotting. (F) Expressions of PD-L1 and FGFR1, in Panc-1 and SW1990 FGFR1 KO cells, were measured by immunoblotting. (G-H) PD-L1 cell surface expression in Panc-1 and SW1990 FGFR1 KO cells was quantified by FACS analysis. (I-J) Correlation between PD-L1 and FGFR1 expression in human pancreatic tumor samples (n=81) using tissue microarray. The protein expression was determined by immunohistochemistry staining. Representative images of immunostaining and H/E staining (scale bars, 50 μm) are shown in (I); the quantitative data are shown in (J). Data are means ± SEM of three separate experiments; Two-tailed unpaired t-test for A, C-D; One-way ANOVA followed by Tukey post hoc test for G-H. Spearman’s rank correlation test for J. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Interestingly, FGF and EGF were also able to regulate PD-L1 expression in Panc-1 and CFPAC-1 cells, but not in SW1990 and Capan-2 cell lines (Fig. 4C–E, Figs. S3A–B, Fig. S7G). Consistent with the observation in T-Rex/K-ras cells, PD-L2 protein was not induced when the pancreatic cells were incubated with the growth factors (Fig. 4D, Figs. S3C–E), further confirming that this regulatory process was specific for PD-L1. It should be noted that Capan-2 cells exhibited the lowest level of FGFR1 that was correlated with a very low PD-L1 protein level (Fig. S3A-B, S3G). Thus, Capan-2 cells did not respond to exogenous FGF was likely due to the intrinsic lack of FGFR1 signaling. In contrast, SW1990 cells exhibited the highest expressions of FGFR1 and PD-L1, and could not be further stimulated by exogenous FGF due to signaling saturation. Importantly, the EGFR inhibitor afatinib, which was effective in inhibiting PD-L1 expression in T-Rex/K-ras cells, was unable to suppress the expression of PD-L1 in any of the pancreatic cell lines (Fig. S3F), suggesting that it was FGFR1 (but not EGFR) that play a key role in mediating K-ras-induced PD-L1 expression in human pancreatic cancer cells.
To further validate the role of FGFR1 in regulating of PD-L1 expression, we generated FGFR1 knock-out cell lines and tested its impact on PD-L1 expression. To this end, we first selected Panc-1 and SW1990 cell lines because they exhibited a high level of basal expression of PD-L1 and FGFR1 and also secrete FGF into their medium (Fig. S3G). When FGFR1 was knocked-out, the expression of PD-L1 was significantly decreased (Fig. 4F–H, Fig. S7H). These data together demonstrated the important role of FGFR1 in the regulation of PD-L1 expression in pancreatic cancer.
We then used tissue microarray to evaluate the potential correlation between PD-L1 expression and FGFR1 in human pancreatic tumor tissues, and found that the expression of PD-L1 was positively correlated with the expression of FGFR1 (Fig. 4I-J), suggesting that the regulation of PD-L1 expression by FGFR1 could occur in vivo. TCGA analyses also revealed a positive correlation between these two genes in human pancreatic ductal adenocarcinoma (PDAC), but only when K-ras is mutated (Fig. S4A). Such correlation was not observed in pancreatic cancer samples with wild-type K-ras (Fig. S4B).
2.3. ROS mediate K-ras-induced PD-L1 expression through activation of FGFR1 pathway
Considering that K-rasG12V promotes ROS generation due in part to mitochondrial dysfunction induced by the oncogenic signal [7,9], we thus tested the possibility that ROS might play a role in the K-rasG12V-induced activation of growth factor signaling for PD-L1 expression. We first examined the direct effect of ROS on PD-L1 expression, and found that exposure of K-rasG12V/Off cells to 50 μM hydrogen peroxide (H2O2) could induce a significant increase of PD-L1 (Fig. 5A). Since the exogenous H2O2 added to cell culture had a very short half-life, we then used glucose oxidase (GLOX), an enzyme known to oxidize glucose in the medium to produce H2O2 [32], as a continuous ROS-generating system to evaluate the impact of chronic ROS stress on PD-L1 expression. As shown in Fig. 5B, addition of GLOX (20 ng/ml) to the culture medium led to a significant increase in cellular ROS in K-rasG12V/Off cells. This ROS stress resulted in an up-regulation of PD-L1 mRNA and protein (Fig. 5C–D, Fig. S7I). Importantly, the antioxidant enzyme catalase (CAT) could decrease ROS in K-rasG12V/On cells to a level comparable to that of K-rasG12V/Off cells (Fig. 5E), and prevented K-rasG12V-induced PD-L1 upregulation (Fig. 5F). Since exogenously added glucose oxidase and catalase were unlikely to cross cell membranes, their direct effect was to alter extracellular H2O2 and create a concentration gradient across the cellular membranes to facilitate the diffusion of H2O2 [33], leading to changes in intracellular H2O2 concentrations. GLOX also enhanced PD-L1 expression on the cell surface and catalase, an enzyme that converts H2O2 to water and oxygen [34], could reverse this process (Fig. 5G). Moreover, incubation with GLOX also increased FGFR1 protein levels correlated with PD-L1 expression (Fig. 5H, Fig. S7J). Catalase was able to abolish the up-regulation of FGFR1 and PD-L1 induced by GLOX, indicating that H2O2 is a key regulator. Since EGFR protein level did not increase after cells were incubated with GLOX, the effect of H2O2 on PD-L1 expression was mainly through FGFR1 signaling in these cells. We also noted that addition of exogenous EGF or FGF1 in the medium did not cause any ROS increase in HEK293T cells (Fig. S5A), indicating that ROS were upstream of the growth factor activation.
Fig. 5.

Role of ROS in regulation of PD-L1 expression. (A) T-Rex/K-ras/Off cells were incubated with hydrogen peroxide (50 μM, added daily) for 72 h, and PD-L1 mRNA was quantified by qRT-PCR. (B) T-Rex/K-ras cells were incubated with or without glucose oxidase (GLOX, 20 ng/ml) for 24 h, ROS were measured by FACS analysis using DCFDH-DA probe. Data are representative of three separate experiments. (C) T-Rex/K-ras cells were incubated for 48 h with GLOX (20 ng/ml, added daily). PD-L1 mRNA was measured by qRT-PCR. (D) T-Rex/K-ras cells were incubated with GLOX for 48 h (added daily). PD-L1 expression was measured by immunoblotting. (E) T-Rex/K-ras cells were incubated for 72 h without (OFF) or with doxycycline (ON) in the presence or absence of catalase (CAT, 50 μg/ml). ROS were measured by FACS analysis. (F) T-Rex/K-ras cells were incubated with or without doxycycline and catalase (50 μg/ml) as indicated, and PD-L1 mRNA was measured by qRT-PCR. (G) T-Rex/K-ras cells were incubated with GLOX (20 ng/ml, added daily) for 48 h and catalase (50 μg/ml) as indicated, and cell surface PD-L1 was quantified by FACS analysis. (H) T-Rex/K-ras cells were incubated with GLOX (20 ng/ml, added daily) with or without catalase (50 μg/ml) for 48 h. EGFR, FGFR1 and PD-L1 were measured by immunoblotting. Data are representative of three separate experiments. (I) T-Rex/K-ras FGFR1 wild-type (WT) and FGFR1 KO cells were incubated for 48 h with GLOX (20 ng/ml, once a day). PD-L1 and FGFR1 were measured by immunoblotting. Data are representative of three separate experiments. (J) Four pancreatic cell lines were incubated with H2O2 (0.75–1 mM, added once) for 48 h. PD-L1 expression was measured by immunoblotting. Data are representative of three separate experiments. (K-L) C57BL/6 mice were inoculated with syngeneic pancreatic cancer cells expressing K-rasG12D (2 × 106 cells per injection). The mice were divided into the control group (treated with PBS) and NAC-treated group (1000 mg/kg, i.p. daily, for 7 days). PD-L1 mRNA levels were measured by qRT-PCR. PD-L1 protein expression was analyzed by IHC and scored as described in methods. Data are means ± SEM of seven biological replicates. Statistical analyses: Data are means ± SEM of three separate experiments; Two-tailed unpaired t-test for A, C, K-L; one-way ANOVA followed by Tukey post hoc test for F-G. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further test the important role of FGFR1 signaling pathway in mediating ROS-induced PD-L1 expression, we used CRISPR/Cas9 technology to abolish FGFR1 gene in K-rasG12V/Off cells, and then tested the effect of ROS on PD-L1 expression. As shown in Fig. 5I, ROS generated by GLOX could increase PD-L1 expression only in the K-rasG12V/Off cells with normal FGFR1, but this effect was almost completely abolished when FGFR1 was knocked-out (Fig. 5I, Fig. S7K).
In order to test if this redox regulation of PD-L1 is a general mechanism, various human cell lines were incubated with hydrogen peroxide. All four human pancreatic cancer cells showed an increase in their PD-L1 expression after exposure to oxidative stress (various concentrations of H2O2), and catalase could again abolish this PD-L1 induction (Fig. 5J, Figs. S5B–E, Fig. S7L). In contrast, PD-L2 protein levels did not change when the cancer cells were incubated with H2O2 (Fig. S5F). When the cells were treated with high concentrations of H2O2 (0.75–1.0 mM, added once), there was a moderate cytotoxicity with a loss of up to 25% viable cells and their proliferation ability, as measured by MTT assay at 48 h. It is noteworthy that in Capan-2 cells with low FGFR1 protein level, PD-L1 expression could not be induced by FGF (Fig. 4E) but could be enhanced after exposure to H2O2 (Fig. 5J), suggestion that ROS might also promote PD-L1 expression in a FGFR1-independent manner.
Consistent with above observations, incubation of HPNE cells with the ROS-generating enzyme GLOX also induced PD-L1 expression at both mRNA and protein levels (Figs. S6A–C), which were prevented by catalase (Figs. S6D–F). The expression level of FGFR1 was also significantly enhanced by GLOX, whereas that of EGF and EGFR remained unchanged (Fig. S6G). These data together revealed a novel role of ROS in mediating upregulation of PD-L1 expression via activation of the FGFR1 signaling. Moreover, hydrogen peroxide also increased PD-L1 protein levels in mouse CT26 colon cancer cells (Fig. S6H) and mouse pancreatic cancer cells (Fig. S6I). Likewise the antioxidant N-acetyl cysteine (NAC) decreased the expression of PD-L1 in vitro (Fig. S6I) and in vivo in the mouse pancreatic cancer model expressing mutated K-ras (Fig. 5K-L, Fig. S6J).
To further test the hypothesis that ROS regulate PD-L1 expression, we generated Nrf2 knockout cells from T-Rex/K-ras cells with compromised antioxidant capacity and elevated ROS [35]. The expression of PD-L1 was significantly higher in the Nrf2-KO cells compared to wild-type Nrf2 cells (Figs. S5G–H), consistent with the role of ROS in promoting PD-L1 expression.
2.4. FGFR1 knock-out differentially impacted tumor growth in immune competent and immunodeficient mice
In order to further evaluate the role of FGFR1 in promoting PD-L1 expression in vivo, we used CRISPR/Cas9 technology to generate FGFR1 knockout cells from the mouse pancreatic cancer KPC cell line (K-rasG12D/p53R172H) (Fig. 6A). In line with the findings obtained with human FGFR1 KO cell lines (Fig. 4F–H), a knockout of FGFR1 in mouse PDAC cells also led to a decrease in PD-L1 expression in vitro (Fig. 6B). Importantly, a knockout of FGFR1 severely impaired the ability of the KPC cells to grow tumor in the immune competent C57BL/6 mice (Fig. 6C). In contrast, the same KPC cells grew faster when they were inoculated in the immunodeficient nude mice, while knocking out of FGFR1 only moderately retarded tumor growth in the nude mice (Fig. 6D). These data indicate that the mouse immune system played a significant role in inhibiting KPC tumor growth, and suppression of PD-L1 expression by knocking out FGFR1 further enhanced the immune function against cancer growth. The moderate retardation in tumor growth observed with FGFR1-null cells in the nude mice (Fig. 6D) likely reflected the loss of FGFR1 signaling. Analysis of tumor tissues isolated for the immune competent mice revealed that the PD-L1 expression decreased by approximately 30% in FGFR1 KO tumors (Fig. 6E). Since tumor PD-L1 expression may impact T cell function, we then quantified CD8+ T cell infiltration in the tumor tissues. Compared to the wild-type FGFR1 tumors, the FGFR1 KO1 and FGFR1 KO2 tumors contained a significantly higher number of infiltrating T cells with an increase of 1.85-fold and 1.41-fold, respectively (Fig. 6F). Consistently, the expression of effector T cell marker genes such as interferon-gamma, granzyme B and perforin were significantly increased in FGFR1 KO tumors (Fig. 6G–I). These data together suggest that the knockout of FGFR1 in K-ras-driven cancer cells could decrease the expression of PD-L1 in vivo, promote immune response and thus inhibit tumor growth.
Fig. 6.

Increased CD8+ T cell infiltration and impaired tumor growth in FGFR1 KO tumors. (A) Expression of FGFR1, in mouse PDAC FGFR1 WT and KO cells, was measured by immunoblotting. (B) In vitro comparison of PD-L1 cell surface expression in mouse PDAC cells and their FGFR1-KO cells, quantified by FACS analysis. (C) C57BL/6 mice were inoculated with syngeneic pancreatic cancer cells (FGFR1 WT or KO; n=7) expressing K-rasG12D (2 × 106 cells per injection). Tumor size data (means ± SD) were log-transformed before statistical analyses using generalized linear model. (D) BALB/c nude mice were inoculated with syngeneic pancreatic cancer cells (FGFR1 WT or KO; n=7) expressing K-rasG12D (1 × 106 cells per injection). Tumor size data (means ± SD) were log-transformed before statistical analyses using generalized linear model. (E) C57BL/6 mice were inoculated with syngeneic pancreatic cancer cells as described in (C). The mice were sacrificed at day 21 after tumor inoculation. PD-L1 protein expression was evaluated by IHC and scored as described in methods. (F) Tumor tissues were processed for immunostaining of CD8+ T cells (same tumor samples than E). The number of CD8+ lymphocytes was quantified in each group. (G-I) Quantitative RT-PCR analysis of IFNγ (IFNG), Granzyme B (GZMB) and Perforin (PRF1B) mRNA expression in tumor tissues from mice (same tumor samples than E). Statistical analysis: Data are mean ± SEM of three independent experiments (B) or three biological replicates (E–I); One-way ANOVA followed by Tukey post hoc test for B, E-I. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
3. Discussion
Pancreatic ductal adenocarcinoma is one of the most deadly cancers [1], and the presence of K-ras mutation is a major molecular hallmark of this disease. Oncogenic mutations of KRAS gene are frequently found in multiple cancers, and are associated with more aggressive disease phenotype [2,5]. A previous study of EGFR-driven cancer suggested an association between K-ras mutation and PD-L1 expression in lung cancer [30]. In lung cancer, PD-L1 expression seems to be associated with oncogenic K-ras through MAPK [36,37] or Akt signaling [38]. Another study suggested that oncogenic K-ras might enhance PD-L1 expression by increasing its mRNA stability via the modulation of the tristetraprolin enzyme activity [39]. However, the causal role of K-ras in regulating PD-L1 expression still remained elusive. Using multiple experimental models including an inducible K-rasG12V expression cell system [9], we demonstrated that activation of oncogenic K-ras in cancer cells could induce PD-L1 expression through activation of FGFR1 signaling by a novel ROS-mediated mechanism as illustrated in Fig. 7. This conclusion is supported by both in vitro and in vivo data, including induction of ROS generation and expressions of FGFR1 and PD-L1 by K-rasG12V, direct activation of FGFR1 and PD-L1 expression by ROS, and suppression of PD-L1 by antioxidants or by the FGFR1 inhibitor (PD173074) in vitro and in vivo. Genetic silencing of the expression of ERK, STAT3, AP-1, and NF-κB did not significantly affect the expression of PD-L1, suggesting that these putative regulators did not play a significant role in K-rasG12V-induced PD-L1 expression. In contrast, genetic knockout of FGFR1 significantly decreased the expression of PD-L1 and profoundly suppressed tumor growth in immunocompetent mice. These data together demonstrate that the ROS-FGFR1 pathway seems to be the main regulatory mechanism for PD-L1 expression in K-ras-driven cancers. Neither the expression of PD-1 nor PD-L2 was changed by K-ras activation, suggesting that this regulation is relatively specific for PD-L1. However, it should be noted that the regulatory mechanisms of PD-L1 expression are likely multifactorial processes, and may depend on cell types. Cytokines such as IFN-γ, IL-6 and growth factors could stimulate PD-L1 expression, and a recent study suggests that Akt signaling pathway may also be involved in regulation of PD-L1 expression in non-small cell lung cancer [38].
Fig. 7.

Schematic model illustatrating the redox regulation of PD-L1 expression through growth factor signaling. Activation of K-rasG12V promotes ROS generation due to K-ras-induced mitochondrial dysfunction. The elevated ROS promotes FGFR1 expression at the transcriptional level. The signals from growth factor receptors (FGFR1 and EGFR) activate the Akt signaling pathway, which enhances the transcription of PD-L1 gene, leading to a significant increase of PD-L1 mRNA and protein. The subsequent interaction of PD-L1 with PD-1 on T cells lead to immunosuppression.
Interestingly, the expression of both PD-L1 and FGFR1 are increased in human pancreatic cancer tissues [[40], [41], [42]]. The results from our tissue microarray and TCGA analyses further showed that PD-L1 expression was correlated with FGFR1 expression in tumor tissues with mutant K-ras, suggesting that the regulation of PD-L1 expression by FGFR1 in K-ras-driven cancer is clinically relevant. Consistent with our findings, a strong correlation between FGF2-FGFR1/Akt3/PD-L1 exists in invasive bladder carcinomas in a TCGA analysis [43]. The discovery that ROS could promote PD-L1 expression through activation of growth factor signaling is a significant new finding from this study. Several lines of evidences demonstrate that ROS-modulating agents may also lead to PD-L1 upregulation in cancer cells [44]. It is also worthy of noting that although our data showed that ROS could activate both EGFR and FGFR1 signaling pathways, it seems that FGFR1 signaling plays a key role in upregulation of PD-L1 expression in the K-ras-driven pancreatic cancer cells. This notion is supported by the observations that FGFR1 expression was increased in K-ras-transformed pancreatic epithelial cells (HPNE/K) whereas the EGFR expression was similar in both cell lines. The redox regulation of FGFR1 has been already reported. It has been shown that hydrogen peroxide could regulate FGFR1 expression at the transcriptional level in retinal pigment epithelial cells while FGFR2 expression was not modified under oxidative stress [45], suggesting a specific induction of FGFR1 by ROS. However, the detail mechanism for redox-mediated transcriptional regulation of FGFR1 remains to be elucidated. Beside the effect on gene transcription, ROS could directly modulate the receptor activation. Indeed, it has been shown that oxidative stress could activate the receptor by phosphorylation [46], and could also increase the affinity of FGF for its receptor [47]. The finding that inhibition of FGFR1 (but not EGFR) signaling could suppress K-ras-induced PD-L1 expression further underscores the important role of the FGFR1 pathway, although EGFR signaling is known to induce PD-L1 expression in other cell types such as lung cancer [23,38].
Our study provided several lines of evidence for the significant role of oncogenic K-ras in affecting tumor microenvironment and tumor immunity by upregulating PD-L1 expression and inflammatory cytokines. ROS are generally associated with tumor aggressiveness by activating cell proliferation, migration and invasion. However, the impact of ROS on tumor immunity is still underappreciated. In this study, we demonstrated that ROS/FGFR1 signaling could have a major impact on the immune functions against tumors. The ROS-mediated FGFR1 signaling could promote PD-L1 expression, which in turn alters T cell functions in the tumor tissues. The critical role of ROS and FGFR1 in regulating PD-L1 expression and affecting tumor growth has been demonstrated both in vitro and in vivo in our study. It is of particular interest to note that knockout of FGFR1 severely impaired the ability of the mouse pancreatic cancer cells to form tumors in the immune competent mice, but only moderately retard tumor growth in the immunodeficient nude mice.
4. Conclusions
We have identified a novel mechanism by which K-ras promotes PD-L1 expression through ROS-mediated FGFR1 signaling, and suggests that modulation of ROS or inhibition of the FGFR1 pathway could abrogate PD-L1-mediated immunosuppression. Our results provide a basis for developing novel strategies to overcome cancer immune evasion.
Funding
This work was supported in part by grants from the National Key R&D Program of China (2018YFC0910203), and from the National Natural Science Foundation of China (No.81430060).
Authorship
(1) C.G., X.X., Y-G.H., Y.H., K.C., A.R., Ju.L., F.W., Ji.L., P.J.C. and P.H.: the conception and design of the study, or acquisition of data, or analysis and interpretation of data, (2) C.G., X.X. and P.H.: drafting the article or revising it critically for important intellectual content, (3) C.G. and P.H.: final approval of the version to be submitted.
5. Material and methods
Cell lines: The doxycycline inducible T-Rex/K-rasG12V cells were constructed as previously described [9] and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% tetracycline-free fetal bovine serum (FBS). Nrf2-knockout (KO) cells were generated from T-Rex/K-rasG12V cells as previously described [35]. HEK293T (#CRL-3216), Panc-1 (#CRL-1469), Capan-2 (#HTB-80), SW1990 (#CRL-2172), CFPAC-1 (#CRL-1918), Jurkat (#TIB-152), CT26.WT (#CRL-2638) and the h-TERT immortalized HPNE cell line (#CRL-4023) were from ATCC (Manassas, VA, USA). They were cultured in DMEM with 10% FBS, except for Jurkat and CT26 cultured in RPMI-1640 (Roswell Park Memorial Institute) medium with 10% FBS. HPNE stably transfected with mutant K-rasG12V was cultured as previously described [48]. The mouse pancreatic cancer cell line with K-ras and p53 mutations was derived from KPC mice generated by crossing Pdx1-Cre, LSL-K-rasG12D and LSL-p53R172H mice (from The Jackson Laboratory), according to the procedures described previously [49]. HPNE cell lines were initially mycoplasma positive and treated adequately in order to remove the bacteria. Other cell lines were confirmed to be mycoplasma negative (LookOut mycoplasma PCR detection kit, Sigma) and authentication of cell lines were performed by STR genotyping (Microread Genetics, Beijing, China).
Cell culture reagents: Cells were incubated with various compounds: Doxycycline (#D9891), LY294002 (#L9908), bovine serum catalase (#C9322), glucose oxidase (#G7141), N-acetyl cysteine (#V900429) and hydrogen peroxide (#323381) were from Sigma-Aldrich (Saint Louis, MO, USA). MK-2206 (#S1078), Gefitinib (#S1025), Afatinib (#S1011) and PD173074 (#S1264) were from Selleck Chemicals (Houston, TX, USA). Recombinant human EGF (#BMS320) and IL-1α (#BMS328) were from eBioscience (San Diego, CA, USA). Recombinant human FGF1 (#RP-8639) and FGF2 (#PHG0261) were from Invitrogen (Grand Island, NY, USA).
Glucose oxidase and catalase in cell culture: Glucose oxidase (Sigma, #G7141) was first prepared in PBS at a concentration of 0.2 mg/mL and then was filtered to remove potential bacterial contamination. This solution was diluted 100 times in PBS (concentration: 2 μg/mL) before adding to cell culture medium. Catalase (Sigma, #C9322) was prepared in PBS at a concentration of 1 mg/mL, filtered, and added to the cell culture medium at the indicated concentrations. All solutions were freshly prepared and added to the cell culture every 24 h. The stability of GLOX in culture medium has not been well characterized.
Mice and tumor models: Cohorts of 5–7 weeks old female C57BL/6 and BALB/c nude mice (Vital River Laboratory Animal Technology, Beijing, China) were housed in a controlled environment with free access to food and water. Upon delivery, mice underwent an acclimation period of one week. Body weight was recorded twice weekly. At the beginning of each experiment, mice were randomly assigned to each group to ensure that all groups were matched in terms of body weight, tumor sizes and by drawing lots. The animal experiments were performed in an unblinded manner. Approximately 1-2x106 pancreatic cancer cells (FGFR1 WT or KO) driven by mutant K-ras and mutant p53 (derived from KPC mouse) were injected into the right flank of mice. Tumor volumes were measured twice per week and calculated using a modified ellipsoid formula: tumor volume (mm3) = (length (mm) x width2 (mm))/2. Animals, whose tumors were ulcerated, length ≥20 mm or moribund, were promptly sacrificed to minimize animal distress and suffering. In order to investigate the role of ROS in the regulation of PD-L1 expression in vivo, KPC-bearing mice (as described above) were treated with N-acetyl cysteine (NAC, 1000 mg/kg/daily; i.p.) for seven days. KPC-bearing mice were also treated with PD173074 (20 mg/kg/daily; orally) for two days to study the role of FGFR1 signaling. All animal experiments were conducted in accordance with the institutional guidelines and approved by the Animal Care and Use Committee of Sun Yat-sen University Cancer Center (L102012016010E).
Quantitative reverse transcription-polymerase chain reaction: Total RNA was isolated using Trizol (Invitrogen) according to the manufacturer’s instructions. RNA was reverse-transcribed using Primer Script RT reagent Kit with gDNA Eraser (Takara BIO INC, Kusatsu, Shiga, Japan). Real-time PCR was performed using the SYBR Premix Ex Taq RNAse H+ kit (Takara), and analyzed using the Bio-Rad detection system (Bio-Rad, Hercules, CA, USA). The samples were first incubated 5 min at 95 °C, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The results were calculated (formula: 2-(Ct target-Ct EF1)) and matched to the control samples. The primers sequences were listed in Supplementary Table S1, and produced by Sangon Biotech (Shanghai, China).
Immunoblotting: The procedures for protein sample preparation from cell cultures, protein quantification, immunoblotting and data analyses were performed as previously described [50]. The following antibodies were used for immunoblotting analyses: PD-L1 (#ab174838), EGFR (#ab52894) and β-actin (#ab6276) were from Abcam (Cambridge, UK); K-ras (#sc-30) and Akt1 (#sc-5298) were from Santa-Cruz biotechnology (Santa Cruz, CA, USA); FGFR1 (#9740), phospho-Akt Thr 308 (#2965s) and phospho-Akt Ser 473 (#4060p) were from Cell signaling (Beverly, MA, USA). PD-1 (#AF1086-SP) was from R&D systems (Minneapolis, MN, USA). PD-L2 (#MABC969) was from Millipore (Darmstadt, Germany). Protein bands were detected by chemiluminescence, using an ECL detection kit (Pierce, Thermo Scientific, Rockford, IL, USA). When appropriate, bands obtained via Western blot analysis were quantified, using the ImageJ software (http://rsb.info.nih.gov/ij/). Protein expression was normalized by β-actin of the respective samples.
ELISA: Secretions of EGF, FGF1 and IL-1α in medium were measured by ELISA (enzyme-linked immunosorbent assay) kits according to the protocol provided by the manufacturers. EGF (#BMS2070INST) and IL-1α (#BMS243-2) ELISA kits were from eBioscience; FGF1 (#ab219636) ELISA kit was from Abcam.
Cell transfection: The small interfering RNAs (siRNAs) against Akt1, ERK2, STAT3, JunB and p65 (RelA) were synthesized by RiboBio (Guangzhou, China). Sequences of oligonucleotides are listed in Table S1. Cells were incubated with doxycycline to induce K-ras expression for 48 h before siRNA transfection, using lipofectamine RNAi Max reagents according to the manufacturer (Invitrogen). Transfection was performed for 24 h with a 100 nM siRNA solution in presence of doxycycline. Assays for expression of the target molecules were performed 72 h after the transfection.
Generation of FGFR1 knock-out cells: To product lentiviruses, 2.8 μg of plasmid psPAX2 (Addgene, Watertown, MA, USA; #12260), 1.6 μg of pMD2.G vector (Addgene, #12259) and 2 μg of Lenticrispr V2 plasmid (Addgene, #52961) containing human or mouse FGFR1 sgRNA were co-transfected into HEK293T cells (1.4 million cells) cultured in T-25 flasks using X-tremeGENE HP transfection reagent (Roche, Basel, Switzerland) according to manufacturer’s protocol. The sgRNA sequences are listed in Table S1. After 60 h, the medium containing viruses was collected and centrifuged for 10 min at 3000 rpm at 4 °C and then passed through a 0.45 μm filter. Cells were seeded in 6-well plate and infected with viruses and 8 μg/ml of polybrene containing medium. Viruses were removed 24 h after infection and cells were selected with 1–2 μg/ml puromycin (Invivogen, San Diego, CA, USA). Clones were selected by serial dilution and FGFR1 expression was detected by immunoblotting.
Flow cytometry: For the detection of membrane PD-L1 or PD-L2, cells were fixed with 4% formaldehyde in PBS and stained with primary antibodies with a dilution of 1:100 for 2 h at room temperature. Rabbit anti-human PD-L1 antibody (#13684; Cell Signaling), rat anti-mouse PD-L1 (#11-9971-81; eBioscience), mouse anti-human PD-L2 (#MABC969; Millipore) were used. Cells were then washed and incubated for 30 min at room temperature with PBS containing anti-rabbit IgG (#11-4839; eBioscience), anti-rat IgG (#11-4811-85; eBioscience) or anti-mouse IgG (#ab6785; Abcam) antibody coupled with FITC. Cells were then collected and washed twice with PBS before flow cytometry analysis.
For ROS detection, cells were incubated with 10 μM DCFDH-DA (Molecular Probes, Rockford, IL, USA) for 20 min. The cells were harvested, washed twice with PBS, and analyzed by flow cytometer (Gallios; Beckman Coulter, Brea, CA, USA). For each experiment, at least 10,000 cells per sample were analyzed using FlowJo software (https://www.flowjo.com).
Immunohistochemistry: Mouse tumor tissue sections or human pancreatic cancer tissue microarrays (Shanghai Outdo Biotech, China) were first dried at 58 °C for 1 h, dewaxed and rehydrated before epitope-retrieval by heating at 100 °C in 10 mM sodium-citrate (pH6.0) for 4 min. The sections were cooled down to room temperature for 30 min. To eliminate the endogenous peroxidase and alkaline phosphatase activity in the tissue, the tissue sections were treated with 3% hydrogen peroxide for 20 min. The sections were then incubated with the individual primary antibodies overnight, followed by incubation with secondary antibodies for 1 h. DAB (3,3'-diaminobenzidine) was then applied as a substrate to reveal the antigen. Hematoxylin was used for counterstaining. Primary antibodies used in this study included rabbit anti-PD-L1 (#ab174838, Abcam), rabbit anti-human FGFR1 (#9740, Cell Signaling) and rabbit anti-mouse CD8 (#bs-0648R; Bioss, Woburn, MA, USA). All other reagents were from ZSGB-Bio (Beijing, China). Scoring of the immuno-stained tissue sections was performed in a blind fashion, and recorded as score 0 (no target protein staining), score 1 (low staining), score 2 (intermediate staining), and score 3 (high staining). Results were quantified by multiplying the percentage of positive cells by the staining intensity scores (0-3), with a maximum score of 300 (3-x100). CD8+ lymphocytes and tissue surface were measured using ImageJ software on randomly chosen 20× fields per section.
Bioinformatics: Illumina HiSeq_RNASeqV2 RSEM normalized gene expression profiles for human pancreatic adenocarcinoma were retrieved from TCGA’s Pan-Cancer atlas (paad_tcga_pan_can_atlas_2018) by CGDS-R package. 168 samples having expression data for PD-L1, FGFR1 and the mutation status of KRAS gene were included for further study.
Statistical analysis: All experiments were performed at least three times (3 separate repeats). Q-Q plots were used to compare and determine data distribution. Data are expressed as mean ± S.E.M., unless otherwise specified. Student t-tests were used to evaluate the statistical significance of the difference between two groups of samples with normal distributions. When there were more than two independent groups, ANOVA were used to compare the means if normal distribution could be assumed. Tukey post hoc tests were performed when ANOVA was significant. Despite a large sample size, the relationship between PD-L1 and FGFR1 expression in human pancreatic carcinoma tissues was assessed using a Spearman’s rank correlation because of the nature of data (integer scores). When the standard deviation was increasing with the mean, as for tumor volumes (in vivo animal studies), data were log-transformed before conducting statistical analyses. The log-transformed tumor volume data were analyzed using a generalized linear model, with cell type (FGFR1 WT or KO) as a 3-level between group factor, with time as a within group factor, and with their interaction. Post-hoc comparisons of groups two by two were performed using a Bonferroni correction for P-values. Statistical analyses were performed with GraphPad Prism (San Diego, CA, USA) and SPSS 16.0 (Chicago, IL, USA) software. No statistical method was used to calculate sample sizes, which were determined empirically. No data were excluded. All tests were two-tailed, and a P-value of 0.05 or less was considered statistically significant.
Availability of data and materials
The key raw data have been deposited into the Research Data Deposit (http://www.researchdata.org.cn), with the Approval Number of RDDB2020000996 and the datasets used in this study are publicly available.
Declaration of competing interest
The authors declare that they have no competing interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101780.
Contributor Information
Christophe Glorieux, Email: christophe@sysucc.org.cn.
Peng Huang, Email: huangpeng@sysucc.org.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Bardeesy N., DePinho R.A. Pancreatic cancer biology and genetics. Nat. Rev. Canc. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 3.Herbst R.S., Heymach J.V., Lippman S.M. Lung cancer. N. Engl. J. Med. 2008;359:1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abankwa D., Gorfe A.A., Hancock J.F. Mechanisms of Ras membrane organization and signalling: ras on a rocker. Cell Cycle. 2008;7:2667–2673. doi: 10.4161/cc.7.17.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsushita J., Lambeth J.D., Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Canc. Res. 2004;64:3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 7.Szatrowski T.P., Nathan C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Canc. Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 8.Irani K., Goldschmidt-Clermont P.J. Ras, superoxide and signal transduction. Biochem. Pharmacol. 1998;55:1339–1346. doi: 10.1016/s0006-2952(97)00616-3. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y., Lu W., Chen G., Wang P., Chen Z., Zhou Y. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H., Zhu G., Tamada K., Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 11.Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H., Strome S.E., Salomao D.R., Tamura H., Hirano F., Flies D.B. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 13.Dong H., Zhu G., Tamada K., Flies D.B., van Deursen J.M., Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327–336. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 14.David R. PD-L1 expression by circulating breast cancer cells. Lancet Oncol. 2015;16:e321. doi: 10.1016/S1470-2045(15)00074-1. [DOI] [PubMed] [Google Scholar]
- 15.Gao Q., Wang X.Y., Qiu S.J., Yamato I., Sho M., Nakajima Y. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Canc. Res. 2009;15:971–979. doi: 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- 16.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J., Robert L. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borghaei H., Paz-Ares L., Horn L., Spigel D.R., Steins M., Ready N.E. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brahmer J.R., Tykodi S.S., Chow L.Q., Hwu W.J., Topalian S.L., Hwu P. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsa A.T., Waldron J.S., Panner A., Crane C.A., Parney I.F., Barry J.J. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 21.Gong A.Y., Zhou R., Hu G., Li X., Splinter P.L., O'Hara S.P. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J. Immunol. 2009;182:1325–1333. doi: 10.4049/jimmunol.182.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green M.R., Rodig S., Juszczynski P., Ouyang J., Sinha P., O'Donnell E. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin. Canc. Res. 2012;18:1611–1618. doi: 10.1158/1078-0432.CCR-11-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen N., Fang W., Zhan J., Hong S., Tang Y., Kang S. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J. Thorac. Oncol. 2015;10:910–923. doi: 10.1097/JTO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 24.Wolfle S.J., Strebovsky J., Bartz H., Sahr A., Arnold C., Kaiser C. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011;41:413–424. doi: 10.1002/eji.201040979. [DOI] [PubMed] [Google Scholar]
- 25.Kondo A., Yamashita T., Tamura H., Zhao W., Tsuji T., Shimizu M. Interferon-gamma and tumor necrosis factor-alpha induce an immunoinhibitory molecule, B7-H1, via nuclear factor-kappaB activation in blasts in myelodysplastic syndromes. Blood. 2010;116:1124–1131. doi: 10.1182/blood-2009-12-255125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J., Hamrouni A., Wolowiec D., Coiteux V., Kuliczkowski K., Hetuin D. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- 28.Song S., Yuan P., Wu H., Chen J., Fu J., Li P. Dendritic cells with an increased PD-L1 by TGF-beta induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int. Immunopharm. 2014;20:117–123. doi: 10.1016/j.intimp.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Tamura H., Ishibashi M., Yamashita T., Tanosaki S., Okuyama N., Kondo A. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. 2013;27:464–472. doi: 10.1038/leu.2012.213. [DOI] [PubMed] [Google Scholar]
- 30.Akbay E.A., Koyama S., Carretero J., Altabef A., Tchaicha J.H., Christensen C.L. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Canc. Discov. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santi S.A., Lee H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Cell Physiol. 2010;298:C580–C591. doi: 10.1152/ajpcell.00375.2009. [DOI] [PubMed] [Google Scholar]
- 32.Bankar S.B., Bule M.V., Singhal R.S., Ananthanarayan L. Glucose oxidase--an overview. Biotechnol. Adv. 2009;27:489–501. doi: 10.1016/j.biotechadv.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Antunes F., Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- 34.Glorieux C., Calderon P.B. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017;398:1095–1108. doi: 10.1515/hsz-2017-0131. [DOI] [PubMed] [Google Scholar]
- 35.Shao J., Glorieux C., Liao J., Chen P., Lu W., Liang Z. Impact of Nrf2 on tumour growth and drug sensitivity in oncogenic K-ras-transformed cells in vitro and in vivo. Free Radic. Res. 2018;52:661–671. doi: 10.1080/10715762.2018.1462494. [DOI] [PubMed] [Google Scholar]
- 36.Chen N., Fang W., Lin Z., Peng P., Wang J., Zhan J. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017;66:1175–1187. doi: 10.1007/s00262-017-2005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sumimoto H., Takano A., Teramoto K., Daigo Y. RAS-Mitogen-Activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PloS One. 2016;11 doi: 10.1371/journal.pone.0166626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lastwika K.J., Wilson W., 3rd, Li Q.K., Norris J., Xu H., Ghazarian S.R. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Canc. Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 39.Coelho M.A., de Carne Trecesson S., Rana S., Zecchin D., Moore C., Molina-Arcas M. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–10899 e6. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng L., Huang D., Liu J., Qian Y., Deng J., Li D. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J. Canc. Res. Clin. Oncol. 2008;134:1021–1027. doi: 10.1007/s00432-008-0364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobrin M.S., Yamanaka Y., Friess H., Lopez M.E., Korc M. Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas. Canc. Res. 1993;53:4741–4744. [PubMed] [Google Scholar]
- 42.Wang L., Ma Q., Chen X., Guo K., Li J., Zhang M. Clinical significance of B7-H1 and B7-1 expressions in pancreatic carcinoma. World J. Surg. 2010;34:1059–1065. doi: 10.1007/s00268-010-0448-x. [DOI] [PubMed] [Google Scholar]
- 43.McNiel E.A., Tsichlis P.N. Analyses of publicly available genomics resources define FGF-2-expressing bladder carcinomas as EMT-prone, proliferative tumors with low mutation rates and high expression of CTLA-4, PD-1 and PD-L1. Signal Transduct Target Ther. 2017;2 doi: 10.1038/sigtrans.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailly C. Regulation of PD-L1 expression on cancer cells with ROS-modulating drugs. Life Sci. 2020;246:117403. doi: 10.1016/j.lfs.2020.117403. [DOI] [PubMed] [Google Scholar]
- 45.Alizadeh M., Wada M., Gelfman C.M., Handa J.T., Hjelmeland L.M. Downregulation of differentiation specific gene expression by oxidative stress in ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 2001;42:2706–2713. [PubMed] [Google Scholar]
- 46.Rao G.N. Protein tyrosine kinase activity is required for oxidant-induced extracellular signal-regulated protein kinase activation and c-fos and c-jun expression. Cell. Signal. 1997;9:181–187. doi: 10.1016/s0898-6568(96)00139-8. [DOI] [PubMed] [Google Scholar]
- 47.Herbert J.M., Bono F., Savi P. The mitogenic effect of H2O2 for vascular smooth muscle cells is mediated by an increase of the affinity of basic fibroblast growth factor for its receptor. FEBS Lett. 1996;395:43–47. doi: 10.1016/0014-5793(96)00998-2. [DOI] [PubMed] [Google Scholar]
- 48.Zhuang Z., Ju H.Q., Aguilar M., Gocho T., Li H., Iida T. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-kappaB activation. Clin. Canc. Res. 2016;22:1432–1444. doi: 10.1158/1078-0432.CCR-14-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Canc. Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 50.Glorieux C., Sandoval J.M., Fattaccioli A., Dejeans N., Garbe J.C., Dieu M. Chromatin remodeling regulates catalase expression during cancer cells adaptation to chronic oxidative stress. Free Radic. Biol. Med. 2016;99:436–450. doi: 10.1016/j.freeradbiomed.2016.08.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The key raw data have been deposited into the Research Data Deposit (http://www.researchdata.org.cn), with the Approval Number of RDDB2020000996 and the datasets used in this study are publicly available.