

Abstract

Reaction of the Pt(II) complexes [PtMe2(pbt)], 1a, (pbt = 2-(2-pyridyl)benzothiazole) and [PtMe(C^N)(PPh2Me)] [C^N = deprotonated 2-phenylpyridine (ppy), 1b, or deprotonated benzo[h]quinoline (bhq), 1c] with benzyl bromide, PhCH2Br, is studied. The reaction of 1a with PhCH2Br gave the Pt(IV) product complex [PtBr(CH2Ph)Me2(pbt)]. The major trans isomer is formed in a trans oxidative addition (2a), while the minor cis products (2a′ and 2a″) resulted from an isomerization process. A solution of Pt(II) complex 1a in the presence of benzyl bromide in toluene at 70 °C after 7 days gradually gave the dibromo Pt(IV) complex [Pt(Br)2Me2(pbt)], 4a, as determined by NMR spectroscopy and single-crystal XRD. The reaction of complexes 1b and 1c with PhCH2Br gave the Pt(IV) complexes [PtMeBr(CH2Ph)(C^N)(PPh2Me)] (C^N = ppy; 2b; C^N = bhq, 2c), in which the phosphine and benzyl ligands are trans. Multinuclear NMR spectroscopy ruled out other isomers. Attempts to grow crystals of the cycloplatinated(IV) complex 2b yielded a previously reported Pt(II) complex [PtBr(ppy)(PPh2Me)], 3b, presumably from reductive elimination of ethylbenzene. UV–vis spectroscopy was used to study the kinetics of reaction of Pt(II) complexes 1a–1c with benzyl bromide. The data are consistent with a second-order SN2 mechanism and the first order in both the Pt complex and PhCH2Br. The rate of reaction decreases along the series 1a ≫ 1c > 1b. Density functional theory calculations were carried out to support experimental findings and understand the formation of isomers.
Introduction
There is a great current interest in the chemistry of cyclometalated organometallic compounds on the basis of their applications in stoichiometric and catalytic organic synthesis,1−6 optoelectronic devices,7−9 therapeutic agents,10−12 chemical sensors,13 and luminescent probes for biomolecules because of their photophysical properties.14−16 Investigations of the cycloplatinated compounds have given rise to many interesting complexes and new mechanistic insights into their reactions.17−20 Cyclometalation proceeds by the reaction of Pt(II) precursor complexes, [Pt(aryl)2(SMe2)2] or [PtMe2(μ-SMe2)]2, with ligands such as 2-phenylpyridine or benzo[h]quinoline.17,21−28
The C–halide bonds, along with other C–X bonds with large electronegativity differences, are considered as polar substrates in the oxidative addition reactions.29,30 Among distinct mechanisms suggested for C–X bond activation, the SN2 mechanism is quite common. Because oxidative addition reactions are key steps in many catalytic reactions, they are extensively investigated.17,31−35 Although the kinetics and mechanism of organic halide addition to organoplatinum(II) complexes of the general formula [PtR2(NN)] (R = Me or aryl and NN = 2,2′-bipyridine or 1,10-phenanthroline) are well established,30,34,36,37 related reactions with cycloplatinated(II) complexes have been less studied.29,38−40
C–C bond-forming reductive elimination is well recognized as the last step in many catalytic cycles used in organic synthesis.41−44 Such processes have been studied extensively including Pt(IV) complexes.45−49 Although there are some reports on C–C bond formation from the organoplatinum (IV) complex involving Me–Me or acyl-Me,34,46,50−53 a limited number of studies on the intramolecular C–C reductive elimination from Pt(IV) complexes, especially C–C benzyl-methyl reductive elimination, are reported.52 Goldberg and Crumpton reported C–C reductive elimination reactions from Pt(IV) complexes.54 Also, we have recently reported on homocoupling of benzene.6
Because our interests lie in the reactivity of cycloplatinated complexes, here, we show that although the cycloplatinated(II) complex [PtMe(C^N)(PPh2Me)] (C^N = deprotonated 2-phenylpyridinine (ppy), 1b, or deprotonated benzo[h]quinoline (bhq), 1c) reacts with benzyl bromide, PhCH2Br, to give exclusively the trans addition product, the previously reported complex [PtMe2(pbt)],551a, (pbt = 2-(2-pyridyl)benzothiazole) reacts uniquely with PhCH2Br to give a mixture isomeric products. The reactivity of Pt(II) centers in these complexes as a nucleophile toward PhCH2Br is compared. Our kinetic and mechanistic study suggests that the reactions proceed through a bimolecular SN2 pathway. The resulting cycloplatinated(IV) complex [PtMeBr(CH2Ph)(ppy)(PPh2Me)] undergoes a benzyl-Me C–C bond-forming reductive elimination to give the cycloplatinated(II) complex [PtBr(ppy)(PPh2Me)]. The experimental findings are also computationally investigated, and the optimized structures of the possible transition states and intermediates were determined.
Results and Discussion
Synthesis and Characterization of the Complexes
The routes to prepare the new organoplatinum(IV) complexes are described in Scheme 1.
Scheme 1. C–Br Oxidative Addition and C–C Bond Formation Reductive Elimination at Platinum Complexes.

The reaction of a solution of [PtMe(C^N)(PPh2Me)] (1b; C^N = ppy, 1c; C^N = bhq) with benzyl bromide at room temperature gave the complexes [PtMeBr(CH2Ph)(C^N)(PPh2Me)] (2b; C^N = ppy, 2c; C^N = bhq) in good yields through oxidative addition of PhCH2Br. Both complexes were characterized using multinuclear 1H, 31P, and 13C NMR spectroscopy (see Table 1) and elemental analysis. The characteristic signal in the 1H NMR spectrum of [PtMeBr(CH2Ph)(ppy)(PPh2Me)], 2b (see Figure 1), is the methylene protons of the benzylplatinum group, which are diastereotopic and appeared as two doublets of doublets at δ = 2.77 and 3.88 ppm. In the 13C NMR spectrum of 2b, the signal for the C atom of the Me group in PPh2Me as a doublet appeared in a low field at δ = 11.2 with 1J(PC) and 2J(PtC) values of 29 and 16 Hz, respectively, indicating that they are located trans to the C atom of the benzyl group. A doublet with Pt satellites at δ = 31.6 was assigned to the C atom of CH2 in the benzyl group, which was further confirmed by DEPT 13C NMR analysis (see Figure 1). The coupling constants mentioned above are typical values for Pt(IV) complexes.44,56−60
Table 1. NMR Data (Chemical Shifts in ppm and J in Hz) for Pt(IV) Complexes.

n.r. = Not resolved.
Figure 1.

(A) 1H (aliphatic region), (B) 31P, (C) 13C (aliphatic region), and (D) DEPT 13C (aliphatic region) NMR spectra of complex 2b in CDCl3.
Attempts to grow suitable crystals of the Pt(IV) complex [PtMeBr(CH2Ph)(ppy)(PPh2Me)], 2b, for a single-crystal X-ray diffraction experiment in solvents such as acetone and benzene were not successful. During the crystallization process in CH2Cl2/hexane at room temperature, a benzyl-Me C–C bond reductive elimination from cycloplatinated(IV) complex 2b occurred to give the complex [PtBr(PPh2Me)(ppy)], 3b, whose structure was confirmed by single-crystal analysis. It should be mentioned that 3b had been prepared by direct reaction of the Pt(II) complex [Pt(ppy)(PPh2Me)(CF3COO)] with NaBr.52 The suggested mechanism for this process is depicted in Scheme 2.50,52,61,62
Scheme 2. Suggested Mechanism for Benzyl-Me C–C Bond Reductive Elimination from 2b.

In the reaction of 1a with PhCH2Br, the product [PtBr(CH2Ph)Me2(pbt)] was shown to be a mixture containing all three possible isomers, 2a (PhCH2 being trans to Br), 2a′ (the PhCH2 ligand being trans to the N ligating atom of the benzothiazole group), and 2a″ (the PhCH2 ligand being trans to the N ligating atom of the pyridyl group). When the reaction was performed for 2 h, the ratio of the three isomers 2a/2a′/2a″ was found to be equal to 77:17:6, while after 24 h, the ratio changed to 90:10:0, indicating that the isomers 2a′ (significantly) and 2a″ (completely) returned back to isomer 2a, being both the kinetic and thermodynamic products. Our density functional theory (DFT) calculations (see the Theoretical Investigation of the Suggested Mechanisms section) are consistent with these experimental results. The trend of stability follows 2a > 2a′ > 2a″.
In the 1H NMR spectrum of the isomer mixture of [PtBr(CH2Ph)Me2(pbt)] (see Table 1 and Figure 2), the major isomer 2a displays two doublets at δ = 2.90 and 3.11 ppm for the PtCH2 diastereotopic protons, as observed for similar compounds.37,60 For the minor isomer 2a′, the two singlet signals at δ = 1.71 and 1.98 are assigned to the two different methyl ligands being trans to Br and N of the pyridyl ligand, respectively. The isomer 2a′ shows two doublets for diastereotopic protons of the methylene group at δ = 2.87 and 3.10. In the 1H NMR of isomer 2a″, two singlets at δ = 1.65 and 1.93 are assigned to the Me ligands trans to Br and N of the benzothiazole ligand, respectively.
Figure 2.

(A) 1H (aliphatic region) NMR spectrum of the complex [PtBr(CH2Ph)Me2(pbt)] in CDCl3. The peak labeled # is due to water of the CDCl3 solvent. The signals for the methylene group of isomer 2a″ were overlapped by those for isomers 2a and 2a′ and not resolved.
In the 13C NMR spectrum (see Table 1 and Figure 3), for 2a, two singlets with Pt satellites are observed for the two different methyl groups directly connected to Pt, trans to N of pyridyl and benzothiazole rings.55 The 1J(PtC) values (see Table 1) are smaller than the corresponding values reported for the methyl groups of complex 1a (1J(PtC) = 807 Hz and 844 Hz),55 confirming the oxidation of Pt(II) to Pt(IV) by benzyl bromide. The CH2 of benzyl appears at δ = 22.8 and was confirmed by DEPT 13C analysis.632a′ also shows the two singlet resonances for two different methyl ligands being trans to N of the pyridyl ligand and Br. Similar to 2a, the isomer 2a′ displays a singlet C atom of CH2, which was confirmed by DEPT 13C NMR experiments. The resonances for isomer 2a″ were not resolved in the 13C NMR spectrum.
Figure 3.

Selected aliphatic regions of the 13C NMR spectrum of [PtBr(CH2Ph)Me2(pbt)] in CDCl3. Assignments are given on the spectrum. The resonances for isomer 2a″ were not resolved. The peak labeled # is due to the diethyl ether solvent used for purification.
It should be noted that the reaction of complex 1a with excess of benzyl bromide in toluene at 70 °C for 6 days gave the dibromoplatinum(IV) complex [Pt(Br)2Me2(pbt)], 4a. Its 1H NMR spectrum and X-ray crystal structure (Figure S1) are reported in the Supporting Information.
Kinetic Study
The kinetic study of oxidative addition reactions of Pt(II) complexes 1a–c with PhCH2Br (and in one case with MeI in order to study the effect of alkyl halide on the rate of reaction) was carried out in acetone (and toluene to investigate the solvent effect). The rate of the oxidative addition reactions was studied by dissolving complexes 1a–c in the selected solvent followed by rapid mixing with a known excess of PhCH2Br. Typical examples of the observed spectral changes and kinetic traces (Abs–time curves) are shown in Figures 4 and 5, respectively. The kinetic traces recorded for these reactions displayed excellent fits to eq 1 (for the pseudo-first-order condition) or eq 2 (for the 1:1 stoichiometric condition).
Figure 4.

Changes in the UV–visible spectrum during the reaction of [PtMe(PPh2Me)(ppy)], 1b, (3 × 10–4 M) with PhCH2Br in acetone(A) and [PtMe2(pbt)], 1a, (3 × 10–4 M) with PhCH2Br in acetone (B) and in toluene (C) at 25 °C.
Figure 5.

(A) Abs–time curves for the reaction of [PtMe2(pbt)], 1a, (3 × 10–4 M) with PhCH2Br (0.006–0.015 M, concentration increases reading downward) in toluene at 25 °C. (B) Plots of first-order rate constants (kobs/s–1) versus concentration of benzyl bromide for the reaction of (a) [PtMe2(pbt)], 1a, with PhCH2Br in toluene; (b) [PtMe(PPh2Me)(ppy)], 1b, with MeI in acetone; (c) [PtMe(PPh2Me)(ppy)], 1b, with PhCH2Br in acetone; and (d) [PtMe(PPh2Me)(bhq)], 1c, with PhCH2Br in acetone at T = 40 °C.
The activation parameters (enthalpy and entropy of activation) were obtained from the temperature dependence of k2 by applying Eyring plots (see Figure 6), and the kinetic data are collected in Table 2. The large negative values of entropy of activation for the reactions studied in the present work are typical of oxidative addition by a common SN2 mechanism29,30,64,65 which involves nucleophilic attack of the Pt center at the methylene group of PhCH2Br and the formation of a five-coordinate cationic intermediate (see the next section).
Figure 6.

Eyring plots for the reactions of (a) [PtMe2(pbt)], 1a, with PhCH2Br, in acetone; (b) [PtMe2(pbt)], 1a, with PhCH2Br, in toluene; (c) [PtMe(PPh2Me)(ppy)], 1b, with MeI in acetone; (d) [PtMe(PPh2Me)(bhq)], 1c, with PhCH2Br in acetone; and (e) [PtMe(PPh2Me)(ppy)], 1b, with PhCH2Br in acetone.
Table 2. Second-Order Rate Constantsa and Activation Parameters for the Reactions of [PtMe2(pbt)], 1a, [PtMe(PPh2Me)(ppy)], 1b, and [PtMe(PPh2Me)(bhq)], 1c, with PhCH2Br in Acetone.
|
k2/L mol–1 s–1 at different
temperatures |
||||||||
|---|---|---|---|---|---|---|---|---|
| complex | λmax/nm | 10 °C | 20 °C | 25 °C | 30 °C | 40 °C | ΔH‡/kJ mol–1 | ΔS‡/J Kmol–1 |
| 1ab | 520, (562) | 7.66, (0.38) | 10.44, (0.47) | 11.95, (0.63) | 14.29, (0.75) | 21.47, (1.17) | 22.5 ± 1.7, (31.5 ± 1.1) | –148 ± 6, (−143 ± 4) |
| 102 k2/L mol–1 s–1 at different temperatures | ||||||||
| 15 °C | 20 °C | 25 °C | 30 °C | 40 °C | ||||
| 1bc | 358 | 0.10 | 0.15, [3.75] | 0.20, [5.05] | 0.31, [6.55] | 0.51, [11.00] | 45.9 ± 2.0, [38.2 ± 0.4] | –142 ± 7, [−141 ± 2] |
| 1c | 390 | 0.51d | 0.83e | 0.31 | 0.39 | 0.61 | 35.8 ± 1.7 | –172 ± 6 |
Estimated errors in k2 values are ±5%.
Values in parenthesis are for toluene.
Values in brackets are for MeI.
At 35 °C.
At 45 °C.
The order of the reaction rates of PhCH2Br with Pt(II) complexes in acetone is 1a ≫ 1c > 1b. The slightly observed increase in rate on going from [PtMe(PPh2Me)(ppy)], 1b, to [PtMe(PPh2Me)(bhq)], 1c, by a factor of about 1.5 is probably due to more electron releasing character of the bhq ligand as compared to that of the ppy ligand, which makes the Pt(II) center in 1c more electron rich than the Pt(II) center in 1b, toward oxidative addition reactions. The same behavior has been reported for the reactions of H2O2,66 PhI(OAc)2,45 and MeI67 with complexes 1b and 1c. The rate of reaction of dimethylplatinum(II) complex 1a with PhCH2Br was considerably higher than those obtained for complexes 1b and 1c. For example, at 25 °C, the values of k2 are 11.95 and 0.31 × 10–2 L mol–1 s–1, respectively, for complexes 1a and 1b. Thus, 3 orders of magnitude difference (103×) can be attributed to the presence of an extra methyl group (a very strong σ-donor) in 1aversus an electron-withdrawing phosphine ligand in 1b and 1c.
As shown in Table 2, the rates of reaction of complex 1a in toluene were some 18–20 times slower than those in acetone. We found that probably because the intermediate is ionic, as expected in the classical SN2 mechanism, reactions are sensitive to the solvent polarity, and consequently, the reactions are faster in acetone than toluene.
The rates of reaction of complex 1b with MeI in acetone are faster than those with PhCH2Br. Two factors can explain this observation. First, the halogen effect in which the rate of the reactions decreases in the order I > Br (generally ascribed to “iodide being a better leaving group than bromide”).68 Second, the effect of the R group. In SN2 reactions, the rate of reaction is dependent on the steric of the R group. The bulkier R group (in this instant, benzyl vs Me) has a slower reaction rate.69 With due attention to the R group and the halogen effect, the Pt(II) complex 1b must react faster with MeI as compared to PhCH2Br. Also, given that these reactions follow second-order kinetics with remarkable reproducibility and the fact that radical scavengers did not affect the rate would rule out the possibility of any radical mechanism.30,70 Thus, the abovementioned observations strongly suggest an SN2 mechanism of oxidative addition of the benzyl bromide to Pt(II) complexes 1a–1c. Also, the large negative entropies of activation are typical values for oxidative addition by the SN2 mechanism (see Table 2). It should be noted that PhCH2Br at 25 °C reacted nearly 1.3 times slower with the unsymmetric benzothiazol complex 1a (k2 = 11.95 L mol–1 s–1) than the symmetric 2,2′-bipyridine (bpy) derivative, [PtMe2(bpy)]37 (k2 = 15.60 L mol–1 s–1). The same mechanism for the reaction of benzyl bromide with [PtMe2(bpy)] has been suggested and supported by the kinetic isotope effect.37
Theoretical Investigation of the Suggested Mechanisms
To get more insight into the suggested mechanism and perform a reliable molecular modeling of the new Pt complexes containing the pbt ligand, a suitable DFT method should be used. DFT calculations have proven to be useful methods for calculations of structures of transition-metal complexes.71−73 Among methods and basis sets used for metal complexes, the B3LYP/6-31G(d) level (LANL2DZ potential for Pt) is shown to be a good candidate between accuracy and CPU time of calculations,74−76 and therefore, they have been used for mechanistic study and structural optimizations of the Pt complexes.45,77,78 The reactions studied in the present work, shown in Scheme 1, have been considered with an emphasis on the differences between the Pt complexes (1a, 1b, and 1c), alkyl halides (MeI and PhCH2Br), and the reaction medium, solvent. The suggested mechanism is presented in Scheme 3.
Scheme 3. Suggested Mechanisms for Oxidative Addition of Complexes 1a–1c with PhCH2Br.

The reaction of PhCH2Br with the Pt(II) complex 1a in acetone is initiated by nucleophilic attack37,69,79 by the 5dz2 HOMO of 1a on the σ* LUMO of benzyl bromide (Figure 7) to give transition state TSa. The most significant changes in bond distances of TSa are computed for the Br–CH2 and Pt–CH2 bonds. The computed bond length of 2.108 Å for Br–CH2Ph increases to 2.589 Å in TSa, while the Pt–CH2 distance decreases from far apart in the reactants to 2.788 Å. The formation of TSa is followed by completely breaking the Br–CH2 bond and forming the Pt–CH2 bond, to give intermediate IMa, which can abstract bromide to form the isomer 2a or undergo pseudorotation to give IMa′ and IMa″. The coordination of bromide to these intermediates can then give isomers 2a′ and 2a″ with an octahedral geometry. As expected, the bond lengths of the starting cycloplatinated(II) complex 1a are shorter than those of the corresponding Pt(IV) complex 2a. For example, the Pt–Npy and Pt–Nbz bonds in 1a are shorter (2.200 and 2.250 Å, respectively) than those in 2a (with the values of 2.280 and 2.308 Å, respectively). The energy barrier for the formation of TSa in acetone is calculated by DFT to be 23.8 kJ mol–1 (see Figure 8), which is in excellent agreement with the experimental value of 22.5 kJ mol–1 (see Table 2). The oxidative addition of similar Pt(II) complexes had been also performed using DFT calculations by us and others.33,34,38,66,80−82 For example, the computed energy barrier for oxidative addition of [PtMe2(bpy)] with benzyl bromide was found to be 23.0 kJ mol–1,69 which is lower than the calculated value of 1a. This is in agreement with experimental finding where the rate of the reaction of benzyl bromide with 1a is lower than that for [PtMe2(bpy)].
Figure 7.
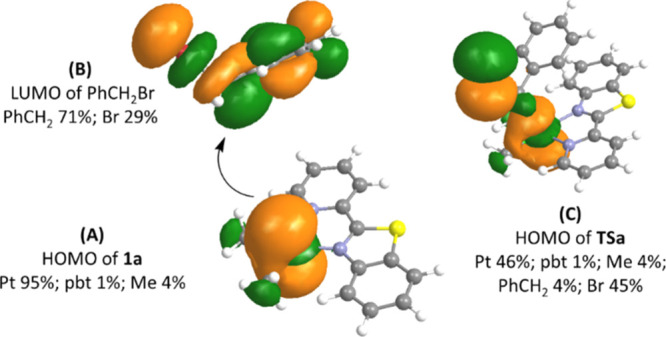
Initiation of SN2 oxidative addition by interaction of HOMO of 1a (A) and LUMO of PhCH2Br (B) to form TSa (C). The main compositions (%) of the relevant frontier orbitals of species are also shown. The calculations were performed at the B3LYP/6-31G(d)-LANL2DZ level. See Figure S2 for qualitative frontier molecular orbitals for 1a, TSa, and 2a.
Figure 8.

Enthalpy profile for oxidative addition of 1a with PhCH2Br in acetone at the B3LYP/6-31G(d)-LANL2DZ level. The optimized structures of the species involved in the reactions are shown. The summation of the energies of the 1a and PhCH2Br was considered to be zero, and the other energy levels vary relative to this.
There are, in principle, seven possible isomers for the complex [PtBr(CH2Ph)Me2(pbt)]. We can quickly delete four of the possible isomers, that is, those having two C atoms in trans positions to one another, because we would have seen only one resonance at the same chemical shift for the two Pt–Me groups (when they are in trans arrangement) or two resonances for the two Pt–Me groups with very different coupling constants (one trans to the PhCH2 group and another one cis because of different trans influences of C and other atoms) in the 1H NMR, whereas only two signals with close 2J(PtH) values (see Figure 2) are observed. A series of DFT calculations was performed on the remaining three isomers, 2a, 2a′, and 2a″, depicted in Figure 8 with their relative energies. The enthalpy values obtained from the calculations show the order 2a < 2a′ < 2a″ with the lowest lying isomer being that the larger PhCH2 group is located in the axial position as compared with the equatorial position in 2a′ and 2a″ isomers. This agrees with NMR experimental findings where the product ratio of 2a/2a′/2a″ is 77:17:6.
The reaction of 1a with PhCH2Br was also computationally investigated in toluene with lower polarity compared to acetone to understand the effect of the solvent on the energy barrier of oxidative addition reaction. The calculated enthalpy of activation in toluene is 40.5 kJ mol–1 (in good agreement with the experimental value of 45.9 kJ mol–1). This solvent effect once again is consistent with an SN2 mechanism.
It was experimentally found that complex 1b reacts with MeI faster than benzyl bromide (see Table 2). As shown in Figure 9, the computational investigations show that the ΔH‡ for the oxidative addition reactions of complex 1b with PhCH2Br and MeI in acetone are 41.2 and 36.1 kJ mol–1, respectively. These observations are also consistent with the experimental values of 45.9 and 38.2 kJ mol–1. As explained above, one possible interpretation for the higher energy barrier observed for PhCH2Br versus MeI is that in SN2-type reactions, the rate of reaction is dependent on the sterics of the R group: the bulkier the R group, the slower the reaction. Also, the iodide is a better leaving group than bromide.
Figure 9.

Enthalpy profile for oxidative addition of 1b with (A) PhCH2Br and (B) MeI in acetone at the B3LYP/6-31G(d)-LANL2DZ level. The optimized structures of the species involved in the reactions are shown. The summation of the energies of 1b and PhCH2Br or MeI was considered to be zero, and the other energy levels vary relative to this.
Experimental Section
General Remarks
1H, 13C, and 31P NMR spectra in CDCl3 were recorded using a Bruker Ultrashield 400 spectrometer (with TMS or 85% H3PO4 as references). The chemical shifts and coupling constants are in ppm and Hz, respectively. The microanalyses were performed using a ThermoFinigan Flash EA-1112 CHNSO rapid elemental analyzer, and melting points were recorded on a Buchi 530 apparatus. Kinetic studies were carried out using a Perkin-Elmer Lambda 25 spectrophotometer with temperature control using an EYELA NCB-3100 constant-temperature bath. Benzyl bromide and 2-(2-pyridyl)benzothiazole (abbreviated as pbt) were purchased from commercial sources, and the precursor complexes [Pt2Me4(μ-SMe2)],83 [PtMe2(pbt)], 1a,55 [PtMe(ppy)(PPh2Me)], 1b,67 [PtMe(bhq)(PPh2Me)],1c,67 and [PtIMe2(ppy)(PPh2Me)], 4b,67 were prepared as reported.
Synthesis of Platinum Complexes
Preparation of [PtBr(CH2Ph)Me2(pbt)], 2a + 2a′ + 2a″
Benzyl bromide (0.014 mL, 0.12 mmol) was added to a solution of [PtMe2(pbt)], 1a, (0.05 g, 0.11 mmol) in dichloromethane, and the mixture was stirred at room temperature for 2 h. The solvent was evaporated from the solution, and the residue was washed with ether and n-hexane. The product as a light green solid was dried under vacuum. Yield: 0.063 g; 91%, mp 204 °C (decomp.). Anal. Calcd for C21H21BrN2SPt: C, 41.4; H, 3.5; N, 4.6; S, 5.3. Found: C, 41.5; H, 3.1; N, 4.7; S, 5.6. NMR data in CDCl3; 1H NMR data: 2a (major isomer): δ 1.78 [s, 2J(PtH) = 76.0 Hz, 3H, Me trans to N of 2–pyridyl ring], 2.05 [s, 2J(PtH) = 76.1 Hz, 3H, Me trans to N of benzothiazole ring], 2.90 [d, 2J(PtHa) = 85.0 Hz, 2J(HaHb) = 9.6 Hz, 1H, H of CH2Ph]; 3.11 [d, 2J(PtHb) = 96.3 Hz, 2J(HaHb) = 9.6 Hz, 1H, H of CH2Ph]; 6.29–8.65 [m, H of aromatic region], 8.68 [d, 3J(PtH) = 12.0 Hz, 3J(HH) = 5.6 Hz, 1H, CH group adjacent to coordinated 2-pyridyl N atom]. 13C NMR: δ −6.4 [s, 1J(PtC) = 680 Hz, Me trans to N of 2-pyridyl ring], −0.2 [s, 1J(PtC) = 712 Hz, Me trans to N of benzothiazole ring], 22.8 [s, 1J(PtC) = 639 Hz, PtCH2 of benzyl], 120–170 [m, C of 2-(2-pyridyl) benzothiazole ligand]. 2a′: 1H NMR 1.71 [s, 2J(PtH) = 72.0 Hz, 3H, Me trans to Br], 1.98 [s, 2J(PtH) = 72.2 Hz, 3H, Me trans to N of benzothiazole ring], 2.87 [d, 2J(PtHa) = 85.2 Hz, 2J(HaHb) = 9.4 Hz, 1H, H of CH2Ph], 3.10 [d, 2J(PtHb) = 97.3 Hz, 2J(HaHb) = 9.6 Hz, 1H, H of CH2Ph], 8.66 [d, 3J(PtH) = not resolved, 3J(HH) = 5.6 Hz, 1H, CH group adjacent to coordinated 2-pyridyl N atom]. 13C NMR: δ −5.8 [s, 1J(PtC) = 718 Hz, PtMe]; −0.2 [s, 1J(PtC) = not resolved, PtMe], 18.6 [s, 1J(PtC) = 648 Hz, PtCH2 of benzyl]. 13C dept NMR: δ 18.6 [s, 1J(PtC) = 654, PtCH2 of benzyl]. 1H NMR data for 2a″: 1.65 [s, 2J(PtH) = 72.4 Hz, 3H, Me trans to N of benzothiazole ring], 1.93 [s, 2J(PtH) = 74.0 Hz, 3H, Me trans to Br].
Preparation of [PtMeBr(CH2Ph)(ppy)(PPh2Me)], 2b
This compound as a white solid was made similarly using benzyl bromide (0.014 mL, 0.12 mmol) and [PtMe(ppy)(PPh2Me)], 1b, (0.05 g, 0.09 mmol) for 24 h. Yield: 77%. mp 226 °C (decomp.). Anal. Calcd for C32H31BrNPPt: C, 52.2; H, 4.2; N, 1.9; Found: C, 51.8; H, 3.8; N, 2.2. 1H NMR data in CDCl3: δ 1.36 [d, 3J(PH) = 7.9 Hz, 2J(PtH) = 68.1 Hz, 3H, Pt–Me], 1.81 [d, 2J(PH) = 9.1 Hz, 3J(PtH) = 11.6 Hz, 3H, Me of PPh2Me], 2.77 [dd, 3J(PH) = 8.3 Hz, 2J(HaHb) = 12.8 Hz, 2J(PtHa) = 49.8 Hz, 1H, H of CH2Ph], 3.88 [dd, 3J(PH) = 8.3 Hz, 2J(HaHb) = 12.8 Hz, 2J(PtHb) = 102.5 Hz, 1H, H of CH2Ph]. 31P NMR: δ −18.5 [s, 1J(PtP) = 1063 Hz]. 13C NMR: δ −3.4 [d, 2J(PC) = 4 Hz, 1J(PtC) = 645 Hz, Me trans to N of 2-pyridyl ring], 11.2 [d, 1J(PC) = 29 Hz, 2J(PtC) = 16 Hz, Me of PPh2Me], 31.6 [d, 2J(PC) = 106 Hz, 1J(PtC) = 447 Hz, CH2 of benzyl].
Preparation of [PtMeBr(CH2Ph)(bhq)(PPh2Me)], 2c
This compound as a white solid was made similarly using benzyl bromide (0.014 mL, 0.12 mmol) and [PtMe(bhq)(PPh2Me)] (0.04 g, 0.08 mmol) for 24 h. Yield: 62%. mp 237 °C(decomp.). Anal. Calcd for C34H31BrNPPt: C, 53.7; H, 4.1; N, 1.8, Found: C, 53.5; H, 4.1; N, 2.1. 1H NMR data in CDCl3: δ 1.59 (d, 2J(PtH) = 68.6 Hz, 3J(PH) = 7.7 Hz, 3H, Me group), 1.70 (d, 3J(PtH) = 11.8 Hz, 2J(PH) = 9.2 Hz, Me group of the PPh2Me ligand), 2.74 (dd, 3J(PH) = 9.3 Hz, 2J(HaHb) = 13.6 Hz, 2J(PtHa) = 51.4 Hz, 1H, H of CH2Ph group), 3.92 (dd, 3J(PH) = 9.3 Hz, 2J(HaHb) = 13.6 Hz, 2J(PtHa) = 101.2 Hz, 1H, H of CH2Ph group), aromatic protons: 8.93 (d, 3J(PtH) = 10.7 Hz, 3J(HH) = 4.2, 1H, H1), 7.82 (d, 3J(HH) = 7.8 Hz, 1H, H2), 7.66 (t, 3J(HH) = 16.7 Hz, 2H, H3 and H6), 7.51 (d, 3J(HH) = 3.8, 2H, H4 and H5), 7.37 (d, 3J(HH) = 8.72, 1H, H7), 7.09 (m, 4H, meta H of phenyl group of the PPh2Me ligands), 6.97 (d, 3J(HH) = 6.2, 4H, ortho H of phenyl group of the PPh2Me ligands), 6.83 (t, 3J(HH) = 17.2, 2H, para H of phenyl group of the PPh2Me ligands), 6.03–6.25 (5H, H of phenyl group of the Ph of benzyl ligand); 31P NMR: δ −18.0 [s, 1J(PtP) = 1064 Hz].
Preparation of [Pt(Br)2Me2(pbt)], 4a
To a solution of [PtMe2(pbt)] (0.03 g, 0.069 mmol) in toluene was added an excess of benzyl bromide (0.207 mmol, 25 μL). The reaction was refluxed at 70 °C for 6 days. The solvent was evaporated, and the residue was washed with cold diethyl ether. Yield: 77%. mp 290 °C(decomp.). Anal. Calcd for C14H14Br2N2SPt: C, 28.2; H, 2.4; N, 4.7. Found: C, 27.9; H, 2.2; N, 4.9. 1H NMR data in CDCl3: δ 2.35 (s, 2J(PtH) = 75.0 Hz, 3H, Me), 2.61 (s, 2J(PtH) = 75.0 Hz, 3H, Me), aromatic protons: 8.97 (d, 3J(PtH) = 13.1 Hz, 3J(HH) = 6.6, 1H), 8.52 (d, 3J(HH) = 7.2 Hz, 1H), 8.20 (m, 3J(HH) = 17.0 Hz, 2H), 7.60–7.77 (m, 4H).
Kinetic Studies of the Oxidative Addition Reactions
In a typical experiment, a solution of the Pt(II) complex in a cuvette was thermostated at 25 °C in acetone, and a known concentration of PhCH2Br was added using a microsyringe. After rapid stirring, the absorbance at the corresponding wavelength was collected with time. The Abs–time curves were analyzed by pseudo-first-order methods ([PhCH2Br]0 ≫ [1b] or [1c]) or under second-order 1:1 stoichiometric conditions ([PhCH2Br]0 = [1a]). Under pseudo-first-order conditions, the pseudo-first-order rate constants (kobs) were evaluated by nonlinear least-squares fitting of the absorbance–time profiles to a first-order equation (eq 1). Then, the slope of the linear plot of kobsversus [PhCH2Br] gave the second-order rate constant (k2). In the case of second-order 1:1 stoichiometric conditions, the Abs–time data fit to eq 2 to give k2 values.
| 1 |
| 2 |
The same method was used at other temperatures, and activation parameters were obtained from the Eyring equation (eq 3).
| 3 |
Computational Details
Gaussian 09 was used84 to fully optimize the compounds using the B3LYP level of DFT. The starting structures were created by the GaussView program and optimized using the CPCM solvation method85 considering acetone and toluene as solvents, as implemented in the Gaussian program. The effective core potential of Hay and Wadt with a double-ξ valence basis set (LANL2DZ) was chosen to describe Pt and Br.86 The 6-31G(d) basis set was used for all other atoms.87 Frequency calculations were carried out at the same level of theory to identify whether the calculated stationary point is a minimum (zero imaginary frequency) or a transition-state structure (one imaginary frequency). All data were calculated at standard temperature and pressure (298.15 K and 1.0 atm.). We have also checked that imaginary frequencies exhibit the expected motion.
Acknowledgments
We acknowledge support from Shiraz University, the Iran National Science Foundation (Grants no. 97012633 and 98019717), and the Department of Chemistry and Biochemistry at UCSB.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03573.
The authors declare no competing financial interest.
Supplementary Material
References
- Liu P.; Liang R.; Lu L.; Yu Z.; Li F. Use of a Cyclometalated Iridium(III) Complex Containing a N∧C∧N-Coordinating Terdentate Ligand as a Catalyst for the α-Alkylation of Ketones and N-Alkylation of Amines with Alcohols. J. Org. Chem. 2017, 82, 1943–1950. 10.1021/acs.joc.6b02758. [DOI] [PubMed] [Google Scholar]
- Boyaala R.; Touzani R.; Roisnel T.; Dorcet V.; Caytan E.; Jacquemin D.; Boixel J.; Guerchais V.; Doucet H.; Soulé J.-F. Catalyst-Controlled Regiodivergent C-H Arylation Site of Fluorinated 2-Arylpyridine Derivatives: Application to Luminescent Iridium(III) Complexes. ACS Catal. 2018, 9, 1320–1328. 10.1021/acscatal.8b04553. [DOI] [Google Scholar]
- Blons C.; Mallet-Ladeira S.; Amgoune A.; Bourissou D. (P,C) Cyclometalated Gold(III) Complexes: Highly Active Catalysts for the Hydroarylation of Alkynes. Angew. Chem., Int. Ed. 2018, 57, 11732–11736. 10.1002/anie.201807106. [DOI] [PubMed] [Google Scholar]
- Ito J.-i.; Ishihara T.; Fukuoka T.; Binti Mat Napi S. R.; Kameo H.; Nishiyama H. Modulation of the coordination geometries of NCN and NCNC Rh complexes for ambidextrous chiral catalysts. Chem. Commun. 2019, 55, 12765–12768. 10.1039/c9cc06520b. [DOI] [PubMed] [Google Scholar]
- Ranieri A. M.; Burt L. K.; Stagni S.; Zacchini S.; Skelton B. W.; Ogden M. I.; Bissember A. C.; Massi M. Anionic cyclometalated platinum(II) tetrazolato complexes as viable photoredox catalysts. Organometallics 2019, 38, 1108–1117. 10.1021/acs.organomet.8b00913. [DOI] [Google Scholar]
- Nabavizadeh S. M.; Niroomand Hosseini F.; Park C.; Wu G.; Abu-Omar M. M. Discovery and mechanistic investigation of Pt-catalyzed oxidative homocoupling of benzene with PhI(OAc)2. Dalton Trans. 2020, 49, 2477–2486. 10.1039/c9dt04261j. [DOI] [PubMed] [Google Scholar]
- Skórka Ł.; Filapek M.; Zur L.; Małecki J. G.; Pisarski W.; Olejnik M.; Danikiewicz W.; Krompiec S. Highly phosphorescent cyclometalated iridium(III) complexes for optoelectronic applications: fine tuning of the emission wavelength through ancillary ligands. J. Phys. Chem. C 2016, 120, 7284–7294. 10.1021/acs.jpcc.6b01663. [DOI] [Google Scholar]
- Lu G.; Yao J.; Chen Z.; Ma D.; Yang C. Saturated red iridium(III) complexes containing a unique four-membered Ir–S–C–N backbone: mild synthesis and application in OLEDs. J. Mater. Chem. C 2020, 8, 1391. 10.1039/c9tc05806k. [DOI] [Google Scholar]
- Shao J.-Y.; Gong Z.-L.; Zhong Y.-W. Bridged cyclometalated diruthenium complexes for fundamental electron transfer studies and multi-stage redox switching. Dalton Trans. 2018, 47, 23–29. 10.1039/c7dt04168c. [DOI] [PubMed] [Google Scholar]
- Aseman M. D.; Aryamanesh S.; Shojaeifard Z.; Hemmateenejad B.; Nabavizadeh S. M. Cycloplatinated(II) Derivatives of Mercaptopurine Capable of Binding Interactions with HSA/DNA. Inorg. Chem. 2019, 58, 16154–16170. 10.1021/acs.inorgchem.9b02696. [DOI] [PubMed] [Google Scholar]
- Fereidoonnezhad M.; Ramezani Z.; Nikravesh M.; Zangeneh J.; Golbon Haghighi M.; Faghih Z.; Notash B.; Shahsavari H. R. Cycloplatinated(II) complexes bearing an O,S-heterocyclic ligand: search for anticancer drugs. New J. Chem. 2018, 42, 7177–7187. 10.1039/c8nj01332b. [DOI] [Google Scholar]
- Zhang P.; Sadler P. J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. 10.1016/j.jorganchem.2017.03.038. [DOI] [Google Scholar]
- Graf M.; Gothe Y.; Metzler-Nolte N.; Czerwieniec R.; Sünkel K. Bis-cyclometalated rhodium- and iridium-complexes with the 4,4′-dichloro-2,2′-bipyridine ligand. Evaluation of their photophysical properties and biological activity. Inorg. Chim. Acta 2017, 463, 36–43. 10.1016/j.ica.2017.04.006. [DOI] [Google Scholar]
- Barzegar-Kiadehi S. R.; Golbon Haghighi M.; Jamshidi M.; Notash B. Influence of the Diphosphine Coordination Mode on the Structural and Optical Properties of Cyclometalated Platinum(II) Complexes: An Experimental and Theoretical Study on Intramolecular Pt···Pt and π···π Interactions. Inorg. Chem. 2018, 57, 5060–5073. 10.1021/acs.inorgchem.8b00137. [DOI] [PubMed] [Google Scholar]
- Leopold H.; Císařová I.; Strassner T. Phosphorescent C∧C* Cyclometalated Thiazol-2-ylidene Iridium(III) Complexes: Synthesis, Structure, and Photophysics. Organometallics 2017, 36, 3016–3018. 10.1021/acs.organomet.7b00421. [DOI] [Google Scholar]
- Rajabi S.; Jamali S.; Naseri S.; Jamjah A.; Kia R.; Samouei H.; Mastrorilli P.; Shahsavari H. R.; Raithby P. R. Pt-M (M = Au and Tl) Dative Bonds Using Bis(cyclometalated)platinum(II) Complexes. Organometallics 2019, 38, 1709–1720. 10.1021/acs.organomet.8b00907. [DOI] [Google Scholar]
- Nahaei A.; Nabavizadeh S. M.; Hosseini F. N.; Hoseini S. J.; Abu-Omar M. M. Arene C-H bond activation and methane formation by a methylplatinum(II) complex: experimental and theoretical elucidation of the mechanism. New J. Chem. 2019, 43, 8005–8014. 10.1039/c9nj01968e. [DOI] [Google Scholar]
- Zucca A.; Petretto G. L.; Stoccoro S.; Cinellu M. A.; Manassero M.; Manassero C.; Minghetti G. Cyclometalation of 2,2′-Bipyridine. Mono- and Dinuclear C,N Platinum(II) Derivatives. Organometallics 2009, 28, 2150–2159. 10.1021/om801033g. [DOI] [Google Scholar]
- Calvet T.; Crespo M.; Font-Bardia M.; Gómez K.; González G.; Martinez M. Kinetico-Mechanistic Insight into the Platinum-Mediated C–C Coupling of Fluorinated Arenes. Organometallics 2009, 28, 5096–5106. 10.1021/om9004934. [DOI] [Google Scholar]
- Jamali S.; Nabavizadeh S. M.; Rashidi M. Binuclear Cyclometalated Organoplatinum Complexes Containing 1,1′-Bis(diphenylphosphino)ferrocene as Spacer Ligand: Kinetics and Mechanism of MeI Oxidative Addition. Inorg. Chem. 2008, 47, 5441–5452. 10.1021/ic701910d. [DOI] [PubMed] [Google Scholar]
- Niknam F.; Hamidizadeh P.; Nabavizadeh S. M.; Niroomand Hosseini F.; Hoseini S. J.; Ford P. C.; Abu-Omar M. M. Synthesis, structural characterization, and luminescence properties of mono- and di-nuclear platinum(II) complexes containing 2-(2-pyridyl)-benzimidazole. Inorg. Chim. Acta 2019, 498, 119133–119142. 10.1016/j.ica.2019.119133. [DOI] [Google Scholar]
- Nahaei A.; Rasekh A.; Rashidi M.; Hosseini F. N.; Nabavizadeh S. M. Phenylpyrazolate cycloplatinated(II) complexes: Kinetics of oxidation to Pt(IV) complexes. J. Organomet. Chem. 2016, 815–816, 35–43. 10.1016/j.jorganchem.2016.05.005. [DOI] [Google Scholar]
- Aghakhanpour R. B.; Nabavizadeh S. M.; Rashidi M. Newly designed luminescent di- and tetra-nuclear double rollover cycloplatinated(II) complexes. J. Organomet. Chem. 2016, 819, 216–227. 10.1016/j.jorganchem.2016.07.007. [DOI] [Google Scholar]
- Solomatina A. I.; Chelushkin P. S.; Krupenya D. V.; Podkorytov I. S.; Artamonova T. O.; Sizov V. V.; Melnikov A. S.; Gurzhiy V. V.; Koshel E. I.; Shcheslavskiy V. I.; Tunik S. P. Coordination to imidazole ring switches on phosphorescence of platinum cyclometalated complexes: the route to selective labeling of peptides and proteins via histidine residues. Bioconjugate Chem. 2017, 28, 426–437. 10.1021/acs.bioconjchem.6b00598. [DOI] [PubMed] [Google Scholar]
- Leopold H.; Tronnier A.; Wagenblast G.; Münster I.; Strassner T. Photoluminescence of a New Material: Cyclometalated C∧C* Thiazole-2-ylidene Platinum(II) Complexes. Organometallics 2016, 35, 959–971. 10.1021/acs.organomet.5b00991. [DOI] [Google Scholar]
- Moussa J.; Loch A.; Chamoreau L.-M.; Degli Esposti A.; Bandini E.; Barbieri A.; Amouri H. Luminescent Cyclometalated Platinum Complexes with π-Bonded Catecholate Organometallic Ligands. Inorg. Chem. 2017, 56, 2050–2059. 10.1021/acs.inorgchem.6b02731. [DOI] [PubMed] [Google Scholar]
- Fereidoonnezhad M.; Kaboudin B.; Mirzaee T.; Babadi Aghakhanpour R.; Golbon Haghighi M.; Faghih Z.; Faghih Z.; Ahmadipour Z.; Notash B.; Shahsavari H. R. Cyclometalated Platinum(II) Complexes Bearing Bidentate O,O′-Di(alkyl)dithiophosphate Ligands: Photoluminescence and Cytotoxic Properties. Organometallics 2017, 36, 1707–1717. 10.1021/acs.organomet.7b00054. [DOI] [Google Scholar]
- Jamshidi M.; Babaghasabha M.; Shahsavari H. R.; Nabavizadeh S. M. The influence of thiolate ligands on the luminescence properties of cycloplatinated(II) complexes. Dalton Trans. 2017, 46, 15919–15927. 10.1039/c7dt03599c. [DOI] [PubMed] [Google Scholar]
- Crespo M.; Martínez M.; Nabavizadeh S. M.; Rashidi M. Kinetico-mechanistic studies on CX (X=H, F, Cl, Br, I) bond activation reactions on organoplatinum(II) complexes. Coord. Chem. Rev. 2014, 279, 115–140. 10.1016/j.ccr.2014.06.010. [DOI] [Google Scholar]
- Rendina L. M.; Puddephatt R. J. Oxidative addition reactions of organoplatinum(II) complexes with nitrogen-donor ligands. Chem. Rev. 1997, 97, 1735–1754. 10.1021/cr9704671. [DOI] [PubMed] [Google Scholar]
- Diccianni J. B.; Katigbak J.; Hu C.; Diao T. Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc. 2019, 141, 1788–1796. 10.1021/jacs.8b13499. [DOI] [PubMed] [Google Scholar]
- Kehoe R.; Mahadevan M.; Manzoor A.; McMurray G.; Wienefeld P.; Baird M. C.; Budzelaar P. H. M. Reactions of the Ni(0) Compound Ni(PPh3)4 with Unactivated Alkyl Halides: Oxidative Addition Reactions Involving Radical Processes and Nickel(I) Intermediates. Organometallics 2018, 37, 2450–2467. 10.1021/acs.organomet.8b00244. [DOI] [Google Scholar]
- Hamidizadeh P.; Nabavizadeh S. M.; Hoseini S. J. Effects of the number of cyclometalated rings and ancillary ligands on the rate of MeI oxidative addition to platinum(II)-pincer complexes. Dalton Trans. 2019, 48, 3422–3432. 10.1039/c9dt00205g. [DOI] [PubMed] [Google Scholar]
- Azizpoor Fard M.; Behnia A.; Puddephatt R. J. Models for Cooperative Catalysis: Oxidative Addition Reactions of Dimethylplatinum(II) Complexes with Ligands Having Both NH and OH Functionality. ACS Omega 2019, 4, 257–268. 10.1021/acsomega.8b03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichaandi K. R.; Kabalan L.; Amini H.; Zhang G.; Zhu H.; Kenttämaa H. I.; Fanwick P. E.; Miller J. T.; Kais S.; Nabavizadeh S. M.; Rashdi M.; Abu-Omar M. M. Mechanism of Me-Re Bond Addition to Platinum(II) and Dioxygen Activation by the Resulting Pt-Re Bimetallic Center. Inorg. Chem. 2017, 56, 2145–2152. 10.1021/acs.inorgchem.6b02801. [DOI] [PubMed] [Google Scholar]
- Fard M. A.; Behnia A.; Puddephatt R. J. Cycloneophylplatinum Chemistry: A New Route to Platinum(II) Complexes and the Mechanism and Selectivity of Protonolysis of Platinum-Carbon Bonds. Organometallics 2018, 37, 3368–3377. 10.1021/acs.organomet.8b00650. [DOI] [Google Scholar]
- Aseman M. D.; Rashidi M.; Nabavizadeh S. M.; Puddephatt R. J. Secondary kinetic isotope effects in oxidative addition of benzyl bromide to dimethylplatinum(II) complexes. Organometallics 2013, 32, 2593–2598. 10.1021/om400084b. [DOI] [Google Scholar]
- Niroomand Hosseini F.; Nabavizadeh S. M.; Abu-Omar M. M. Which is the Stronger Nucleophile, Platinum or Nitrogen in Rollover Cycloplatinated(II) Complexes?. Inorg. Chem. 2017, 56, 14706–14713. 10.1021/acs.inorgchem.7b02678. [DOI] [PubMed] [Google Scholar]
- Maidich L.; Zucca A.; Clarkson G. J.; Rourke J. P. Oxidative Addition of MeI to a Rollover Complex of Platinum(II): Isolation of the Kinetic Product. Organometallics 2013, 32, 3371–3375. 10.1021/om400300n. [DOI] [Google Scholar]
- Whitfield S. R.; Sanford M. S. Reactions of platinum(II) complexes with chloride-based oxidants: routes to Pt(III) and Pt(IV) products. Organometallics 2008, 27, 1683–1689. 10.1021/om070095e. [DOI] [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis; Univ Science Books, 2010. [Google Scholar]
- Grubbs R. H.Organometallic Chemistry in Industry: A Practical Approach; John Wiley & Sons, 2020. [Google Scholar]
- Albrecht M.; Gossage R. A.; Spek A. L.; van Koten G. Metal-Mediated C–C Bond Making and Breaking: First Direct Evidence for a Reversible Migration of a Benzyl Group along a Metal–Carbon Bond. J. Am. Chem. Soc. 1999, 121, 11898–11899. 10.1021/ja992494+. [DOI] [Google Scholar]
- McCready M. S.; Puddephatt R. J. Supramolecular Organoplatinum(IV) Chemistry: Dimers and Polymers Formed by Intermolecular Hydrogen Bonding. ACS Omega 2018, 3, 13621–13629. 10.1021/acsomega.8b01860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aseman M. D.; Nabavizadeh S. M.; Niroomand Hosseini F.; Wu G.; Abu-Omar M. M. Carbon-Oxygen Bond Forming Reductive Elimination from Cycloplatinated(IV) Complexes. Organometallics 2018, 37, 87–98. 10.1021/acs.organomet.7b00745. [DOI] [Google Scholar]
- Anderson C. M.; Brown G.; Greenberg M. W.; Yu D.; Bowen N.; Ahmed R.; Yost-Bido M.; Wray A. Stereoselective CX and regioselective CH activation to, and selective C(sp2)-C(sp3) reductive elimination from, platinum compounds with thiophene-derived ligands. Tetrahedron Lett. 2019, 60, 151156. 10.1016/j.tetlet.2019.151156. [DOI] [Google Scholar]
- Sarju J. P.; Dey D.; Torroba J.; Whitwood A. C.; Redeker K.; Bruce D. W. Catalytic Activation of Unstrained, Nonactivated Ketones Mediated by Platinum(II): Multiple C-C Bond Cleavage and CO Extrusion. Organometallics 2019, 38, 4539–4542. 10.1021/acs.organomet.9b00650. [DOI] [Google Scholar]
- Shiba Y.; Inagaki A.; Akita M. C-C Bond Forming Reductive Elimination from Diarylplatinum Complexes Driven by Visible-Light-Mediated Photoredox Reactions. Organometallics 2015, 34, 4844–4853. 10.1021/om501080v. [DOI] [Google Scholar]
- Liberman-Martin A. L.; Bergman R. G.; Tilley T. D. A remote Lewis acid trigger dramatically accelerates biaryl reductive elimination from a platinum complex. J. Am. Chem. Soc. 2013, 135, 9612–9615. 10.1021/ja404339u. [DOI] [PubMed] [Google Scholar]
- Brown M. P.; Puddephatt R. J.; Upton C. E. E.; Lavington S. W. Thermal decomposition of some acyl(dialkyl)-, dialkyl(allyl)-, dialkyl(benzyl)-, and trialkyl-halogenobis(dimethylphenylphosphine)platinum(IV) complexes. J. Chem. Soc., Dalton Trans. 1974, 1613–1618. 10.1039/dt9740001613. [DOI] [Google Scholar]
- Shahsavari H. R.; Babadi Aghakhanpour R.; Biglari A.; Niazi M.; Mastrorilli P.; Todisco S.; Gallo V.; Lalinde E.; Moreno M. T.; Giménez N.; Halvagar M. R. C(sp2)–C(sp2) Reductive Elimination from a Diarylplatinum(II) Complex Induced by a S–S Bond Oxidative Addition at Room Temperature. Organometallics 2020, 39, 417–424. 10.1021/acs.organomet.9b00771. [DOI] [Google Scholar]
- Zanganeh M.; Hoseini S. J.; Rashidi M.; Nabavizadeh S. M.; Halvagar M. R. Reaction of allyl bromide with cyclometallated platinum(II) complexes: Unusual kinetic behavior and a novel case of methyl and allyl C-C bond reductive elimination. J. Organomet. Chem. 2018, 856, 1–12. 10.1016/j.jorganchem.2017.12.016. [DOI] [Google Scholar]
- van Asselt R.; Rijnberg E.; Elsevier C. J. Rigid bidentate nitrogen ligands in organometallic chemistry and homogeneous catalysis. 7. Stabilization of high oxidation states by rigid bidentate nitrogen ligands: synthesis and characterization of diorgano- and triorganopalladium(IV) and cationic triorganoplatinum(IV) complexes. Organometallics 1994, 13, 706–720. 10.1021/om00014a049. [DOI] [Google Scholar]
- Crumpton D. M.; Goldberg K. I. Five-Coordinate Intermediates in Carbon–Carbon Reductive Elimination Reactions from Pt(IV). J. Am. Chem. Soc. 2000, 122, 962–963. 10.1021/ja9912123. [DOI] [Google Scholar]
- Nabavizadeh S. M.; Raoof F.; Pakpour F.; Shafiei Sarvestani L.; Niknam F.; Niroomand Hosseini F.; Hoseini S. J. Facile activation of the C-I bond of primary alkyl halides by Pt(II) complexes having a benzothiazole ligand. Inorg. Chim. Acta 2020, 506, 119535–119543. 10.1016/j.ica.2020.119535. [DOI] [Google Scholar]
- Fard M. A.; Behnia A.; Puddephatt R. J. Supramolecular Polymer and Sheet and a Double Cubane Structure in Platinum(IV) Iodide Chemistry: Solution of a Longstanding Puzzle. ACS Omega 2018, 3, 10267–10272. 10.1021/acsomega.8b01367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzaki Y.; Kiho M.; Osakada K. Fluoroalkylation of a Methylplatinum(II) Complex under Photoirradiation. Organometallics 2017, 36, 1391–1397. 10.1021/acs.organomet.7b00098. [DOI] [Google Scholar]
- Azizpoor Fard M.; Behnia A.; Puddephatt R. J. Activation of Dioxygen by Dimethylplatinum(II) Complexes. Organometallics 2017, 36, 4169–4178. 10.1021/acs.organomet.7b00614. [DOI] [Google Scholar]
- Niazi M.; Shahsavari H. R. Cycloplatinated(II) complex bearing 2-vinylpyridine and monodentate phosphine ligands: Optical properties and kinetic study. J. Organomet. Chem. 2016, 803, 82–91. 10.1016/j.jorganchem.2015.12.005. [DOI] [Google Scholar]
- Thompson K. A.; Kadwell C.; Boyle P. D.; Puddephatt R. J. Reactivity of organoplatinum complexes containing appended alcohol functional groups: Activation of dioxygen and hydrogen peroxide. J. Organomet. Chem. 2017, 829, 22–30. 10.1016/j.jorganchem.2016.11.027. [DOI] [Google Scholar]
- Shahsavari H. R.; Rashidi M.; Nabavizadeh S. M.; Habibzadeh S.; Heinemann F. W. A Tetramethylplatinum(IV) Complex with 1,1′-Bis(diphenylphosphanyl)ferrocene Ligands: Reaction with Trifluoroacetic Acid. Eur. J. Inorg. Chem. 2009, 3814–3820. 10.1002/ejic.200900454. [DOI] [Google Scholar]
- Brown M. P.; Puddephatt R. J.; Upton C. E. E. Mechanism of reductive elimination of ethane from some halogenotrimethylbis(tertiary phosphine)platinum(IV) complexes. J. Chem. Soc., Dalton Trans. 1974, 2457–2465. 10.1039/dt9740002457. [DOI] [Google Scholar]
- Momeni B. Z.; Hadi S.; Biglari A. Design of alkyl- and haloalkyl complexes of dimethylplatinum(IV) via oxidative addition: Reactivity, characterization and crystal structures. Inorg. Chim. Acta 2017, 455, 262–270. 10.1016/j.ica.2016.10.029. [DOI] [Google Scholar]
- Momeni B. Z.; Rashidi M.; Jafari M. M.; Patrick B. O.; Abd-El-Aziz A. S. Oxidative addition of some mono, di or tetra haloalkanes to organoplatinum(II) complexes. J. Organomet. Chem. 2012, 700, 83–92. 10.1016/j.jorganchem.2011.11.018. [DOI] [Google Scholar]
- Hoseini S. J.; Nasrabadi H.; Nabavizadeh S. M.; Rashidi M.; Puddephatt R. J. Reactivity and Mechanism in the Oxidative Addition of Allylic Halides to a Dimethylplatinum(II) Complex. Organometallics 2012, 31, 2357–2366. 10.1021/om201289e. [DOI] [Google Scholar]
- Hadadi E.; Nabavizadeh S. M.; Niroomand Hosseini F. Platinum-oxygen Bond Formation: Kinetic and Mechanistic Studies. Inorg. Chem. Res. 2019, 2, 117–128. 10.22036/ICR.2019.210340.1057. [DOI] [Google Scholar]
- Nabavizadeh S. M.; Amini H.; Jame F.; Khosraviolya S.; Shahsavari H. R.; Niroomand Hosseini F.; Rashidi M. Oxidative addition of MeI to some cyclometalated organoplatinum(II) complexes: Kinetics and mechanism. J. Organomet. Chem. 2012, 698, 53–61. 10.1016/j.jorganchem.2011.10.028. [DOI] [Google Scholar]
- Nabavizadeh S. M.; Hoseini S. J.; Momeni B. Z.; Shahabadi N.; Rashidi M.; Pakiari A. H.; Eskandari K. Oxidative addition of n-alkyl halides to diimine-dialkylplatinum(II) complexes: a closer look at the kinetic behaviors. Dalton Trans. 2008, 2414–2421. 10.1039/b800771c. [DOI] [PubMed] [Google Scholar]
- Sangari M. S.; Rashidi M.; Nabavizadeh S. M.; Askari B.; Niroomand Hosseini F. Reaction of dimethylplatinum(II) complexes with PhCH2 CH2 Br: Comparative reactivity with CH3CH2Br and PhCH2Br and synthesis of Pt(IV) complexes. Appl. Organomet. Chem. 2018, 32, e3954. 10.1002/aoc.3954. [DOI] [Google Scholar]
- Rashidi M.; Nabavizadeh M.; Hakimelahi R.; Jamali S. Kinetics and mechanism of cleavage of the oxygen-oxygen bond in hydrogen peroxide and dibenzoyl peroxide by arylplatinum(II) complexes. J. Chem. Soc., Dalton Trans. 2001, 3430–3434. 10.1039/b103690b. [DOI] [Google Scholar]
- Cramer C. J.; Truhlar D. G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. 10.1039/b907148b. [DOI] [PubMed] [Google Scholar]
- Lin Z. Interplay between theory and experiment: computational organometallic and transition metal chemistry. Acc. Chem. Res. 2010, 43, 602–611. 10.1021/ar9002027. [DOI] [PubMed] [Google Scholar]
- Bencini A. Some considerations on the proper use of computational tools in transition metal chemistry. Inorg. Chim. Acta 2008, 361, 3820–3831. 10.1016/j.ica.2008.03.076. [DOI] [Google Scholar]
- Ghari H.; Li Y.; Roohzadeh R.; Caramenti P.; Waser J.; Ariafard A. Gold-catalyzed domino cyclization-alkynylation reactions with EBX reagents: new insights into the reaction mechanism. Dalton Trans. 2017, 46, 12257–12262. 10.1039/c7dt03154h. [DOI] [PubMed] [Google Scholar]
- Chipman A.; Yates B. F.; Canty A. J.; Ariafard A. Reduction of a platinum(IV) prodrug model by sulfur containing biological reductants: computational mechanistic elucidation. Chem. Commun. 2018, 54, 10491–10494. 10.1039/c8cc05682j. [DOI] [PubMed] [Google Scholar]
- Fiuza S. M.; Amado A. M.; Marques M. P. M.; Batista de Carvalho L. A. E. Use of Effective Core Potential Calculations for the Conformational and Vibrational Study of Platinum(II) Anticancer Drugs.cis-Diamminedichloroplatinum(II) as a Case Study. J. Phys. Chem. A 2008, 112, 3253–3259. 10.1021/jp710868p. [DOI] [PubMed] [Google Scholar]
- Nabavizadeh S. M.; Niroomand Hosseini F.; Nejabat N.; Parsa Z. Bismuth-Halide Oxidative Addition and Bismuth-Carbon Reductive Elimination in Platinum Complexes Containing Chelating Diphosphine Ligands. Inorg. Chem. 2013, 52, 13480–13489. 10.1021/ic4018745. [DOI] [PubMed] [Google Scholar]
- Hosseini F. N.; Ariafard A.; Rashidi M.; Azimi G.; Nabavizadeh S. M. Density functional studies of influences of Ni triad metals and solvents on oxidative addition of MeI to [M(CH3)2(NH3)2] complexes and C–C reductive elimination from [M(CH3)3(NH3)2I] complexes. J. Organomet. Chem. 2011, 696, 3351–3358. 10.1016/j.jorganchem.2011.07.024. [DOI] [Google Scholar]
- Safa M.; Jennings M. C.; Puddephatt R. J. Reactivity of a Dimethylplatinum(II) Complex with the Bis(2-pyridyl)dimethylsilane Ligand: Easy Silicon-Carbon Bond Activation. Organometallics 2012, 31, 3539–3550. 10.1021/om3000136. [DOI] [Google Scholar]
- Gilbert T. M.; Hristov I.; Ziegler T. Comparison between Oxidative Addition and σ-Bond Metathesis as Possible Mechanisms for the Catalytica Methane Activation Process by Platinum(II) Complexes: A Density Functional Theory Study. Organometallics 2001, 20, 1183–1189. 10.1021/om0007264. [DOI] [Google Scholar]
- Zeng G.; Sakaki S. Noble Reaction Features of Bromoborane in Oxidative Addition of B-Br σ-Bond to [M(PMe3)2] (M = Pt or Pd): Theoretical Study. Inorg. Chem. 2011, 50, 5290–5297. 10.1021/ic200749w. [DOI] [PubMed] [Google Scholar]
- Ateşin T. A.; Jones W. D. A Deeper Look into Thiophene Coordination Prior to Oxidative Addition of the C-S Bond to Platinum(0): A Computational Study Using DFT and MO Methods. Organometallics 2008, 27, 53–60. 10.1021/om700679j. [DOI] [Google Scholar]
- Scott J. D.; Puddephatt R. J. Ligand dissociation as a preliminary step in methyl-for-halogen exchange reactions of platinum(II) complexes. Organometallics 1983, 2, 1643–1648. 10.1021/om50005a028. [DOI] [Google Scholar]
- Frisch M.; Trucks G.; Schlegel H. B.; Scuseria G.; Robb M.; Cheeseman J.; Scalmani G.; Barone V.; Mennucci B.; Petersson G.. Gaussian 09, revision D. 01; Gaussian, Inc.: Wallingford CT, 2009.
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/bf00533485. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.