

Abstract
Cardiovascular disease is the primary driver of morbidity and mortality associated with diabetes. Hyperglycaemia is implicated in driving endothelial dysfunction that might underpin the link between diabetes and cardiovascular disease. This study was designed to determine the impact of chronic preconditioning of cells to hyperglycaemia and transient switching of cultured endothelial cells between hyper- and normo-glycaemic conditions on bioenergetic and functional parameters. Immortalised EA.hy926 endothelial cells were cultured through multiple passages under normoglycaemic (5.5 mM) or hyperglycaemic (25 mM) conditions. Cells were subsequently subjected (48 h) to continued normo- or hyperglycaemic exposure, or were switched to the alternative glycaemic condition, or to an intermediate glucose concentration (12.5 mM) and metabolic activity, together with key markers of function were measured. Cells habituated to hyperglycaemia were energetically quiescent. Functional activity, characterised by the measurement of nitric oxide, endothelin-1, tissue plasminogen activator and plasminogen activator inhibitor-1, was depressed by exposure to high glucose, with the reduction in nitric oxide production being the most notable. Function was more responsive to acute changes in extracellular glucose than were bioenergetic changes. We conclude that glucose is a key determinant of endothelial function. The study highlights the importance of chronic glucose exposure on cell phenotype and emphasises the need to pay close attention to glucose preconditioning in interpreting results under culture conditions.
Subject terms: Biochemistry, Biological techniques, Cell biology
Introduction
Cardiovascular disease (CVD) is the major cause of morbidity and mortality in individuals with diabetes. Whilst Type 1 and Type 2 Diabetes Mellitus are pathologically distinct diseases, hyperglycaemia is common to both and is considered to be the cornerstone of diabetic complications, including vascular disease.
Chronic hyperglycaemia is known to induce cellular metabolic adaptation or changes in endothelial cell functionality. This phenomenon is described by some as metabolic memory1–4, a term first used to describe the impact of early glycaemic control on the reversibility or progression of complications associated with diabetes in human5 and animal6–9 studies.
A wide range of mechanisms have been proposed to explain hyperglycaemia-driven cellular dysfunction, including oxidative stress, advanced glycation end product (AGE) formation, epigenetic modifications and inflammation1–4,10–13. Central to many of these processes is the impact of glucose on cellular respiration, either directly on account of carbohydrate overload or indirectly through epigenetic modifications in mitochondrial and nuclear DNA and/or post-translational modifications of metabolic enzymes.
Changes to the function of endothelial cells induced by hyperglycaemia are also recognised in a number of studies14–24, but the relationship to bioenergetic modulation is poorly defined. Nitric oxide (NO) is one of the key players in endothelial function and its protective qualities are well-known to be impacted upon by several key processes thought to be related to hyperglycaemic challenge, including inflammation, oxidative stress and cofactor availability (e.g. tetrahydrobiopterin, nicotinamide adenine dinucleotide phosphate (NAD(P)H))25. However, NO is only one of a number of important modulators of both vasomotion, thrombosis and fibrinolysis, some of which have received much less attention in this respect.
The need for chronic exposure of cells to drive some of these processes presents a problem in traditional cell culture experiments because standard procedure typically involves cells grown in hyperglycaemic conditions (25 mM); treatments are often fairly brief and really represent deprivation of glucose from cells habituated to hyperglycaemia21,26–30. In order to gain a more informative picture of the impact of hyperglycaemia on cell function, cell culture models need to be considered afresh, with cells grown under normo-glycaemic conditions through multiple passages to habituate them to glucose at concentrations similar to those found in vivo.
The aim of this study was to test the hypothesis that chronic hyperglycaemia drives important changes in metabolism, redox balance and function in endothelial cells. A supplementary hypothesis was that the key changes to cell function were resistant to reversal by acute glucose normalisation.
Results
Preconditioning in high and normal glucose does not alter cell growth/death parameters
Cells preconditioned in high glucose (HG) and normal glucose (NG) conditions through many (n > 4) passages were indistinguishable under light microscopy (Supplementary Fig. S1A online) and grew at similar rates (Supplementary Fig. S1B online). The lack of impact of glucose concentration on these base parameters is vital knowledge prior to quantifying and interpreting subsequent experimental data.
Effect of glucose concentration on bioenergetic parameters
HG preconditioning (HGPC) and subsequent continued exposure to 25 mM glucose for a further 48 h (HGPC → HG48h) generated a more quiescent phenotype compared to the NG control (NGPC → NG48h), represented as a vertical reduction on the energy map (Fig. 1). The impact of exposing NGPC cells to higher glucose was to instigate a slide down the energetic/quiescent axis, whereas reducing glucose in HGPC cells caused a transition in the opposite direction, towards a more energetic state. There was no detectable impact of changing glucose concentrations on the aerobic-glycolytic axis of the energy map.
Figure 1.
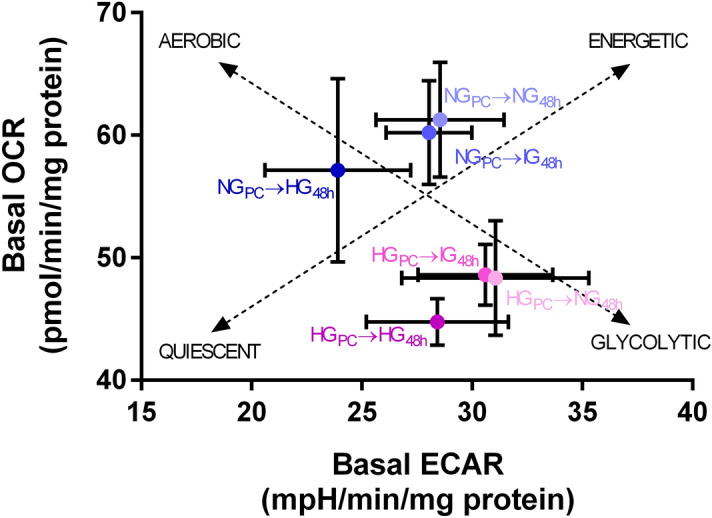
Energy map for cells cultured under various glucose conditions. HGPC cells exhibit a more quiescent phenotype than NGPC cells. 48 h exposure of HGPC cells to lower glucose concentrations (IG or NG) drove a partial reversal of the effect, just as 48 h exposure of NGPC cells with IG or HG medium partially induced the quiescent phenotype. Dotted lines represent the aerobic-glycolytic axis and the quiescent-energetic axis. Data are expressed as mean ± SEM (n = 10–12 for each mean depicted). Raw data were analysed using Seahorse Wave Software (version 2.3.0.19) prior to graphical presentation using Graphpad Prism version 6.00.
Complex V activity, glucose depletion from the medium, and maximum and spare capacity
Oxygen consumption rate (OCR) related to complex V activity was significantly higher (+ 45%) in NGPC cells than in HGPC cells (Fig. 2A) and there was a trend towards a reduction in complex V-mediated OCR in NGPC cells upon 48 h exposure to increased glucose concentrations (Fig. 2B). Complex V-mediated OCR was unaffected by normalisation of glucose in HGPC cells (Fig. 2C). The variation in complex V-mediated OCR across different treatments was directly correlated to glucose depletion from the medium, most likely entirely due to uptake by cells (Fig. 2D).
Figure 2.

(A) Effect of glucose preconditioning on complex V-associated OCR (**P < 0.01; Mann Whitney U test). (B) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells (P > 0.05; Kruskal–Wallis). (C) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells (P > 0.05; Kruskal–Wallis). (D) Correlation of mean complex V-associated OCR with mean glucose depletion from medium under each HG and NG condition. Raw data for glucose depletion are found in Supplementary Fig. S3. Raw data were analysed using Seahorse Wave Software (version 2.3.0.19) prior to graphical presentation using Graphpad Prism version 6.00.
The original data relating to glucose depletion from media is found in Supplementary Fig. S2 online; glucose depletion was significantly lower in HGPC cells, despite the theoretical concentration gradient being substantially higher (Supplementary Fig. S2A online). The effect was not induced by acute exposure (Supplementary Fig. S2B online) and not reversed by 48 h incubation with lower glucose concentrations (Supplementary Fig. S2C online).
Maximum capacity (the maximum OCR obtained following FCCP injection) and derived spare capacity (maximum OCR minus basal OCR) were both significantly lower in HGPC cells (Supplementary Fig. S3A, D online). The effect was not induced by 48 h exposure of NGPC cells to higher glucose concentrations (Supplementary Fig. S3B,E online) and was not reversed by exposure of HGPC cells to lower glucose concentrations (Supplementary Fig. S3C,F online).
Effect of glucose on measures of oxidative stress-associated parameters
Both non-mitochondrial OCR (NMOCR; − 58%) and ROS-induced fluorescence (an indicator of total ROS/RNS; − 21%) were significantly depressed in HGPC cells compared to NGPC cells (Fig. 3A,D). The effect of HGPC on NMOCR and ROS detection was not reversed by exposure to lower glucose concentrations for 48 h (Fig. 3B,E) and was not induced in NGPC cells by exposure to higher concentrations for 48 h (Fig. 3C,F).
Figure 3.

(A) Effect of glucose preconditioning on NMOCR (***P < 0.001; Student’s t-test). (B) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells (P > 0.05; One-way ANOVA). (C) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells (P > 0.05; One-factor ANOVA). (D) Effect of glucose preconditioning on DFDCA fluorescence (**P < 0.01; Student’s t-test). (E) Effect of 48 h exposure of increasing glucose concentrations in NGPC cells (P > 0.05; one-factor ANOVA). (F) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells (P > 0.05; one-factor ANOVA). (G) Effect of glucose preconditioning on SOD activity (***P < 0.001; Mann Whitney U test). (H) Effect of 48 h exposure of increasing glucose concentrations on SOD activity in NGPC cells (P > 0.05; one-factor ANOVA). (I) Effect of 48 h exposure of decreasing glucose concentrations on SOD activity in HGPC cells (P > 0.05; one-factor ANOVA). Raw data were analysed using Seahorse Wave Software (version 2.3.0.19) prior to graphical presentation using Graphpad Prism version 6.00.
SOD activity was substantially reduced in HGPC cells (− 38%; Fig. 3G; there was no significant impact of switching NGPC cells to IG or HG media for 48 h (Fig. 3H) or of switching HGPC cells to lower glucose concentrations for 48 h (Fig. 3I).
Effect of glucose concentration on endothelial cell function
Nitrite generation
Nitrite was measured in the medium as a surrogate marker of basal nitric oxide (NO) generation. Accumulation of basal nitrite was significantly inhibited in HGPC cells (Fig. 4A; − 67%), but the maximum eNOS-derived nitrite generated in cells treated with the calcium ionophore, A23187 was not affected (Fig. 4B). The source of nitrite was confirmed to be NOS on account of the substantial inhibitory effect of the NOS inhibitor, l-NAME (Fig. 4B). Accumulation of basal nitrite was significantly inhibited by 48 h incubation of NGPC cells with higher glucose concentrations (Fig. 4C). Conversely, basal nitrite was increased in HGPC cells exposed to NG conditions for 48 h (Fig. 4D).
Figure 4.

(A) Effect of glucose preconditioning on nitrite accumulation (***P < 0.001; Mann Whitney U test). (B) Effect of activation (A23187 1 μM, 1 min calcium ionophore) and inhibition (l-NAME 100 μM, 24 h) on nitrite accumulation in medium) in NGPC and HGPC cells (P > 0.05; Student’s t-tests between glucose conditions). (C) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells on nitrite (***P < 0.001; Kruskal–Wallis test with Dunn’s post-test). (D) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells on nitrite (***P < 0.001; Kruskal–Wallis test with Dunn’s post-test). Data were analysed using Liquid Software (version 2) and presented using Graphpad Prism version 6.00 Software.
ET-1 secretion
ET-1 secretion was modestly inhibited (− 20%) in HGPC cells compared to NGPC cells (Fig. 5A). A trend towards this effect was seen when NGPC cells were switched to IG and HG conditions for 48 h (Fig. 5B), but HGPC cells were recalcitrant to any effect of NG in this regard (Fig. 5C).
Figure 5.

(A) Effect of glucose preconditioning on ET-1 (***P < 0.001; Student’s t-test). (B) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells on ET-1 (*P < 0.05, **P < 0.01; One-factor ANOVA with Bonferroni post-test). (C) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells on ET-1 (P > 0.05; One-factor ANOVA). Data were collected using SkanIt Software 2.4.5 and further analysed and presented using Graphpad Prism version 6.00 Software.
t-PA and PAI-1 antigen
t-PA antigen secretion into the medium was significantly reduced (-28%) under basal conditions in HGPC cells compared to the NG equivalents (Fig. 6A). Treatment of NGPC cells with higher glucose for 48 h showed a trend towards inhibition (Fig. 6B), while the trend was reversed in HGPC cells treated with reduced glucose, reaching significance in those treated with NG (Fig. 6C).
Figure 6.

(A) Effect of glucose preconditioning on t-PA antigen (**P < 0.001; Student’s t-test). (B) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells on t-PA antigen (P > 0.05; One-factor ANOVA). (C) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells on t-PA antigen (*P < 0.05; One-factor ANOVA with Bonferroni post-test). (D) Effect of glucose preconditioning on PAI-1 antigen (**P < 0.001; Student’s t-test). (E) Effect of 48 h exposure of increasing glucose concentrations to NGPC cells on PAI-1 antigen (P > 0.05; One-factor ANOVA). (F) Effect of 48 h exposure of decreasing glucose concentrations in HGPC cells on PAI-1 antigen (*P < 0.05, **P < 0.01; One-factor ANOVA with Bonferroni post-test). Data were collected using SkanIt Software 2.4.5 and further analysed and presented using Graphpad Prism version 6.00 Software.
PAI-1 was depressed (− 20%) in HGPC cells compared to NGPC cells (Fig. 6D). The effect was not recapitulated in NGPC cells exposed to higher glucose conditions for 48 h (Fig. 6E), nor was the reverse seen in HGPC cells exposed to IG or NG (Fig. 6F). Interestingly, a reduction of glucose to 12.5 mM in HGPC cells resulted in a significant increase in PAI-1 (Fig. 6F). Taken in conjunction with the t-PA results, while both antigens are reduced in hyperglycaemia, the impact is marginally greater on t-PA. Given that the net fibrinolytic activity of t-PA is modulated by PAI-1 through 1:1 binding, it is reasonable to deduce that the overall effect of hyperglycaemia is a modest shift away from fibrinolytic capacity. The effect is particularly clear in cells exposed to intermediate glucose after hyperglycaemic preconditioning (Fig. 6F).
Discussion
The data presented demonstrate that an immortalised endothelial cell line habituated to hyperglycaemic conditions through many passages display a bioenergetically and functionally distinct phenotype from cells habituated to normoglycaemic conditions. Cells habituated to hyperglycaemic conditions are substantially less metabolically active than normoglycaemic counterparts, an effect that is not replicated with acute (48 h) treatment of normoglycaemic cells with intermediate (12.5 mM) or high (25 mM) glucose. Likewise, 48 h treatment of hyperglycaemic cells with lower (intermediate or normal) glucose conditions fails to fully reverse the effect. The bioenergetic changes in hyperglycaemia (HGPC cells) are associated with reduced glucose depletion from the medium in HGPC cells, a depression of ROS generation and SOD activity. Several key endothelium-derived factors were assessed and all were reduced under hyperglycaemic conditions, with NO showing the most marked depression. Unlike the bioenergetic parameters, functional changes were found to bear a more dynamic association with glucose concentration; 48 h treatments were sufficient to initiate at least partial switching between the quiescent HG state and the more active NG state; this effect was bi-directional in the case of NO and ET-1. The different timescales involved for glucose-induced modulation of function, compared to bioenergetic changes, might suggest that there is no causal association between function and metabolism. However, it cannot be excluded that extended glucose exposure (in this specific case, greater than 48 h) might also alter cell bioenergetics as well as cell function over the longer time scale.
Bioenergetics
The overwhelming impact of chronic preconditioning of EA.hy926 endothelial cells in HG media is to drive them towards a more quiescent phenotype; the glycolysis element of the effect is not induced in NGPC cells following 48 h exposure to HG, but quiescence is partially induced with this acute exposure. Maximal and spare capacity, markers of metabolic flexibility, were similarly depressed by HGPC but not 48 h exposure and were not reversed by acute normalization of glucose in the medium. The temporal aspect of these effects is potentially important: glycolytic alteration is clearly much slower to develop and aligns more closely with the counter-intuitive reduction in glucose uptake in HGPC cells. The reason behind the substantial reduction in glucose uptake is not known, but likely involves a range of counter-regulatory measures within the cell in an effort to prevent glucose overload. Switching to energetically less efficient glycolysis might be part of this strategy, which would accelerate utilization of excess glucose and contribute to the observed reduction in maximum and spare capacity, which also are not seen with acute exposure to HG. By contrast, the migration of cells along the energetic/quiescent axis is a far more dynamic effect that is partially induced and reversed in the 48 h treatment experiments.
Oxidative stress
The results obtained for measures associated with oxidative stress largely run counter to previous reports. NMOCR is an indicator of oxygen consumption related to all cellular oxygen consumption that persists after respiration is abolished by complex I and 3 inhibition. The sources of NMOCR are multiple, including various enzymes (NAD(P)H oxidases, xanthine oxidase, cyclo-oxygenases)31–34. The observed substantial and persistent reduction of NMOCR in HGPC cells could be interpreted as a depression of oxidative stress in these cells, which is supported by the fluorescence measure of total ROS production and perhaps partially explained by the reduction in SOD activity. However, we cannot rule out a downregulation of various oxidases and peroxidases under HG conditions, which would resonate with the overall reduction of metabolic activity seen. What is clear is that these results are at odds with published results, that indicate hyperglycaemic exposure as the stressor that leads to COX-2 upregulation33,35,36 or NOX37–39. The precise reason for this difference is unclear at this stage, but could be due to the preconditioning element of our experiments, which is unusual compared to much of the literature, but in our view more appropriate. In addition, it is worth noting that the glucose concentration in the medium of NG-treated cells fell to what might be considered hypoglycaemic levels over 24 h, perhaps inducing glycaemic stress of a different kind.
Function
While many of the effects of HG in terms of bioenergetics and oxidative stress are slow to develop and irreversible with acute normalisation of glucose, the impacts on key endothelial cell functions are altogether more dynamic, perhaps reflecting transient changes to physiological needs. Of the measures we undertook in this study, nitrite (a surrogate marker of NO generation) experienced the most profound effect, with ~ 70% reduction seen with both acute (48 h) and preconditioning HG treatments. eNOS regulation occurs at a range of levels, from gene expression (e.g. via the diacylglycerol protein kinase C (DAG-PKC) pathway), through substrate (l-arginine) and cofactor (NADPH, tetrahydrobiopterin) availability and phosphorylation40–45. The data we obtained using A23187 as a maximal activator of eNOS suggests that, given sufficient stimulus, HG cells were still fully capable of generating the same amount of NO, irrespective of glucose conditions. Equally, NOS was found to be the sole source of the NO measured via the surrogate nitrite; taken together, it seems unlikely that eNOS expression was substantially altered by glucose, but rather that the impact is downstream of synthesis. NO activity is influenced by interaction with off-target molecules, including superoxide40,46–49, but our data would suggest that increased interaction with superoxide under HG conditions is unlikely, given the reduced oxidative stress in these cells. Direct inhibition by glucose50, on the other hand, cannot be excluded. What is certain, however, is that a similar effect driven by hyperglycaemia in vivo would constitute endothelial dysfunction and contribute to a pro-atherogenic and pro-thrombotic state.
There was a HG-induced reduction in the vasoconstrictor and pro-mitogenic mediator ET-1 and the fibrinolytic modulators t-PA and PAI-1. That all three proteins are down-regulated, taken together with the apparent depression of SOD, chimes with an overall dampening of cellular activity in line with reduced metabolism. While it is not possible to reach a conclusion as to the net effect of NO/ET-1 changes on constriction and atherogenic processes, the greater impact of HG on t-PA compared to PAI-1 is likely to result in a net reduction in fibrinolytic activity, given that PAI-1 binds to t-PA in a 1:1 stoichiometry to inactivate it. Should such an effect be replicated in vivo, there would be a reduced capability to eliminate potentially harmful micro-thrombi.
Limitations
Accurate mimicking of in vivo glycaemic exposure is all but impossible in cell culture format. While we often consider in vitro glucose treatments to be “constant”, in truth they will gradually decline as glucose is utilised. Our experimental measurements confirmed, for example, that while our NG treatment regimen was set up at 5.5 mM, the glucose concentration remaining after 24 h was often below concentrations that might be considered to be hypoglycaemic from a clinical standpoint.
It could be argued that primary human endothelial cells would have been a more appropriate cell model to use, rather than the immortalised endothelial cell line studied. However, the preconditioning stage requires numerous cell passages and primary endothelial cells are known to lose their endothelial cell characteristics over repeated passages. This would have proved to be a profound limitation to this cell model so, on balance, the EA.hy926 cell line was considered a more suitable option than primary human endothelial cells.
Detection of ROS is fraught with difficulties and no method should be considered perfect. We chose to use a fluorescent marker for ROS, which is fairly unspecific in terms of establishing the nature of the ROS generated or their source. However, the positive control used (pyocyanin) generated a substantial signal, suggesting that the probe is at least capable of detecting ROS related to mitochondrial dysfunction.
Conclusions
Pre-conditioning cultured cells with HG through multiple passages has a profound effect on bio-energetics, antioxidant adaptation and cell function. Collectively, the effect of HGPC is to establish a quiescent phenotype; the impact on bioenergetics is not replicated with relatively acute (48 h) exposure to HG and is not reversed by acute normalisation of glucose in the media. This has important implications for interpretation of results from studies conducted using standard HGPC protocols—it is important to recognise that cells cultured under these conditions are likely to have assumed a different energetic phenotype to those in vivo under homeostatic control. Even the unusually high glucose conditions experienced in the diabetic state are not reflected by sustained exposure to glucose > 20 mM, as found in standard cell culture. Traditional cell culture conditions could, therefore, generate a cell phenotype that is a mis-representation of the diabetic state.
The impact on endothelial cell function is more dynamic. The detrimental effect on eNOS-derived NO is particularly profound, with important implications in terms of potential for increased vasoconstriction, mitogenesis and risk of thrombosis.
Methods
Materials
Unless stated otherwise, all chemicals were purchased from Sigma Aldrich (Dorset, UK). Cell culture consumables were purchased from VWR (Lutterworth, UK).
Cell culture and growth curves
The endothelial cell line EA.hy926 (ATCC CRL-2922) retains an endothelial phenotype over repeated passages and was used in all cellular experiments. The EA.hy926 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HyClone) containing glucose at either 5.5 mM (normal glucose; NG) or 25 mM (high glucose; HG), in standard conditions of a humidified atmosphere at 37 °C and 5% CO2. Both media were supplemented with 1 mM sodium pyruvate, 2 mM l-glutamine, 5% penicillin/streptomycin (10,000 Units/ml penicillin and 10 mg/ml streptomycin) and 10% fetal bovine serum. Stock NG and HG cells at 90% confluence had been passaged by trypsinization (0.05% trypsin-ethylenediaminetetraacetic acid (EDTA) solution; Gibco, Thermo-Fisher Scientific), re-seeded and pre-conditioned (PC) in their respective media a minimum of five times to generate two separate EA.hy926 cell populations, namely NGPC and HGPC.
To determine whether pre-conditioning affected cell growth or survival, growth curves were established for both NGPC and HGPC cells. HGPC and NGPC cells (1.9 × 105) were seeded into multiple T-25 flasks, with replicate flasks trypsinised and counted using a haemocytometer (Mariennfeld, Germany) every day for 6 days. Concurrently, a daily visual inspection of the cells was made by light microscopy to assess and record cell morphology and adherence. A schematic diagram of the cell culture protocol is illustrated in Fig. 7.
Figure 7.

Schematic diagram depicting glucose exposure regimen and outcome measures.
Measurement of glucose depletion
A OneTouch Verio Flex glucose meter (LifeScan) was used to determine the concentration of glucose in medium added to NGPC and HGPC cells seeded at 2 × 104 cells/well in a 96-well plate and treated separately with either NG, HG or an intermediate glucose concentration of 12.5 mM (IG) for 48 h. Glucose concentration in the media was measured in triplicate at the start and end of the incubation period.
Metabolic analysis
The cellular bioenergetic phenotype of both NGPC and HGPC cells was determined by using a Seahorse Bioscience XFe96 Extracellular Flux Analyzer (Agilent) to measure the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) in the mito-stress test (MST; Agilent). Each assay was performed according to the manufacturer’s instructions and all Seahorse-related kits, reagents and consumables were purchased from Agilent unless stated.
Briefly, the MST assay was used to measure cellular basal and then real-time changes in OCR and ECAR following three sequential injections of mitochondrial electron transport chain (ETC) modulators, namely oligomycin (1 µM; complex V inhibitor), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 µM; an uncoupler of mitochondrial oxidative phosphorylation) and finally a combination of rotenone and antimycin A (both 0.5 µM), to inhibit complex I and complex III respectively. For this assay, both NGPC and HGPC cells were plated into a Seahorse XF96 cell culture 96-well microplate (2 × 104 cells/well) and left for 12 h to adhere at 37 °C, 5% CO2 in a humidified atmosphere. Treatment consisted of addition of NG, IG or HG medium to both NGPC and HGPC cells, followed by incubation under standard conditions for 48 h. The treated cells reached full confluence by conclusion of the incubation period.
On the day of the MST assay, Seahorse assay medium was prepared by adding 1 mM pyruvate, 2 mM l-glutamine and 10 mM glucose to Seahorse XF Base Medium, then adjusted to pH 7.4, filtered and warmed to 37 °C. Medium treatments were removed from each well, washed twice and replaced with assay medium prior to incubation at 37 °C for 45 min in a CO2-free incubator. All injectable treatments were freshly prepared, loaded into the injection ports and the sample plate loaded into the Seahorse XFe96 for analysis. Data was acquired starting from the third measurement of each cycle and analysed using Seahorse Wave Software (Version 2.6.0.31).
At the end of the assay, the protein content of each well was quantified using a Coomassie-BradfordProtein Assay Kit (Merck, Sigma Aldrich) according to the manufacturer’s instructions, to allow for normalisation of data. All cells were lysed with distilled water and repeated freeze–thaw cycles. This protein estimate assay was subsequently used throughout as a surrogate for the density of treated cells where removal of cells for counting was not practical.
Total reactive oxygen species/reactive nitrogen species (ROS/RNS) assay
The total ROS/RNS and superoxide detection fluorescent assay (Enzo Life Sciences, UK) was used to estimate the ROS/RNS production of HGPC and NGPC cells seeded at 2 × 104 cells/well in a 96-well plate and treated for 48 h with NG, IG or HG medium. Pyocyanin was used as a positive control for induction of ROS (200 µM for 20 min) and all treatments were made in triplicate. The assay was performed according to the kit instructions. The nature of fluorescent probe for ROS is proprietary information not released by the manufacturer; fluorescence was detected at excitation/emission wavelengths of λ = 488/520 nm (green) using a Varioskan plate reader (Thermo-Fisher) with SkanIt Software 2.4.5 (Varioskan Flash). The results were normalized to the protein content of each well using the method described above.
SOD activity assay
The Superoxide Dismutase Assay kit (Cayman) was used for the detection of total SOD activity of HGPC and NGPC cells seeded at 5.4 × 105 cells/well in a 6-well plate and treated for 48 h with NG, IG or HG medium to the three test-glucose conditions. The assay utilises a tetrazolium salt for the detection of superoxide anion, generated by xanthine oxidase and hypoxanthine. Results are expressed as U/ml, where 1 unit is the amount of enzyme necessary to dismutate 50% of the superoxide produced. The SOD Assay kit reagents were prepared following the manufacturer’s instructions. Following treatment, NGPC and HGPC cells were lysed in cold lysis buffer (20 mM HEPES buffer, pH 7.2, containing 1 mM egtazic acid (EGTA), 210 mM mannitol and 70 mM sucrose) with the aid of a sonicator, and the lysate solution centrifuged at 1500 g (5 min, 4 °C). The absorbance of the supernatant was measured at λ = 450 nm using the Varioskan platereader. After calculating the average absorbance for each sample or standard, and subtracting the background absorbance, the linearised rate (LR) was calculated. Together with the standard curve, LR allowed the calculation of SOD activity using the following equation:
Nitrite assay
Nitrite accumulation was measured as a marker of NO production using a Sievers 280i nitric oxide analyser (NOA). NGPC and HGPC were seeded in triplicate in a 96-well plate at 2 × 104 cells/well and treated with NG, IG or HG medium for 48 h under standard conditions. The eNOS-inhibitor, Nω-nitro-l-arginine methyl ester (l-NAME; 100 µM for 24 h) and calcium ionophore A23187 (1 µM for 1 min) were used as negative and positive controls respectively, to confirm the role of eNOS in the nitrite measured. The medium from each triplicate was collected into a single tube and stored at – 80 °C for batch measurement. Test samples were defrosted on ice, diluted 1 in 4 in deionized water and 100 µl injected into the NOA chamber containing 4.5 ml of glacial acetic acid (Fisher Scientific), 100 µl of diluted antifoam B emulsion and 0.5 ml of sodium iodide (25 mg/ml; Fisher Scientific). Data were acquired using Liquid Software (version 2), integrated and then sample nitrite values extrapolated from the standard curve.
Measurement of endothelin-1 (ET-1), tissue plasminogen activator (t-PA) and plasminogen activator inhibitor-1 (PAI-1)
Secretion of ET-1, t-PA and PA-1 by NGPC and HGPC exposed to NG, IG or HG medium (48 h) was measured by ELISA. For each ELISA, cell supernatants were prepared by seeding cells in triplicate in 96-well plates (2 × 104 cells/well) and exposing them to NG, IG or HG medium for 48 h under standard conditions. Medium from each triplicate was collected into a single tube and stored at − 80 °C for batch measurement. Samples were processed according to the manufacturer’s instructions for ET-1 (Abcam), t-PA antigen (Zymutest; Hyphen BioMed) and PAI-1 antigen (Zymutest; Hyphen BioMed), with absorbance measured at 450 nm using a Varioskan platereader with with SkanIt Software 2.4.5 (Varioskan Flash, Thermo).
Statistics
All data are expressed as mean ± SD unless otherwise stated; individual points on each graph indicate independent experiments. Data were analysed using appropriate parametric and non-parametric tests, as defined by normality (Kolmorov–Smirnov test) and equal variance testing. Significant difference between means was accepted at P < 0.05. Statistical tests were performed using GraphPad Prism version 6.00 software (GraphPad Software, San Diego, CA). Details of tests used are included in each figure legend; data were treated as unpaired in all data sets.
Ethics approval and consent to participate
This was exclusively an in vitro project so consent to participate was not required. The project had internal university ethics approval to proceed.
Supplementary information
Acknowledgements
We thank Lifescan Scotland for the donation of a meter for measurement of glucose in culture media.
Abbreviations
- ATP
Adenosine triphosphate
- CVD
Cardiovascular disease
- COX
Cyclo-oxygenase
- DMEM
Dulbecco’s Modified Eagle’s Medium
- EGTA
Egtazic acid
- ETC
Electron transport chain
- eNOS
Endothelial nitric oxide synthase
- ET-1
Endothelin-1
- FCCP
Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone
- HG
High glucose
- IG
Intermediate glucose
- MST
Mitochondrial stress test
- l-NAME
Nω-Nitro-l-arginine methyl ester
- NOX
NADPH oxidase
- NO
Nitric oxide
- NMOCR
Non-mitochondrial oxygen consumption rate
- NG
Normal glucose
- OGT
O-GlcNAc transferase
- OCR
Oxygen consumption rate
- PAI-1
Plasminogen activator inhibitor-1
- PC
Pre-condition
- PTMs
Post-translational modifications
- t-PA
Tissue plasminogen activator
- ROS
Reactive oxygen species
- RNS
Reactive nitrogen species
- SOD
Superoxide dismutase
Author contributions
M.L.F.—involved in design of the study, conducted the study, analysed results and involved in writing and editing the manuscript. A.T.T.—involved in design of the study and editing the manuscript. D.P.M.—helped design the study and editing the manuscript. I.L.M. – secured funding, analysed results, wrote and edited the manuscript.
Funding
MLF was funded by EU ESF funding.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-76505-4.
References
- 1.Ceriello A, Ihnat MA, Thorpe JE. The ‘Metabolic memory’: Is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009;94:410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- 2.Reddy MA, Natarajan R. Role of epigenetic mechanisms in the vascular complications of diabetes. Subcell. Biochem. 2013;61:435–454. doi: 10.1007/978-94-007-4525-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misra A, Bloomgarden Z. Metabolic memory: Evolving concepts. J. Diabetes. 2018;10:186–187. doi: 10.1111/1753-0407.12622. [DOI] [PubMed] [Google Scholar]
- 4.Drzewoski J, Kasznicki J, Trojanowski Z. The role of ‘metabolic memory’ in the natural history of diabetes mellitus. Pol. Arch. Med. Wewn. 2009;119:493–500. [PubMed] [Google Scholar]
- 5.Nathan DM. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: The epidemiology of diabetes interventions and complications (EDIC) study. J. Am. Med. Assoc. 2003;290:2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowluru RA. Effect of reinstitution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- 7.Kowluru RA, Kanwar M, Kennedy A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007;2007:21976. doi: 10.1155/2007/21976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowluru RA, Chakrabarti S, Chen S. Re-institution of good metabolic control in diabetic rats and activation of caspase-3 and nuclear transcriptional factor (NF-kappaB) in the retina. Acta Diabetol. 2004;41:194–199. doi: 10.1007/s00592-004-0165-8. [DOI] [PubMed] [Google Scholar]
- 9.Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- 10.Roberto T, et al. The “metabolic memory” theory and the early treatment of hyperglycemia in prevention of diabetic complications. Nutrients. 2017;9:437. doi: 10.3390/nu9050437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wegner M, Neddermann D, Piorunska-Stolzmann M, Jagodzinski PP. Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res. Clin. Pract. 2014;105:164–175. doi: 10.1016/j.diabres.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 12.Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2015;58:443–455. doi: 10.1007/s00125-014-3462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao F, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014;63:1460. doi: 10.2337/db13-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest. Ophthalmol. Vis. Sci. 2003;44:5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 15.Du Y, Miller CM, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Lorenzi M, Cagliero E, Toledo S. Glucose toxicity for human endothelial cells in culture: Delayed replication, disturbed cell cycle, and accelerated death. Diabetes. 1985;34:621–627. doi: 10.2337/diab.34.7.621. [DOI] [PubMed] [Google Scholar]
- 17.Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu TS, et al. Oscillating high glucose enhances oxidative stress and apoptosis in human coronary artery endothelial cells. J. Endocrinol. Invest. 2014;37:645–651. doi: 10.1007/s40618-014-0086-5. [DOI] [PubMed] [Google Scholar]
- 19.Wu N, et al. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016;15:109. doi: 10.1186/s12933-016-0427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: Inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–167. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- 21.Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am. J. Physiol. Hear. Circ. Physiol. 2008;294:H1530–H1540. doi: 10.1152/ajpheart.00823.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson RJ, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease1-3. Am. J. Clin. Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 23.Zhou QG, et al. Vascular insulin resistance related to endoplasmic reticulum stress in aortas from a rat model of chronic kidney disease. Am. J. Physiol. Circ. Physiol. 2012;303:H1154–H1165. doi: 10.1152/ajpheart.00407.2012. [DOI] [PubMed] [Google Scholar]
- 24.Targosz-Korecka M, Brzezinka GD, Malek KE, Stepień E, Szymonski M. Stiffness memory of EA.hy926 endothelial cells in response to chronic hyperglycemia. Cardiovasc. Diabetol. 2013;12:96. doi: 10.1186/1475-2840-12-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brocq ML, Leslie SJ, Milliken P, Megson IL. Endothelial dysfunction: From molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxidants Redox Signal. 2008;10:1631–1674. doi: 10.1089/ars.2007.2013. [DOI] [PubMed] [Google Scholar]
- 26.Hoshiyama M, et al. Effect of high glucose on nitric oxide production and endothelial nitric oxide synthase protein expression in human glomerular endothelial cells. Nephron Exp. Nephrol. 2004;95:e62–e68. doi: 10.1159/000073673. [DOI] [PubMed] [Google Scholar]
- 27.Quijano C, Castro L, Peluffo G, Valez V, Radi R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: Direct measurements and formation of hydrogen peroxide and peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H3404–H3414. doi: 10.1152/ajpheart.00761.2007. [DOI] [PubMed] [Google Scholar]
- 28.Westerweel PE, et al. Impaired endothelial progenitor cell mobilization and dysfunctional bone marrow stroma in diabetes mellitus. PLoS ONE. 2013;8:e60357. doi: 10.1371/journal.pone.0060357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du XL, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fink BD, Herlein JA, O’Malley Y, Sivitz WI. Endothelial cell and platelet Bioenergetics: Effect of glucose and nutrient composition. PLoS One. 2012;7:e39430. doi: 10.1371/journal.pone.0039430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chacko BK, et al. The Bioenergetic Health Index: A new concept in mitochondrial translational research. Clin. Sci. 2014;127:367–373. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheu ML, et al. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler. Thromb. Vasc. Biol. 2005;25:539–545. doi: 10.1161/01.ATV.0000155462.24263.e4. [DOI] [PubMed] [Google Scholar]
- 34.Hill BG, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madonna R, et al. High glucose-induced hyperosmolarity contributes to COX-2 expression and angiogenesis: Implications for diabetic retinopathy. Cardiovasc. Diabetol. 2016;15:18. doi: 10.1186/s12933-016-0342-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosentino F, et al. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: Role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 37.Chen F, et al. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of nadph oxidase activation-driven oxidative stress. CNS Neurosci. Ther. 2013 doi: 10.1111/cns.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011 doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci. 2019;20:3775. doi: 10.3390/ijms20153775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Förstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du XL, et al. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimmeler S, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Vanhoutte PM, Leung SWS. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015;129:83–94. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Wu G. Nitric oxide synthesis and the effect of aminoguanidine and arginine on the onset of diabetes in the spontaneously diabetic BB rat. Diabetes. 1995;44:360–364. doi: 10.2337/diab.44.3.360. [DOI] [PubMed] [Google Scholar]
- 45.Pitocco D, et al. Role of asymmetric-dimethyl-l-arginine (ADMA) and nitrite/nitrate (NOx) in the pathogenesis of oxidative stress in female subjects with uncomplicated type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 2009;86:173–176. doi: 10.1016/j.diabres.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 46.Chiueh CC. Neuroprotective properties of nitric oxide. Ann. N. Y. Acad. Sci. 1999;890:301–311. doi: 10.1111/j.1749-6632.1999.tb08007.x. [DOI] [PubMed] [Google Scholar]
- 47.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai S, Khoo J, Mussa S, Alp NJ, Channon KM. Endothelial nitric oxide synthase dysfunction in diabetic mice: Importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia. 2005;48:1933–1940. doi: 10.1007/s00125-005-1857-5. [DOI] [PubMed] [Google Scholar]
- 49.Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA. 2018;115:5839–5848. doi: 10.1073/pnas.1804932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brodsky SV, Morrishow AM, Dharia N, Gross SS, Goligorsky MS. Glucose scavenging of nitric oxide. Am. J. Physiol. Renal Physiol. 2017;280:F480–F486. doi: 10.1152/ajprenal.2001.280.3.F480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.