

Abstract
BACKGROUND:
Type 2 diabetes is a dementia risk factor, but its relation to Alzheimer’s disease (AD), the most common cause of dementia, is unclear.
OBJECTIVE:
Our primary objective was to examine the association of pre-diabetes and type 2 diabetes with brain amyloid β (Aβ), the putative main culprit of AD. Our secondary objective was to examine the association of pre-diabetes and type 2 diabetes with neurodegeneration, cerebrovascular disease (CVD), and memory performance.
METHODS:
We conducted a cross-sectional study of 350 late middle-aged Hispanics without dementia in New York City. We classified diabetes status as normal glucose tolerance (NGT]), pre-diabetes, and type 2 diabetes following American Diabetes Association Criteria. Brain Aβ was ascertained as global Aβ standardized value uptake ratio (SUVR) using positron emission tomography (PET) with 18F-Florbetaben. Neurodegeneration was operationalized as cortical thickness in regions affected by AD using magnetic resonance imaging (MRI). CVD was operationalized as white matter hyperintensity volume (WMH) on MRI, and memory as performance with the selective reminding test (SRT).
RESULTS:
Mean age was 64.15 ± 3.34 years, 72.00% were women, and 35.43% were APOE-ε4 carriers. Pre-diabetes, but not type 2 diabetes, was associated with higher Aβ compared with NGT. Type 2 Diabetes treatment was related to lower Aβ. Type 2 diabetes was related to lower cortical thickness, higher WMH, and lower SRT score.
CONCLUSION:
Pre-diabetes, but not type 2 diabetes, is associated with higher brain Aβ in late middle age, and this observation could be explained by the relation of diabetes treatment with lower brain Aβ. Whether Type 2 diabetes treatment lowers brain Aβ requires further study.
Keywords: Type 2 diabetes, amyloid, middle age, Hispanic
INTRODUCTION
Eleven percent of people aged 65 years and older have dementia in the United States, and the most common cause is Alzheimer’s disease (AD), followed by vascular dementia [1]. One of the most consistent risk factors reported for dementia, including AD and vascular dementia, is type 2 diabetes and its antecedent, pre-diabetes [2]. The role of type 2 diabetes and pre-diabetes in dementia is of significant public health importance because approximately 12% of the United States (US) adult population has type 2 diabetes (30.2 million) and 33% has pre-diabetes (84.1 million) [3]. Moreover, the majority of the US population aged 65 years and older, the most susceptible to dementia, has pre-diabetes or type 2 diabetes [3]. Type 2 diabetes is known to be a cerebrovascular risk factor, but neuropathology [4–10] and biomarker studies [11–14] are conflicting on whether type 2 diabetes is related to AD neuropathology. Neuropathology studies may be limited by selection bias, and most biomarkers studies have been conducted in elderly subjects, which may be limited by selection and survival bias. Decades of advances in AD research, particularly in AD biomarkers [15, 16], have led to the dominance of 3 neuropathological constructs: brain amyloid, brain tau, and neurodegeneration[15]. Current understanding of the natural history leading to dementia due to AD can be summarized as follows [15]: the 2 main proteinopathies underlying AD, amyloid and tau, are separate processes, but amyloid deposition is the main driver of AD and accelerates tau deposition; amyloid and tau deposition precede and cause neurodegeneration, which leads to the clinical syndromes of amnestic mild cognitive impairment (MCI) and dementia. The constructs of amyloid, tau, and neurodegeneration feature prominently in the recent National Institute on Aging (NIA)/Alzheimer’s Association (AA) 2018 research framework [17]. This framework proposes to conduct research in which biomarkers of amyloid, tau, and neurodegeneration are used as outcomes in research for the purpose of better understanding the mechanisms and sequence of neuropathology. Cerebrovascular disease and cognitive performance are secondary constructs in this framework. Our primary objective was to examine whether type 2 diabetes and pre-diabetes are related to brain Amyloid β (Aβ) burden, the main pathological hallmark of AD. Our secondary objective was to examine the relation of type 2 diabetes and pre-diabetes with neurodegeneration, cerebrovascular disease and memory performance in late middle age. We hypothesized that pre-diabetes and type 2 diabetes compared with normal glucose tolerance (NGT), would be related to higher brain Aβ burden, neurodegeneration, cerebrovascular disease, and memory performance in late middle age.
METHODS
Study Design and Population:
This was a cross-sectional analysis of 350 participants of a study focusing on the relation of type 2 diabetes and brain amyloid in middle-aged Hispanics conducted at Columbia University Irving Medical Center (CUIMC) in New York City, recruited between 03/01/2016 and 07/31/2019. We targeted Hispanics because they are the most common ethnic group in the community surrounding CUIMC [18] and because there is a paucity of AD biomarkers studies in Non-Whites [15]. In addition, Hispanics have a higher prevalence of pre-diabetes and type 2 diabetes compared with Non-Hispanic Whites [3]. Inclusion criteria included ages between 55 and 69 years, men and women, able to undergo phlebotomy, clinical and neuropsychological assessments, 3T brain magnetic resonance imaging (MRI), and Positron Emission Tomography (PET) with the Aβ radioligand 18F-Florbetaben. Exclusion criteria included dementia diagnosis, cancer other than non-melanoma skin cancer, and MRI contraindications. We screened 659 potential participants; 114 (17.30%) declined to participate, 178 (27.01%) were ineligible, 16 (2.43%) and did not complete study procedures (Supplemental Figure 1). One additional participant (0.15%) was excluded from analyses due to incomplete data on APOE genotype, the most important predictor of in-vivo brain amyloid burden [19]. The interval between amyloid PET and MRI was 15.79 ± 33.41 days. The Institutional Review Board and the Joint Radiation Safety Commission at CUIMC approved this study. Participants provided written informed consent. Funding sources had no role in study design, data collection, data analyses or interpretation.
Study measures:
although this is a cross-sectional study in which temporality can only be assumed, we refer to the independent variables as exposure and the dependent variables as outcomes.
Exposures.
The main exposure variable was type 2 diabetes category. Type 2 diabetes categories were normal glucose tolerance (NGT; defined by hemoglobin A1c [HbA1c] < 5.7%), pre-diabetes (HbA1c 5.7 to 6.4%), and unknown type 2 diabetes (HbA1c ≥ 6.5%) following American Diabetes Association guidelines [20]; known type 2 diabetes was ascertained by self-report. We focused on type 2 diabetes categories (NGT, pre-diabetes, and type 2 diabetes) as the exposure because these are clinical entities diagnosed in clinical practice and used in epidemiologic research. HbA1c was measured using a turbidimetric inhibition immunoassay on the automated analyzer Cobas Integra 400 plus (Roche Diagnostics, Indianapolis, IN). We examined HbA1c continuously as a secondary exposure variable.
Outcomes.
The primary outcome was brain Aβ burden ascertained as global standardized uptake value ratio (SUVR) measured with 18F-Florbetaben PET. The secondary outcomes were neurodegeneration, focusing on cortical thickness in areas affected by AD [21] obtained from 3T MRI, cerebrovascular disease, a known complication of type 2 diabetes [3], ascertained as white matter hyperintensity volume (WMH) on MRI, and memory performance (verbal learning), the cognitive domain affected earliest in AD [1].
Amyloid PET:
Participants underwent 18F-Florbetaben PET imaging in a Siemens Biograph64 mCT/PET scanner (target dose: 8.1 mCi; 4×5 minute frames; iterative reconstruction algorithm; voxel size: 1.6×1.6×1mm3. Images were acquired over 20 minutes starting 90 minutes post-injection. Dynamic PET frames (4 scans) were aligned to the first frame using rigid-body registration and a static PET image was obtained by averaging the four registered frames. The static PET image was then registered with the CT scan obtained for attenuation correction during PET imaging reconstruction by rigid-body registration with information theoretic cost function to generate a fused image with skull. The structural T1 image in FreeSurfer space was registered to CT/PET fused image using normalized mutual information and six degrees of freedom. A combination of the two transformation matrices obtained from the two rigid-body registrations was used to transfer all Freesurfer regional masks and the cerebellar gray matter from FreeSurfer space to static PET image space using nearest neighbor interpolation [22]. The standardized uptake value (SUV), defined as the decay-corrected brain radioactivity concentration normalized for injected dose and body weight, was calculated in all FreeSurfer regions. The SUV in each region as well as each voxel was normalized to the SUV in cerebellar gray matter to derive the regional and voxel-wise SUVR. Overall mean Aβ burden was calculated from voxel-based, individual region of interests (ROI), including lateral temporal cortex, parietal cortex, cingulate cortex, and frontal cortex.
The 2018 NIA/AA research framework recommends categorizing AD biomarkers as positive or negative (or high or low), but there is uncertainty about the right approach for categorization [23]. Brain Aβ SUVR has a bimodal distribution and Aβ positivity is usually examined using cutoffs that vary by study [24], but usually represent the point of separation in the bimodal distribution (see supplemental Figure 2). Aβ positivity in the age group we examined has a prevalence of less than 15% [25], but Aβ levels under the positivity threshold are also clinically significant [26] and should be taken into account. We categorized Aβ as high using a SUVR threshold of 1.34, determined using the K-means clustering method, which identifies the partition between the 2 peaks in the Aβ SUVR distribution, which is usually used to determine Aβ positivity quantitatively (supplemental Figure 2). Values under this threshold were categorized as intermediate and low using a median split (SUVR = 1.127) of the range of SUVR values under the positivity threshold (SUVR =1.34), which had a normal distribution. We conducted sensitivity analyses categorizing SUVR by the median (SUVR = 1.134), and examining amyloid SUVR as a continuous outcome for all participants, and restricting analyses to participants with Aβ levels under the positivity threshold.
MRI:
MR images were acquired in a General Electric Signa Premier 3T scanner and processed with FreeSurfer (v6.0 http://surfer.nmr.mgh.harvard.edu/). The measure of neurodegeneration was the average cortical thickness from regions typically affected by AD [21], including entorhinal cortex, parahippocampus, inferior parietal lobule, pars opercularis, pars orbitalis, pars triangularis, inferior temporal pole, supramarginal gyrus, superior parietal lobe, and superior frontal lobe. White matter hyperintensity volume was derived with in-house developed software. Briefly, each participant’s T2-weighted FLAIR MRI scan was intensity normalized and brain extracted. A Gaussian mixture model was fit to the log-transformed distribution of voxel intensity values. The voxels included in the Gaussian distribution that comprised the highest intensity values were labeled, summed, and multiplied by voxel dimensions to yield total WMH volumes in cm^3. Labeled images were visually inspected and edited if necessary. We used the ratio of total WMH and total cranial volume (TCV) for analyses. Total WMH in cm3 was defined as the sum of the number of voxels that are labeled multiplied by voxel dimensions. We used the ratio of total WMH and total cranial volume (TCV) for analyses.
Verbal learning was ascertained using total recall in the Buschke Selective Reminding test (SRT) [27].
Covariates.
We examined age, education, Hispanic subgroup, APOE-ε4 genotype, body mass index (BMI), lipids (high density lipoprotein [HDL] and low-density lipoprotein [LDL]), and diastolic and systolic blood pressure, and medications for diabetes, hypertension, and dyslipidemia. Hispanic subgroup was classified following the format of the 2010 Census by country or region of origin (e.g. Mexican, Puerto Rican, Cuban, Dominican) [28]. The rationale for the covariates is as follows. Age, sex, and education are important predictors of dementia. APOE-ε4 genotype is the strongest risk factor for sporadic dementia due to AD [1], and also the strongest determinant of in-vivo amyloid burden [25]. Dyslipidemia (high LDL and low HDL), obesity (high BMI), and hypertension (high blood pressure) tend to cluster with type 2 diabetes [20]. BMI was estimated as weight in kg divided by height in meters squared. Cholesterol, HDL, and triglycerides were measured on an automated immunochemistry analyzer, Integra 400 plus (Roche Diagnostics, Indianapolis, IN) using an enzymatic colorimetric assay with a lower limit of quantitation of 3.09 mg/dL for HDL and 0.1mmol/L for cholesterol and triglycerides. LDL was calculated using the Friedewald formula [29]. APOE-ε4 genotyping was conducted by LGC genomics (Beverly, MA) using single nucleotide polymorphisms rs429358 and rs7412. Medication for diabetes included metformin, sulfonylureas, dipeptidyl peptidase 4 (DPP-4) inhibitors, peroxisome proliferator-activator γ (PPAR-γ) agonists, Glucagon Like Peptid 1 (GLP1) agonists, and sodium glucose transport 2 (SGLT2) inhibitors; none of the study participants reported using insulin. Blood pressure medications included betablockers, Angiotensin Converting Enzyme (ACE) inhibitors, Angiotension Receptor Blockers (ARB), diuretics, calcium channel blockers, and vasodilators. Lipid medications included HMG-CoA reductase inhibitors (statins), bile acids, ezetimibe, and fibrates.
Statistical analyses:
We examined the distribution of all variables. Global Aβ SUVR and the WMH/TCV ratio were not normally distributed. Aβ SUVR had a bimodal distribution as expected, and no transformation approximated a normal distribution, but the SUVR values under the positivity threshold resembled a normal distribution (supplemental Figure 2). WMH/TCV ratio required a logarithmic transformation to approximate a normal distribution. Bivariate comparisons were made using analysis of variance for continuous variables, and chi-squared for categorical variables. For Aβ categories (low, intermediate, and high) as the outcome, we used multinomial logistic regression adjusting for covariates. For multivariate analyses examining continuous outcomes, we used linear regression when examining glycemia as a continuous exposure and analyses of covariance when examining type 2 diabetes categories as the exposure.. We show results for 3 models. Model 1 was un-adjusted, model 2 adjusted for, age, sex, and APOE-ε4, and model 3 was adjusted for risk factor variables that were different among diabetes subgroups. The rationale for Model 2 was to examine adjustment for demographics and for the most important genetic determinant of amyloid burden in sporadic AD. The rationale for model 3 was to adjust for the metabolic, vascular, and treatment factors that accompany type 2 diabetes. This third model has important caveats. First, the covariates are correlated with the main exposure and can be difficult to disentangle independent effects, and second, they may be in the causal pathway between type diabetes and any outcome. Thus, attenuation of results in model 3 could be interpreted as evidence of potential mediation, not confounding, or interpreted as being the result of over adjustment in the face of multicollinearity. Statistical significance was considered at p < 0∙05. Analyses were conducted using SAS version 9.4m5 and R version 3.6.0.
RESULTS
The mean age of participants was 64.15 ± 3.34 years, 72% were women (Table 1), and 35.43% were APOE-ε4 carriers. All participants were Hispanics and most were Caribbean Hispanics of Dominican descent. The distribution of type 2 diabetes categories was as follows: 39.71% had NGT, 28.57 % had pre-diabetes, 3.42% had unknown type 2 diabetes, and 28.28% had known type 2 diabetes. Given the low proportion of persons with unknown type 2 diabetes, they were grouped with known type 2 diabetes. There were no differences between type 2 diabetes categories in age, sex, Hispanic subgroups and education (Table 1). As expected, use of blood pressure and lipid lowering medications was higher in persons with pre-diabetes and type 2 diabetes compared with persons with NGT. HDL was higher in persons with NGT as compared with persons with pre-diabetes and type 2 diabetes. LDL was lowest among persons with type 2 diabetes, the group with the highest prevalence of use of lipid lowering medications. Use of diabetes medications were reported by 72.97% of persons with type 2 diabetes. Metformin was the most commonly used medication (62.16% of all persons with type 2 diabetes, 85.18% of all diabetes medications).
Table 1.
Demographic and other characteristics for the entire sample and by diabetes status (normal glucose tolerance [NGT], pre-diabetes, type 2 diabetes) defined by American Diabetes Association criteria using Hemoglobin A1c (HbA1c).
| Characteristics | Entire sample (n = 350) |
NGT (n = 139) |
Pre-diabetes (n = 100) |
Type 2 Diabetes (n = 111) |
P value |
|---|---|---|---|---|---|
| Age in years, mean (SD) | 64.15 (3.34) | 63.69 (3.41) | 64.20 (3.04) | 64.68 (3.45) | 0.067 |
| Women, Number (%) | 252 (72.00%) | 106 (76.26%) | 72 (72.00%) | 74 (66.67%) | 0.24 |
| Hispanic subgroup Number (%) | |||||
| Dominican | 299 (85.43%) | 115 (82.73%) | 86 (86.00%) | 98 (88.29%) | 0.46 |
| Other Caribbean Hispanic | 21 (6.00%) | 9 (6.47%) | 6 (6.00%) | 6 (5.41%) | |
| South American | 18 (5.14%) | 9 (6.47%) | 5 (5.00%) | 4 (3.60%) | |
| Unspecified Hispanic | 7 (2.00%) | 5 (3.60%) | 2 (2.00%) | 0 (0.00%) | |
| Central American | 4 (1.14%) | 1 (0.72%) | 1 (1.00%) | 2 (1.80%) | |
| Education in years, mean (SD) | 10.54 (3.94) | 11.16 (3.43) | 10.35 (4.29) | 9.93 (4.12) | 0.042 |
| Medication Use, Number (%) | |||||
| Diabetes medications | 85 (24.29%) | 1 (0.72%) | 3 (3.00%) | 81 (72.97%) | <0.0001 |
| Metformin | 73 (20.86%) | 1 (0.72%) | 3 (3.00%) | 69 (62.16%) | <0.0001 |
| Sulfonylurea | 20 (5.71%) | 0 (0.00%) | 0 (0.00%) | 20 (18.02%) | |
| DPP-4i | 16 (4.57%) | 0 (0.00%) | 1 (1.00%) | 15 (13.51%) | |
| PPAR | 6 (1.71%) | 0 (0.00%) | 0 (0.00%) | 6 (5.41%) | |
| GLP-1a | 4 (1.14%) | 0 (0.00%) | 0 (0.00%) | 4 (3.60%) | |
| SGLT2i | 2 (0.57%) | 0 (0.00%) | 0 (0.00%) | 2 (1.80%) | |
| Blood pressure medications | 151 (43.14%) | 39 (28.06%) | 40 (40.00%) | 72 (64.86%) | <0.0001 |
| Lipid lowering medications | 131 (37.43%) | 36 (25.90%) | 33 (33.00%) | 62 (55.86%) | <0.0001 |
| APOE-ε4, Number (%) | 124 (35.43%) | 49 (35.25%) | 33 (33.00%) | 42 (37.84%) | 0.39 |
| HbA1c (%), mean (SD) | 6.25 (1.32) | 5.41 (0.20) | 5.94 (0.21) | 7.55 (1.67) | <0.0001 |
| Body mass index, mean (SD) | 28.86 (4.96) | 27.48 (4.05) | 29.75 (5.48) | 29.80 (5.13) | 0.0001 |
| HDL mg/dL, mean (SD) | 54.33 (14.65) | 57.82 (15.14) | 53.94 (15.12) | 50.32 (12.49) | 0.0003 |
| LDL mg/dL , mean (SD) | 110.12 (34.46) | 120.94 (32.41) | 111.47 (31.64) | 95.25 (34.29) | <0.0001 |
| SBP mm Hg, mean (SD) | 134.16 (17.80) | 133.32 (18.26) | 134.05 (16.89) | 135.31 (18.13) | 0.68 |
| DBP mm Hg, mean (SD) | 82.65 (10.20) | 83.32 (9.99) | 82.57 (10.55) | 81.88 (10.18) | 0.54 |
SD = Standard deviation; DPP-4i = dipeptidyl peptidase 4 inhibitors; SGLT2i = sodium-glucose co-transporter 2 inhibitors; GLP-1a = glucagon-like peptide receptor 1 agonist; PPAR = Peroxisome proliferator-activated receptor gamma agonist; HbA1c =Hemoglobin A1c; HDL = High Density Lipoprotein; LDL= low density lipoprotein; SBP = Systolic Blood pressure; DBP = diastolic blood pressure.
APOE-ε4 genotype was a strong predictor of Aβ burden examined categorically and continuously. As compared with non-carriers, APOE-ε4 carriers had elevated odds of intermediate (OR: 1.97, 95% CI: 1.18, 3.30, p = 0.0097) and high Aβ (OR: 10.12, 95% CI: 3.90, 26.21, p <0.0001), adjusting for age and sex. Global Aβ SUVR examined as a continuous outcome was also higher in APOE-ε4 carriers (age and sex adjusted means, 1.21 ± 0.01 vs. 1.13 ± 0.09; p<0.0001) for all subjects, and among the 322 individuals under the Aβ positivity threshold (SUVR 1.14 ± 0.006 in carriers vs. 1.12 ± 0.004 in non-carriers; p=0.0017).
We compared Aβ categories across type 2 diabetes categories (Figure 1). Persons with pre-diabetes had a higher proportion of high Aβ (10.00%) and intermediate Aβ (53.00%) as compared with both persons with NGT (8.63% positive, 41.73% intermediate) and type 2 diabetes (5.41% positive, 45.05% intermediate). Persons with pre-diabetes, but not type 2 diabetes, had higher odds of high and intermediate Aβ as compared with persons with NGT, and this result was significant for intermediate Aβ after adjustment for age, sex, and APOE-ε4, but was modestly attenuated in the model adjusting for other risk factors (Table 2). HbA1c examined continuously was not related to intermediate Aβ (OR =1.00, 95% CI: 0.84–1.19), but had an inverse relation with high Aβ (OR =0.57; 95% CI 0.31, 1.03; p = 0.062) that was close to statistical significance after adjustment for age, sex, and APOE-ε4, consistent with the findings for diabetes categories.
Figure 1.
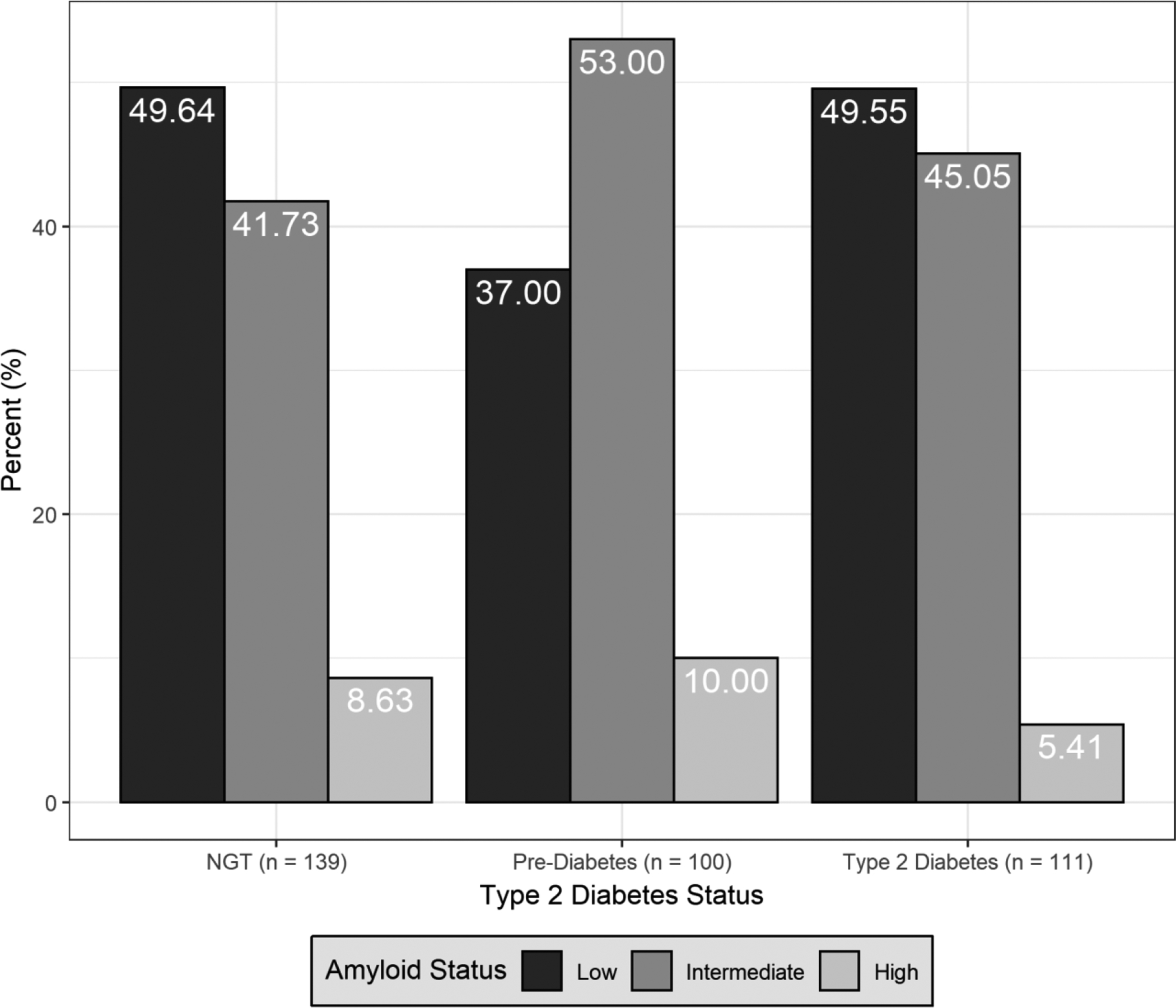
Comparison by type 2 diabetes status (normal glucose tolerance [NGT], pre-diabetes, diabetes), of the proportion of brain amyloid categories (low, intermediate, high).
Table 2.
Relation of diabetes status categories (normal glucose tolerance [NGT; reference category], pre-diabetes, type 2 diabetes [T2D]), and of glycemia ascertained as hemoglobin A1c (HbA1c), with global brain amyloid level categories (low, intermediate, and high). Odds ratios (OR) were estimated with multinomial logistic regression. OR report the odds of the designated amyloid group compared to low amyloid/Model 1 is unadjusted; Model 2 is adjusted for age, sex, and APOE-ε4; Model 3 is adjusted for lipids, body mass index, and differences in medication.
| Model 1 | Model 2 | Model 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| No. | Amyloid Group | OR (95% CI) | p value | OR (95% CI) | p value | OR (95% CI) | P value | |
| Type 2 diabetes categories | ||||||||
| NGT | 58 | Intermediate | 1.0 | 1.0 | 1.0 | |||
| 12 | High | 1.0 | 1.0 | 1.0 | ||||
| Pre-diabetes | 53 | Intermediate | 1.70 (0.99, 2.94) | 0.056 | 1.92 (1.06, 3.47) | 0.031 | 1.68 (0.91, 3.11) | 0.098 |
| 10 | High | 1.55 (0.61, 3.94) | 0.35 | 1.81 (0.67, 4.91) | 0.25 | 1.58 (0.56, 4.44) | 0.38 | |
| T2D | 50 | Intermediate | 1.08 (0.64, 1.82) | 0.77 | 1.15 (0.65, 2.03) | 0.63 | 1.21 (0.50, 2.95) | 0.67 |
| 6 | High | 0.63 (0.22, 1.78) | 0.38 | 0.53 (0.18, 1.62) | 0.27 | 0.90 (0.20, 4.03) | 0.89 | |
| p (global) | 0.24 | 0.11 | 0.55 | |||||
| Hemoglobin A1c (HbA1c) | ||||||||
| HbA1c | 161 | Intermediate | 0.96 (0.82, 1.13) | 0.63 | 1.00 (0.84, 1.19) | 0.99 | 1.02 (0.81, 1.28) | 0.89 |
| 28 | High | 0.57 (0.32, 1.01) | 0.054 | 0.5 (0.31, 1.03) | 0.062 | 0.59 (0.28, 1.26) | 0.18 | |
| p (global) | 0.15 | 0.17 | 0.37 | |||||
CI = Confidence interval
We also conducted sensitivity analyses examining high Aβ levels defined by the median SUVR (1.134), and found that prediabetes was related to high Aβ levels after adjustment for age, sex, and APOE-ε4 as compared with NGT, but type 2 diabetes was not (Table 3).
Table 3.
Relation of diabetes status categories (normal glucose tolerance [NGT], pre-diabetes, type 2 diabetes [T2D]), and of glycemia ascertained as hemoglobin A1c (HbA1c), with high global brain amyloid level by the median standardized uptake value ratio (SUVR). Odds ratios (OR) and 95% confidence intervals (CI) estimated defined by multinomial logistic regression. ORs report the odds of high vs. low amyloid defined by the median SUVR. Model 1 is unadjusted; Model 2 is adjusted for age, sex, and APOE-ε4; Model 3 is adjusted for lipids, body mass index, and differences in medication. For analyses with diabetes categories, NGT is the reference category.
| Model 1 | Model 2 | Model 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| High Amyloid | OR (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | ||
| Type 2 diabetes categories | ||||||||
| NGT | 65 | 1.0 | 1.0 | 1.0 | ||||
| Pre-diabetes | 58 | 1.57 (0.94, 2.64) | 0.087 | 1.78 (1.01, 3.13) | 0.045 | 1.60 (0.89, 2.86) | 0.12 | |
| T2D | 52 | 1.00 (0.61, 1.69) | 0.99 | 1.06 (0.61, 1.84) | 0.84 | 1.00 (0.43, 2.34) | 0.99 | |
| P (global) | 0.17 | 0.10 | 0.25 | |||||
| Hemoglobin A1c (HbA1c) | ||||||||
| HbA1c | 175 | 0.94 (0.80, 1.10) | 0.46 | 0.99 (0.83, 1.18) | 0.91 | 0.94 (0.73, 1.22) | 0.66 | |
We also compared Aβ SUVR examined continuously among diabetes groups (Table 4). Persons with pre-diabetes had higher SUVR as compared with persons with NGT in all models, and Aβ SUVR in persons with diabetes were similar to NGT, although these results were not statistically significant. In analyses restricted to individuals under the positivity threshold (SUVR = 1.34), persons with pre-diabetes (N=90) still showed higher Aβ SUVR, and the p-values in the model adjusted for age, sex, and APOE-ε4 was statistically significant (Table 4), supporting our observation that the relation between pre-diabetes and Aβ is driven mostly by intermediate Aβ levels.
Table 4.
Relation of diabetes status categories (normal glucose tolerance [NGT], pre-diabetes, type 2 diabetes [T2D]), and of glycemia ascertained as hemoglobin A1c (HbA1c), with brain amyloid standardized uptake value ratio (SUVR). Amyloid SUVR adjusted means and standard deviations (SD) are compared across T2D categories using analysis of covariance. Coefficients (β) and 95% confidence intervals (CI) relating HbA1c and amyloid SUVR were estimated using linear regression models. Model 1 is unadjusted; Model 2 is adjusted for age, sex, and APOE-ε4; Model 3 is adjusted for lipids, body mass index, and differences in medication (lipid, blood pressure, and diabetes).
| Model 1 | Model 2 | Model 3 | |||||
|---|---|---|---|---|---|---|---|
| No. | mean (SD) or β (95% CI) | P value | mean (SD) or β (95% CI) | P value | mean (SD) or β (95% CI) | P value | |
| Global Brain Amyloid SUVR | |||||||
| Type 2 diabetes categories | |||||||
| NGT | 139 | 1.157 (0.011) | 1.156 (0.011) | 1.156 (0.013) | |||
| Pre-diabetes | 100 | 1.180 (0.013) | 0.19 | 1.182 (0.013) | 0.13 | 1.177 (0.014) | 0.21 |
| T2D | 111 | 1.146 (0.013) | 0.53 | 1.145 (0.012) | 0.47 | 1.150 (0.018) | 0.82 |
| P (global) | 0.17 | 0.099 | 0.36 | ||||
| Hemoglobin A1c (HbA1c) | |||||||
| HbA1c | 350 | −0.10 (−0.20, 0.01) | 0.074 | −0.07 (−0.17, 0.03) | 0.19 | −0.05 (−0.18, 0.08) | 0.46 |
| Global Brain Amyloid SUVR below the positivity threshold | |||||||
| Type 2 diabetes categories | |||||||
| NGT | 127 | 1.121 (0.005) | 1.120 (0.005) | 1.119 (0.006) | |||
| Pre-diabetes | 90 | 1.136 (0.006) | 0.075 | 1.137 (0.006) | 0.030 | 1.133 (0.006) | 0.073 |
| T2D | 105 | 1.126 (0.006) | 0.59 | 1.127 (0.005) | 0.32 | 1.131 (0.008) | 0.32 |
| P (global) | 0.20 | 0.093 | 0.18 | ||||
| Hemoglobin A1c (HbA1c) | |||||||
| HbA1c | 322 | −0.01 (−0.06, 0.04) | 0.68 | 0.004 (−0.04, 0.05) | 0.86 | 0.01 (−0.05, 0.07) | 0.80 |
Since a difference between persons with type 2 diabetes and those with pre-diabetes is the use of diabetes medications, we conducted post-hoc analyses exploring the association of diabetes medication use with Aβ categories among the 211 persons with pre-diabetes and type 2 diabetes using multinomial logistic regression. Three persons in the pre-diabetes group reported using the diabetes medication metformin, which is used for diabetes prevention, and 81 of the persons with type 2 diabetes reported using diabetes medications. The odds of intermediate (OR 0.57; 95% CI 0.30, 1.07; p = 0.079) and high Aβ (OR =0.20; 95% CI 0.05, 0.81; p = 0.024) was lower in persons taking diabetes medications compared to those not taking diabetes medications after adjustment for age, sex, and APOE-ε4. When global Aβ SUVR was examined as the outcome, Aβ SUVR was lower in persons reporting medication use (SUVR 1.14 ± 0.014) vs. those not reporting medication use (SUVR = 1.18 ± 0.011; p = 0.041) after adjustment for age, sex, and APOE-ε4.
In terms of the secondary outcomes of neurodegeneration, WMH, and memory performance, Type 2 diabetes was associated with smaller cortical thickness, higher WMH, and lower total recall of words in the SRT (Table 5) in the unadjusted model and that adjusted for demographics and APOE-ε4. Higher glycemia was also associated with smaller cortical thickness and higher WMH.
Table 5.
Relation of diabetes status categories (normal glucose tolerance [NGT], pre-diabetes, type 2 diabetes [T2D]), and of glycemia ascertained as hemoglobin A1c (HbA1c) with the secondary outcomes of cortical thickness in Alzheimer’s Disease (AD) signature regions, ratio of white matter hyperintensities (WMH) to total cranial volume (TCV) with logarithmic transformation, and total recall of the Selective Reminding Test. Adjusted means and standard deviations (SD) are compared across T2D categories estimated using analysis of covariance. Coefficients (β) and 95% confidence intervals (CI) relating HbA1c and amyloid SUVR were estimated using linear regression models. Model 1 is unadjusted; Model 2 is adjusted for age, sex, and APOE-ε4; Model 3 is adjusted for lipids, body mass index, and differences in medication (lipid, blood pressure, and diabetes).
| Model 1 | Model 2 | Model 3 | |||||
|---|---|---|---|---|---|---|---|
| No. | mean (SD) or β (95% CI) | P value | mean (SD) or β (95% CI) | P value | mean (SD) or β (95% CI) | P value | |
| AD signature region cortical thickness | |||||||
| Type 2 diabetes categories | |||||||
| NGT | 139 | 2.699 (0.009) | 2.695 (0.009) | 2.684 (0.010) | |||
| Pre-diabetes | 100 | 2.683 (0.010) | 0.21 | 2.683 (0.010) | 0.37 | 2.676 (0.010) | 0.53 |
| T2D | 111 | 2.661 (0.010) | 0.0028 | 2.665 (0.009) | 0.018 | 2.689 (0.013) | 0.80 |
| P (global) | 0.012 | 0.061 | 0.73 | ||||
| Hemoglobin A1c (HbA1c) | |||||||
| HbA1c | 350 | −0.21 (−0.31, −0.10) | 0.0001 | −0.17 (−0.28, −0.07) | 0.0009 | −0.08 (−0.21, 0.05) | 0.21 |
| Ln (WMH/TCV) | |||||||
| Type 2 diabetes categories | |||||||
| NGT | 139 | 0.237 (0.102) | 0.276 (0.101) | 0.350 (0.118) | |||
| Pre-diabetes | 100 | 0.319 (0.121) | 0.61 | 0.316 (0.119) | 0.79 | 0.340 (0.131) | 0.95 |
| T2D | 111 | 0.932 (0.115) | <0.0001 | 0.885 (0.114) | <0.0001 | 0.742 (0.166) | 0.098 |
| P (global) | <0.0001 | 0.0001 | 0.20 | ||||
| Hemoglobin A1c (HbA1c) | |||||||
| HbA1c | 350 | 0.19 (0.09, 0.29) | 0.0001 | 0.16 (0.07, 0.26) | 0.0009 | 0.07 (−0.06, 0.19) | 0.30 |
| Selective Reminding Test, Total Recall | |||||||
| Type 2 diabetes categories | |||||||
| NGT | 139 | 40.442 (0.733) | 39.665 (0.680) | 39.579 (0.784) | |||
| Pre-diabetes | 100 | 39.828 (0.865) | 0.59 | 39.893 (0.792) | 0.83 | 39.678 (0.862) | 0.93 |
| T2D | 111 | 36.270 (0.817) | 0.0002 | 37.129 (0.756) | 0.014 | 37.626 (1.077) | 0.21 |
| P (global) | 0.0004 | 0.017 | 0.38 | ||||
| Hemoglobin A1c (HbA1c) | |||||||
| HbA1c | 350 | −0.10 (−0.20, 0.01) | 0.074 | −0.04 (−0.14, 0.06) | 0.41 | 0.05 (−0.07, 0.17) | 0.44 |
DISCUSSION
Contrary to our hypothesis, Aβ burden, the main pathological driver of AD, was higher in persons with pre-diabetes as compared with NGT, while Aβ burden in persons with type 2 diabetes was similar to NGT. Post hoc exploratory analyses showed that use of diabetes medications was related to lower brain Aβ, providing reason to speculate that the lower Aβ seen in type 2 diabetes compared with pre-diabetes could be explained by effects of medication use. Given the cross-sectional nature of our study, the observation that diabetes medication use is related to lower brain Aβ is hypothesis generating, and no causal inferences can be made.
Our secondary outcomes were related to type 2 diabetes as hypothesized. Type 2 diabetes was related related to worse neurodegeneration, cerebrovascular disease, and memory performance as compared with NGT.
Type 2 diabetes and its antecedent, pre-diabetes, are strong risk factors for CVD [3]. It is increasingly accepted that cerebrovascular disease interacts with AD pathology to cause dementia [1], and this seems a likely mechanism that links type 2 diabetes to dementia due to AD. However, type 2 diabetes may promote brain Aβ accumulation [30] or impaired clearance [31], but a causal association of type diabetes with AD has not been proven. Studies addressing this issue are conflicting. Neuropathology studies from Finland [5, 10], and the United States[4, 9, 14] found that type 2 diabetes history was not related to higher AD neuropathology. However, a study of Japanese-Americans found that type 2 diabetes is associated with increased AD neuropathology, [6] and a Japanese autopsy study found that increased glycemia 10 years before autopsy was related to increased AD neuropathology [7]. A study from the Mayo Clinic Study of Aging (MCSA) in the United States among 749 persons without dementia and a mean age of 79 years reported that type 2 diabetes was related to an AD brain metabolism pattern on Fluorodeoxyglucose PET, but not with brain Aβ ascertained by PET [11]. The Baltimore Longitudinal Study of Aging (BLSA) [14] found among 197 persons with autopsy at age 88 years that glucose assessed approximately at age 66 was not related to AD neuropathology. The same study had data on 53 subjects with Aβ PET at age 79 years and found no evidence of a relation of type 2 diabetes and brain Aβ. The Atherosclerosis Risk in Communities Study (ARIC) reported that type 2 diabetes was not related to Aβ positivity among 346 participants with a mean age of 75.8 years [13]. A study in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) among 816 persons with a mean age of 74.3 years reported that type 2 diabetes was not associated with in-vivo Aβ in PET or cerebrospinal fluid (CSF), but type 2 diabetes was associated with higher tau in CSF, and lower cortical thickness [12]. Another analysis from ADNI relating diabetes status to CSF and cortical Aβ found that persons with diabetes had lower cerebral Aβ in anterior cingulate, precuneus, and temporal lobe than persons with diabetes, and higher levels of CSF Aβ 1–42 [32]. Reasons for the discrepancy in the results between these studies and ours is that our study was conducted in a younger age group, differentiated pre-diabetes and NGT among those without type 2 diabetes, and examined intermediate levels of Aβ in addition to examining levels traditionally considered positive.
Few studies have examined the potential relation of diabetes treatment with AD pathology. A recent analysis of data from 900 participants in ADNI who underwent lumbar puncture with a mean age of 73.54 years [33] showed that persons with untreated diabetes had higher biomarkers of AD in the cerebrospinal fluid (phosphorylated-tau, total-tau, and phosphorylated -tau/ Aβ 1–42 levels) than the euglycemic, pre-diabetes, and treated diabetes groups, and these subjects also had a faster progression to dementia. As compared with our study, the ADNI analyses focused on cerebrospinal fluid biomarkers, primarily tau, in participants in their seventies, whereas our study focused on brain Aβ and neurodegeneration ascertained with brain imaging in a cohort that is almost 10 years younger. As in our study, the main diabetes treatment reported by participants was metformin.
We believe that a major issue with studies examining the association of diabetes with AD is that they have been conducted in persons in the 8th and 9th decades of life, who are subject to selection bias and survival bias related to type 2 diabetes related morbidity and mortality, without complete type 2 diabetes category ascertainment, and subject to the fact that Aβ accumulation plateaus in older subjects and those with cognitive impairment [34], which could preclude observing differences between comparison groups when examined late in the lifespan. We believe that our study overcame these limitations by examining a late middle-aged cohort, in whom Aβ accumulation is not plateauing and is accumulating, with ascertainment of type 2 diabetes categories and glycemia, Aβ, neurodegeneration, CVD, and cognitive function.
Our findings clearly support neurodegeneration and CVD as in-vivo neuropathologic correlates of glycemia and type 2 diabetes, and show that these associations are already evident in late middle age. Our finding that prediabetes, but not type 2 diabetes, is associated with increased Aβ burden, was contrary to our hypothesis, and requires further discussion. One clear difference between persons with pre-diabetes and type 2 diabetes is that persons with diabetes are treated with agents that decrease glycemia and persons with pre-diabetes are not. We found that diabetes treatment was related to lower Aβ in post-hoc exploratory analyses. We cannot infer that diabetes treatment is causing the lower Aβ because diabetes treatment itself could be a proxy for another related factor, and because of the cross-sectional nature of our study. The most common type 2 diabetes medication reported in our sample was metformin. Animal and cell culture studies examining the effect of metformin on Aβ have shown conflicting results, some showing an increase in Aβ [35, 36], but metformin has also been found to prevent Aβ accumulation and memory impairment in Alzheimer’s (APP/PS1) mice [37]. It is possible that our unexpected findings for pre-diabetes and diabetes treatment are chance findings. However, there are several arguments against this possibility. First, the comparison of vascular and metabolic variables across diabetes categories showed expected differences (e.g. higher BMI and lipid and blood pressure medication use in persons with pre-diabetes and diabetes). Second, we observed that APOE-ε4 is clearly related higher Aβ burden using our operationalization of Aβ burden, providing face validity for our Aβ measure. Lastly, the relations of diabetes and glycemia with neurodegeneration, cerebrovascular disease, and memory performance followed the expected pattern.
The study’s main limitation is the cross-sectional design. We cannot infer causality. Another relative limitation is the restriction to urban Hispanics in New York City who are able to undergo brain imaging, and the results may not be generalizable to other populations. However, the fact that APOE-ε4 is related to Aβ as reported in non-Hispanic populations [25], suggests that our results may be generalizable. The relatively high prevalence of pre-diabetes and type 2 diabetes in our sample is similar to that reported for Hispanics nationally [3]. Lastly, we did not report on tau, another important hallmark of AD along with Aβ and neurodegeneration [23], which has been related to type 2 diabetes [12, 33] in biomarker studies. We are currently implementing tau PET in the same participants (NCT03389100) and will report results once we achieve a sample size comparable to the present study.
In conclusion, pre-diabetes, but not diabetes, is related to higher Aβ burden in late middle age, while type 2 diabetes is related to worse neurodegeneration, CVD, and memory. Our results need to be replicated in other cohorts. Whether type 2 diabetes treatment or other factors related to type 2 diabetes account for the lower Aβ in persons with diabetes requires further study.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by National Institutes of Health grants R01AG050440, RF1AG051556. Partial support was provided by grants K24AG045334, P30AG059303, and ULT1TR001873. JA Luchsinger receives a stipend from Wolters Kluwer, N.V. as Editor in Chief of the journal Alzheimer’s Disease and Associated Disorders, and has served as a paid consultant to vTv therapeutics, Inc. and Recruitment Partners. The other authors have no interests to declare.
REFERENCES
- [1].(2019) 2019 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 15, 321–387. [Google Scholar]
- [2].Biessels GJ, Strachan MWJ, Visseren FLJ, Kappelle LJ, Whitmer RA (2013) Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes-Endocrinology doi: 10.1016/S2213-8587(13)70088-3. [DOI] [PubMed] [Google Scholar]
- [3].(2017), ed. Centers for Disease Control and Prevention USDoHaHS, Atlanta, GA. [Google Scholar]
- [4].Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA (2006) Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67, 1960–1965. [DOI] [PubMed] [Google Scholar]
- [5].Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M (2010) Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology 75, 1195–1202. [DOI] [PubMed] [Google Scholar]
- [6].Peila R, Rodriguez BL, Launer LJ, Honolulu-Asia Aging S (2002) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262. [DOI] [PubMed] [Google Scholar]
- [7].Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Susuki O, Kanba S, Kiyohara Y, Iwaki T (2010) Insulin resistance is associated with the pathology of Alzheimer’s disease: the Hisayama Study. Neurology. [DOI] [PubMed] [Google Scholar]
- [8].Dos Santos Matioli MNP, Suemoto CK, Rodriguez RD, Farias DS, da Silva MM, Leite REP, Ferretti-Rebustini REL, Farfel JM, Pasqualucci CA, Jacob Filho W, Arvanitakis Z, Naslavsky MS, Zatz M, Grinberg LT, Nitrini R (2017) Diabetes is Not Associated with Alzheimer’s Disease Neuropathology. Journal of Alzheimer’s disease : JAD 60, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J, Purohit DP, Perl DP, Davidson M, Mohs RC, Haroutunian V (2005) Type 2 diabetes is negatively associated with Alzheimer’s disease neuropathology. J Gerontol A Biol Sci Med Sci 60, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alafuzoff I, Aho L, Helisalmi S, Mannermaa A, Soininen H (2009) Beta-amyloid deposition in brains of subjects with diabetes. Neuropathol Appl Neurobiol 35, 60–68. [DOI] [PubMed] [Google Scholar]
- [11].Roberts RO, Knopman DS, Cha RH, Mielke MM, Pankratz VS, Boeve BF, Kantarci K, Geda YE, Jack CR Jr., Petersen RC, Lowe VJ (2014) Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J Nucl Med 55, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moran C, Beare R, Phan TG, Bruce DG, Callisaya ML, Srikanth V, Alzheimer’s Disease Neuroimaging I (2015) Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 85, 1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gottesman RF, Schneider AL, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH (2017) Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. Jama 317, 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thambisetty M, Jeffrey Metter E, Yang A, Dolan H, Marano C, Zonderman AB, Troncoso JC, Zhou Y, Wong DF, Ferrucci L, Egan J, Resnick SM, O’Brien RJ (2013) Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol 70, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, et al. (2018) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luchsinger JA, Cabral R, Eimicke JP, Manly JJ, Teresi J (2015) Glycemia, Diabetes Status, and Cognition in Hispanic Adults Aged 55–64 Years. Psychosom Med 77, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ossenkoppele R, Jansen WJ, Rabinovici GD, Knol DL, van der Flier WM, van Berckel BNM, Scheltens P, Visser PJ, Amyloid PETSG, Verfaillie SCJ, Zwan MD, Adriaanse SM, Lammertsma AA, Barkhof, Jagust WJ, Miller BL, Rosen HJ, Landau SM, Villemagne VL, Rowe CC, Lee DY, Na DL, Seo SW, Sarazin M, Roe CM, Sabri O, Barthel H, Koglin N, Hodges J, Leyton CE, Vandenberghe R, van Laere K, Drzezga A, Forster S, Grimmer T, Sánchez-Juan P, Carril JM, Mok V, Camus V, Klunk WE, Cohen AD, Meyer PT, Hellwig S, Newberg A, Frederiksen KS, Fleisher AS, Mintun MA, Wolk DA, Nordberg A, Rinne JO, Chételat G, Lleo A, Blesa R, Fortea J, Madsen K, Rodrigue KM, Brooks DJ (2015) Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313, 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].(2019) 6. Glycemic Targets: Standards of Medical Care in Diabetes—2019. Diabetes Care 42, S61. [DOI] [PubMed] [Google Scholar]
- [21].Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL (2009) The Cortical Signature of Alzheimer’s Disease: Regionally Specific Cortical Thinning Relates to Symptom Severity in Very Mild to Mild AD Dementia and is Detectable in Asymptomatic Amyloid-Positive Individuals. Cerebral Cortex 19, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tahmi M, Bou-Zeid W, Razlighi QR (2019) A fully automatic technique for precise localization and quantification of Amyloid-β PET scans. Journal of Nuclear Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R (2018) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jansen WJ, Ossenkoppele R, Knol DL, et al. (2015) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313, 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ, Visser PJ, and the Amyloid Biomarker Study G (2015) Prevalence of Cerebral Amyloid Pathology in Persons Without Dementia: A Meta-analysisPrevalence of Cerebral Amyloid Pathology in Persons Without DementiaPrevalence of Cerebral Amyloid Pathology in Persons Without Dementia. JAMA 313, 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Landau SM, Horng A, Jagust WJ (2018) Memory decline accompanies subthreshold amyloid accumulation. Neurology 90, e1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Buschke H, Fuld PA (1974) Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology 24, 1019–1025. [DOI] [PubMed] [Google Scholar]
- [28].Humes KR, Jones NA, Ramirez RR (2011) in 2010 Census Briefs, ed. Bureau UC U.S. Department of Commerce, Washington, DC. [Google Scholar]
- [29].Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, Without Use of the Preparative Ultracentrifuge. Clinical Chemistry 18, 499. [PubMed] [Google Scholar]
- [30].Watson GS, Craft S (2003) The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs 17, 27–45. [DOI] [PubMed] [Google Scholar]
- [31].Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, Tanzi RE, Selkoe DJ (2004) Partial Loss-of-Function Mutations in Insulin-Degrading Enzyme that Induce Diabetes also Impair Degradation of Amyloid {beta}-Protein. Am J Pathol 164, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li W, Risacher SL, Gao S, Boehm SL 2nd, Elmendorf JS, Saykin AJ (2018) Type 2 diabetes mellitus and cerebrospinal fluid Alzheimer’s disease biomarker amyloid beta1–42 in Alzheimer’s Disease Neuroimaging Initiative participants. Alzheimers Dement (Amst) 10, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McIntosh EC, Nation DA (2019) Importance of Treatment Status in Links Between Type 2 Diabetes and Alzheimer’s Disease. Diabetes Care 42, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jack CR Jr., Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC (2013) Brain beta-amyloid load approaches a plateau. Neurology 80, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF (2009) Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A 106, 3907–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Son SM, Shin HJ, Byun J, Kook SY, Moon M, Chang YJ, Mook-Jung I (2016) Metformin Facilitates Amyloid-beta Generation by beta- and gamma-Secretases via Autophagy Activation. J Alzheimers Dis 51, 1197–1208. [DOI] [PubMed] [Google Scholar]
- [37].Ou Z, Kong X, Sun X, He X, Zhang L, Gong Z, Huang J, Xu B, Long D, Li J, Li Q, Xu L, Xuan A (2018) Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain, Behavior, and Immunity 69, 351–363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.