

Abstract
Somatic mutations in the epidermal growth factor receptor (EGFR) gene, such as exon 19 deletion mutations, are important factors in determining therapeutic responses to gefitinib in non‐small‐cell lung cancer (NSCLC). However, some patients have activating mutations in EGFR and show poor responses to gefitinib. In this study, we examined three NSCLC cell lines, HCC827, PC9, and HCC2935, that expressed an EGFR exon 19 deletion mutation. All cells expressed mutant EGFR, but the PC9 and HCC2935 cells also expressed wild‐type EGFR. The HCC827 cells were highly sensitive to gefitinib under both normoxia and hypoxia. However, the PC9 and HCC2935 cells were more resistant to gefitinib under hypoxic conditions compared to normoxia. Phosphorylation of EGFR and ERK was suppressed with gefitinib treatment to a lesser extent under hypoxia. The expression of transforming growth factor‐α (TGFα) was dramatically upregulated under hypoxia, and the knockdown of TGFα or hypoxia‐inducible factor‐1α (HIF1α) reversed the resistance to gefitinib in hypoxic PC9 and HCC2935 cells. Finally, introduction of the wild‐type EGFR gene into the HCC827 cells caused resistance to gefitinib under hypoxia. This phenomenon was also reversed by the knockdown of TGFα or HIF1α. Our results indicate that hypoxia causes gefitinib resistance in EGFR‐mutant NSCLC through the activation of wild‐type EGFR mediated by the upregulation of TGFα. The presence of wild‐type and mutant EGFR along with tumor hypoxia are important factors that should be considered when treating NSCLC patients with gefitinib.
Advanced non‐small‐cell lung cancer (NSCLC) is the leading cause of cancer‐related deaths globally.1 Recent retrospective analyses showed that epidermal growth factor receptor (EGFR) is overexpressed in 62% of NSCLC patients and that its expression is correlated with a poor prognosis.2, 3 In addition to EGFR overexpression, its cognate ligands, such as transforming growth factor‐α (TGFα), are also frequently expressed in NSCLC, and can establish autocrine loops that lead to receptor hyperactivity.4, 5 Thus, EGFR is an attractive target for developing therapeutics in NSCLC.
Activating mutations in EGFR have been found in a certain proportion of NSCLCs.6, 7 Almost 90% of activating mutations in EGFR consist of an in‐frame deletion mutation in exon 19 and an L858R mutation in exon 21. These mutations are associated with favorable responses to the EGFR tyrosine kinase inhibitors (EGFR‐TKIs) gefitinib and erlotinib.4 However, in most reports, the progression‐free survival of patients did not exceed 12 months, and most patients developed acquired resistance.8 In addition, 25–30% of patients are intrinsically resistant to EGFR‐TKIs, although their tumors are diagnosed as harboring activating mutations in EGFR.9, 10 The main mechanisms of resistance identified to date include secondary mutations and an “oncogene kinase switch.” The EGFR T790M mutation accounts for 50% of cases, and hepatocyte growth factor receptor (MET) amplification can be detected in 20% of patients with EGFR‐mutant TKI‐resistant NSCLC.11 Hepatocyte growth factor (HGF) is also an important factor in resistance to EGFR‐TKI.12 However, the mechanisms responsible for intrinsic resistance and other acquired resistance to EGFR‐TKIs in NSCLC with activating mutations in EGFR are not fully understood.
Taniguchi et al.13 examined tumor tissues from NSCLC patients diagnosed as harboring activating mutations in EGFR and treated with gefitinib after surgery. In their studies, the patients with not only EGFR mutation‐positive tumor cells but also mutation‐negative (wild‐type EGFR) cells did not respond well to gefitinib compared to patients with only EGFR‐mutant cells. These clinical findings suggest that genetic heterogeneity in EGFR, that is, the presence of mutations and wild‐type EGFR, within cancer cells is associated with resistance to gefitinib in NSCLC.
Hypoxia, which is defined as subnormal levels of tissue oxygenation, has long been involved in the resistance to chemotherapeutic treatments in several types of tumors.14 Hypoxic cancer cells can be aggressive and exhibit metastatic phenotypes with lower sensitivities to apoptotic signals.14 Hypoxia‐inducible factor‐1α (HIF1α) is a key transcription factor that is induced by hypoxia,15 and it activates the transcription of genes implicated in tumor angiogenesis, cell survival, and resistance to chemotherapeutic drugs.16 The overexpression of HIF1α confers cellular resistance to the EGFR‐blocking mAb cetuximab in A431 epidermoid carcinoma cells, which overexpress wild‐type EGFR. In addition, knocking down HIF1α substantially restores cellular sensitivity to cetuximab‐mediated antitumor activities.17 These findings strongly suggest that HIF1α expression is associated with the therapeutic responses of cancer cells to EGFR‐targeted therapies. However, the involvement of hypoxia in the resistance to EGFR‐TKIs, such as gefitinib and erlotinib, in NSCLC with an EGFR‐sensitive mutation remains unclear.
This evidence prompted us to investigate the role of hypoxia in resistance to gefitinib in NSCLC with activating EGFR mutations. We used three NSCLC cell lines, HCC827, PC9, and HCC2935, each with a different genetic EGFR status, and examined the involvement of tumor hypoxia and wild‐type EGFR in resistance to gefitinib in NSCLC with activating EGFR mutations.
Materials and Methods
Cell culture and reagents
The NSCLC cell lines HCC827 and HCC2935 harboring an EGFR exon 19 deletion mutation were obtained from ATCC (Manassas, VA, USA). A PC‐9 expressing EGFR exon 19 deletion mutation was established at the Tokyo Medical University (Tokyo, Japan).18, 19 A549 cells were obtained from the Riken Bioresource Center (Tokyo, Japan). All the cells were grown in a humidified 5% CO2 atmosphere at 37°C in an incubator, in which the oxygen tension was held at either 21% (normoxia) or 1% (hypoxia). Gefitinib was purchased from Toronto Research Chemicals (North York, ON, Canada). The MEK inhibitor (U0126) was obtained from LC Laboratories (Woburn, MA, USA) and MET inhibitor (PHA‐665752) was obtained from Merck (Darmstadt, Germany).
Detection of exon 19 deletion mutant EGFR and wild‐type EGFR by fragment analysis
An EGFR gene fragment was amplified using the following primer set across the deleted region in exon 19 in EGFR: forward primer, 5′‐CTGGATCCCAGAAGGTGAGA‐3′; reverse primer, 5′‐GATTTCCTTGTTGGCTTTCG‐3′. The forward primer was labeled with Beckman dye D4 (Sigma, Sigma‐Aldrich, St. Louis, MO, USA). The conditions for PCR amplification were 94°C for 2 min; 30 cycles of 94°C for 30 s, 62°C for 30 s, and 72°C for 45 s; and a final extension cycle of 72°C for 10 min. The PCR products were diluted 50‐fold in DNase/RNase‐Free Water (Invitrogen, Carlsbad, CA, USA). One microliter of the PCR dilution was combined with 40 μL sample loading solution (SLS) loading mixture and 0.5 μL DNA Size Standard‐400 (Beckman Coulter, Fullerton, CA, USA). The samples were run on a CEQ8000 DNA sequencer (Beckman Coulter) using standard method FRAG‐3. Fragment analysis was carried out using the software on the CEQ8000 genetic analysis system.
Cell growth inhibition assay
Cells (1 × 103) were seeded onto 96‐well microtiter plates. After 48 h of normoxia or hypoxia, they were grown in the absence or presence of various concentrations of gefitinib. Cell viability was assessed using a Cell Counting Kit‐8 (Wako Pure Chemical Industries, Osaka, Japan) as previously described.20 The results were expressed as the percentage of cell viability. The IC50 value was defined as the concentration needed for a 50% reduction in the absorbance, which was calculated based on survival curves as previously described.21 The experiments were done in triplicate and repeated four times.
Cell apoptosis assay
The PC9 cells were exposed to hypoxia or normoxia for 48 h followed by treatment with 0.1 μM gefitinib for 24 h. The cells were harvested using 0.05% EDTA/PBS, and cell apoptosis was evaluated using an Annexin V‐FITC‐PI Kit (Sigma) as previously described.22 Fluorescence was analyzed with a FACScan (Becton‐Dickson, Mountain View, CA, USA).
Western blot analysis
The PC9, HCC2935, HCC827, and A549 cells were incubated under hypoxia or normoxia for 48 h, then either left untreated or treated with various concentrations of gefitinib for 3 h. After treatment, the cells were lysed with buffer (2% SDS, 50 mM Tris–HCl, pH 6.8, 10% glycerol) containing protease inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor (Roche). Immunoblot analysis was carried out as described previously.23 Equivalent amounts of protein were separated by SDS‐PAGE and transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The membrane was probed with a rabbit mAb against EGFR, a phospho‐EGFR antibody (specific for Tyr1068), p44/42 MAPK, phospho‐p44/42 MAPK (specific for Thr202/Tyr204), AKT, and phospho‐AKT (specific for Ser473) (Cell Signaling Technology, Beverly, MA, USA) as the first antibody, followed by an HRP‐conjugated secondary antibody. The bands were visualized, and images were analyzed with the enhanced chemiluminescence system (Amersham Pharmacia Biotech, Buckinghamshire, UK). The band intensities of Western blots were measured using the software Quantity One version 4.6.7 (Bio‐Rad). The fold change in phospho‐protein expression was calculated by setting the ratio of band intensities of the phospho‐protein/total protein in untreated normoxic cells to unity.
Quantitative RT‐PCR
Quantitative RT‐PCR (qPCR) was carried out using Fast SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer's instructions.
The following program was run: holding at 95°C for 20 s, amplification by 40 cycles (denaturation 95°C for 3 s, annealing and extension at 60°C for 30 s), and melt‐curve analysis. Actin or succinate dehydrogenase complex, subunit A, flavoprotein (SDHA) served as an internal control. The primers that were specific for the genes were: TGFα forward, 5′‐GGCCCTGGCTGTCCTTATC‐3′; TGFα reverse, 5′‐AGCAAGCGGTTCTTCCCTTC‐3′; HIF1α forward, 5′‐ACTAGCCGAGGAAGAACTATGAA‐3′; HIF1α reverse, 5′‐TACCCACACTGAGGTTGGTTA‐3′.
Plasmid construction and transfection
Full‐length cDNA of wild‐type EGFR was amplified by RT‐PCR from the human embryonic kidney cell line HEK293, and exon 19 deletion mutation EGFR (delE746_A750) was amplified from PC9 cells. Wild‐type and mutant EGFR cDNA were cloned into the NheI/XhoI sites of the vector pcDNA3.1/Hygro (Invitrogen), as previously described.24 The empty vector and wild‐type EGFR vector were transfected into HCC827 cells using Lipofectamine LTX (Invitrogen) according to the manufacturer's instructions. The transfectants of the empty vector and wild‐type EGFR were designated as HCC827/Mock and HCC827/Wt EGFR, respectively.
RNA interference
Small interfering RNA targeting HIF1α or TGFα (Stealth Select RNAi siRNA) were custom synthesized by Invitrogen. A negative control was also purchased from Invitrogen. The PC9, HCC2935, HCC827/Mock, and HCC827/Wt EGFR cells were transfected with two different specific siRNAs and one non‐specific control using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. The cells were detached and diluted in complete growth medium without antibiotics then plated in each of the wells. RNAi duplex and Lipofectamine RNAiMAX were mixed in Opti‐MEM I reduced serum medium and incubated for 15 min at room temperature. RNAi duplex‐Lipofectamine RNAiMAX complexes were added to the wells that contained cells. The cells were then incubated for 48 h at 37°C. For the cell growth inhibition assay, 2 × 103 cells/100 μL were used per well in 96‐well plates.
The sequences of the siRNA against HIF1α or TGFα were: HIF1α #1, 5′‐CCAGCCGCUGGAGACACAAUCAUAU‐3′; HIF1α #2, 5′‐GGAAUUAACUCAGUUUGAACUAACU‐3′; TGFα #1, 5′‐UCGCCCUGUUCGCUCUGGGUAUUGU‐3′; and TGFα #2, 5′‐CAGAAGAAGCAGGCCAUCACCGCCU‐3′.
Statistical analysis
Statistical analysis was carried out using anova as previously described.22 The differences between the means were considered to be statistically significant at P < 0.05.
Results
Genetic status of EGFR in NSCLC cell lines
We first extracted RNA and examined the genetic status of EGFR in the NSCLC cell lines HCC827, PC9, HCC2935, and A549. The PCR was set up using a primer set across the deleted region in exon 19 of EGFR. To distinguish the exon 19 deletion mutation EGFR and the wild‐type EGFR, the PCR products were electrophoresed and analyzed by using a DNA sequencer. Wild‐type EGFR showed an 88‐bp peak, and the exon 19 deletion mutation could be discriminated on the basis of 15–18‐bp differences by fragment analysis. We also examined the relative proportions of mutant and wild‐type EGFR expression by fragment analysis. To validate this approach, we mixed mutant EGFR plasmid with wild‐type EGFR plasmid at various ratios and amplified the mixed plasmids as PCR templates for the analyses. The calibration curve is shown in Figure S1. There was clear linearity between the actual mixed proportion of mutant and wild‐type plasmids, thus confirming the validity of our assay.
As shown in Figure 1(A), the HCC827 cells expressed only mutant EGFR with a deletion of 15 nucleotides in exon 19. The expression level of wild‐type EGFR was below the detection limit for both normoxia and hypoxia. However, the PC9 cells expressed not only the exon 19 deletion mutation EGFR but also wild‐type EGFR (Fig. 1B). The relative proportions of mutant and wild‐type EGFR expression were 91.08% and 8.92% for normoxia, and 88.56% and 11.44% for hypoxia, respectively. The HCC2935 cells also expressed both mutant and wild‐type EGFR (Fig. 1C). The ratio of wild‐type EGFR expression to mutant EGFR expression was 1:1. The A549 cells expressed only wild‐type EGFR (Fig. S2A).
Figure 1.
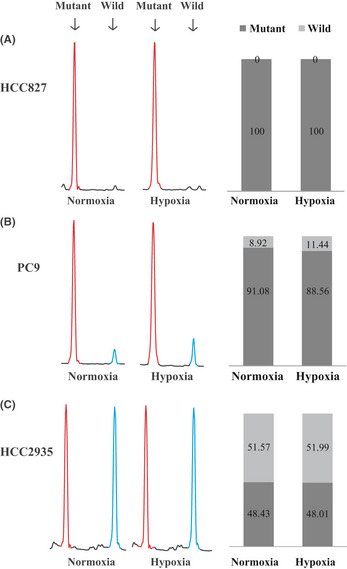
Genetic status of epidermal growth factor receptor (EGFR) in HCC827 (A), PC9 (B), and HCC2935 (C) non‐small‐cell lung carcinoma cell lines. Left: Electropherograms of fragment analysis under normoxia and hypoxia. Red peak, amplified PCR fragment of exon 19 deletion mutation EGFR (73 or 70 bp); blue peak, wild‐type EGFR (88 bp). Right: Relative proportions of mutant and wild‐type EGFR expression determined by fragment analysis. Black bar, mutant EGFR; gray bar, wild‐type EGFR.
The results of fragment analysis for genomic DNA were almost identical to those of cDNA (data not shown). To confirm that the two peaks at 88 and 73 bp observed by fragment analysis for the PC9 cells corresponded to exon 19 deletion mutation EGFR and wild‐type EGFR, the PCR products were cloned into the pCR2.1‐TOPO vector, and 15 clones were randomly selected for direct sequencing. As shown in Figure S3(A), the sequences of the exon 19 deletion mutation EGFR (ΔE746‐A750) and wild‐type EGFR were confirmed. Thus, both the mutant EGFR and wild‐type EGFR were present in the PC9 cells. Copy numbers of the EGFR gene were also examined by qPCR assay. As shown in Figure S3(B), the relative copy number of the EGFR gene in the HCC827 cells was higher than those of the PC9 and HCC2935 cells. The A549 cells had no copy number gain. Taken together, the relative ratios of expression of mutant to wild‐type EGFR or copy numbers were different between the HCC827, PC9, and HCC2935 cells.
Hypoxia induced gefitinib resistance in PC9, HCC2935, and A549 cells, but not in HCC827 cells
To investigate the effect of hypoxia on the response to gefitinib of the NSCLC cell lines, HCC827, PC9, HCC2935, and A549 cells were exposed to hypoxia or normoxia for 48 h followed by treatment with gefitinib. Cell viabilities were evaluated, and IC50 values were estimated. As shown in Figure 2(A–C), HCC827, PC9, and HCC2935 cells were highly sensitive to gefitinib under normoxia. The HCC827 cells, which expressed only mutant EGFR, were also sensitive to gefitinib under hypoxia, and the IC50 value of the hypoxic cells was almost identical to that of the normoxic cells (P = 0.989) (Fig. 2A). In contrast, hypoxia induced gefitinib resistance in the PC9 cells that harbored not only mutant EGFR but also wild‐type EGFR when compared to those under normoxia (Fig. 2B). The IC50 value of the hypoxic PC9 cells was significantly higher than that of the normoxic PC9 cells (P = 0.006). The population of gefitinib‐induced apoptotic cells was not significantly increased under hypoxia for the PC9 cells, although treatment with gefitinib increased the percentage of apoptotic PC9 cells under normoxia compared to the vehicle‐treated cells (P = 0.0001, Fig. 2D). Hypoxia also induced gefitinib resistance in the HCC2935 cells that expressed both mutant and wild‐type EGFR (Fig. 2C). These findings indicated that sensitivity to gefitinib following hypoxia was different among the HCC827, PC9, and HCC2935 cells, all of which were highly sensitive to gefitinib under normoxia. Hypoxia‐induced gefitinib resistance was also observed in the A549 cells that harbored wild‐type EGFR, which were primarily resistant to gefitinib (P = 0.001) (Fig. S2B). Taken together, these findings suggest that the presence of wild‐type EGFR might be involved in hypoxia‐induced resistance to gefitinib in some NSCLC cell lines with activating EGFR mutations.
Figure 2.
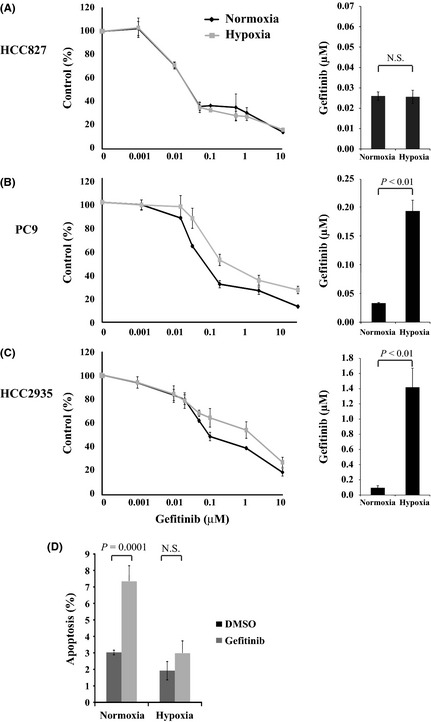
(A–C) Hypoxia induced gefitinib resistance in PC9 (B) and HCC2935 (C) but not in HCC827 (A) non‐small‐cell lung carcinoma cells. Left: Cells were incubated under hypoxia (1% O 2) or normoxia (21% O 2) for 48 h, followed by treatment with the indicated concentrations of gefitinib for 48 h. Each data point represents the average value of four samples and is expressed as a percentage of the surviving cells relative to the untreated controls. Right: Bar graphs showing the IC 50 values of cells resistant to gefitinib. (D) After hypoxia or normoxia for 48 h, PC9 cells were grown in the presence of 0.1% DMSO (control) or 0.1 μM gefitinib for 24 h. Apoptosis was assessed using propidium iodide and annexin‐V staining. The y‐axis denotes the sum of the early and late apoptotic cells as the mean ± standard error of the mean (n = 3). N.S., not significant.
Phosphorylation of EGFR, AKT, and ERK with gefitinib treatment
To examine the effect of gefitinib treatment on the EGFR pathway under normoxia and hypoxia, EGFR, AKT, and ERK phosphorylation was analyzed. As shown in Figure 3, phosphorylation of EGFR was upregulated under hypoxia in the PC9 and HCC2935 cells compared with those under normoxia. In addition, ERK was activated by hypoxia exposure in these cell lines. Phosphorylation of EGFR and ERK was suppressed with gefitinib treatment to a lesser extent under hypoxia than under normoxia, whereas phosphorylated AKT was almost completely suppressed under both conditions. The same results were obtained for the A549 cells that harbored wild‐type EGFR and K‐ras mutations (Fig. S2C). In contrast, EGFR and ERK were not activated in the HCC827 cells that expressed only the mutant EGFR under hypoxia compared to that under normoxia (Fig. 3). The phosphorylation of EGFR and ERK was suppressed with gefitinib treatment in the HCC827 cells, but the inhibitory effect of gefitinib treatment was almost identical between normoxia and hypoxia. These findings suggest that the suppressive effect of gefitinib treatment on the phosphorylation of EGFR and ERK was reduced under hypoxia compared to normoxia in the NSCLC cells with both mutant and wild‐type EGFR, thus resulting in hypoxia‐induced resistance to gefitinib.
Figure 3.
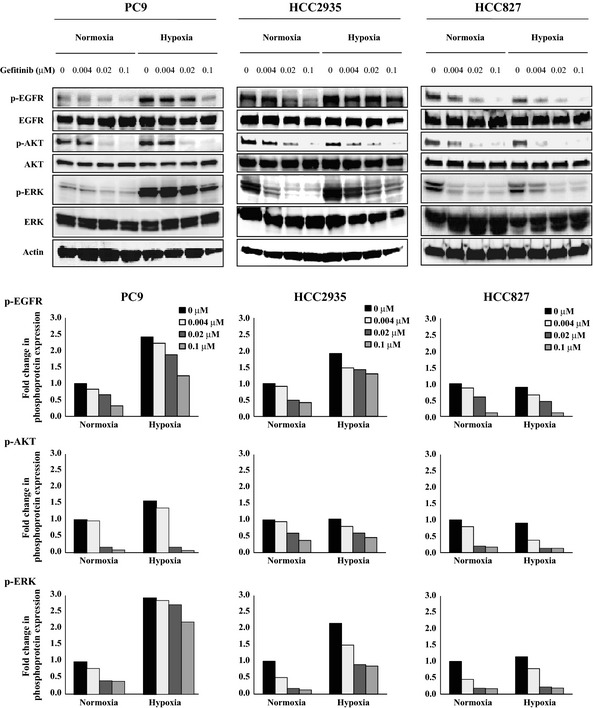
Effects of gefitinib treatment on the levels of phosphorylated epidermal growth factor receptor (EGFR), AKT, and ERK (p‐EGFR, p‐AKT, and p‐ERK, respectively) under normoxia and hypoxia. The PC9, HCC2935, and HCC827 non‐small‐cell lung carcinoma cells were incubated under hypoxia or normoxia for 48 h, then either left untreated or treated with the indicated concentrations of gefitinib for 3 h. After treatment, the cells were lysed and equal amounts of cell lysates were subjected to Western blot analysis using antibodies against total and phosphorylated EGFR (Y‐1068), AKT, and ERK. The levels of actin served as internal controls for equal protein loading in each lane. The bar graph below each blot shows the fold change in phospho‐protein expression due to treatment with the indicated concentrations of gefitinib. The fold changes were calculated by setting the ratios of the phospho‐protein/total protein band intensities for the untreated normoxia cells to unity.
Transforming growth factor‐α upregulated by hypoxia, and TGFα knockdown reversed hypoxia‐induced resistance to gefitinib in PC9 and HCC2935 cells
To investigate the molecular mechanism by which hypoxia‐induced activation of EGFR and resistance to gefitinib occurs, we examined the mRNA expression of ligands for EGFR under normoxia and hypoxia. The expression of EGFR ligands, including EGF, epiregulin, amphiregulin, and TGFα in PC9 cells was determined by qPCR (Fig. S4A). Among them, TGFα mRNA expression was dramatically upregulated by exposure to hypoxia. The TGFα protein expression was also upregulated in PC9 (Fig. 4A), HCC2935, A549, and HCC827 cells (Fig. S4B). Downregulation of TGFα expression by RNAi significantly reversed the resistance to gefitinib in hypoxic PC9 cells (Fig. 4B,C) and hypoxic HCC2935 cells (Fig. S4C). These results indicated that the upregulation of TGFα was involved in hypoxia‐induced resistance to gefitinib in PC9 and HCC2935 cells.
Figure 4.
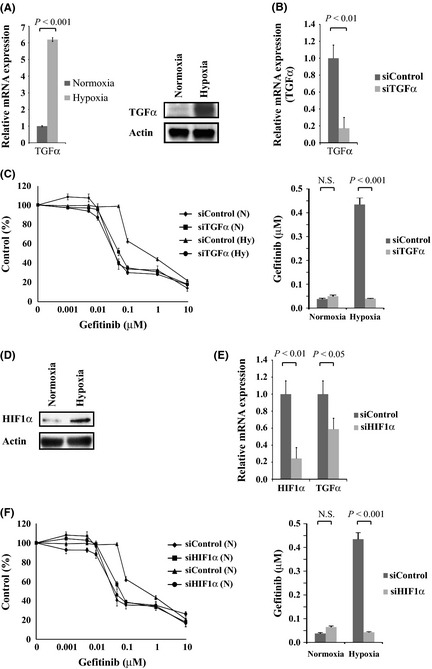
Transforming growth factor‐α (TGFα) and hypoxia‐inducible factor‐1α (HIF1α) were upregulated by hypoxia, and TGFα or HIF1α knockdown reversed hypoxia‐induced resistance to gefitinib in PC9 non‐small‐cell lung carcinoma cells. (A) TGFα messenger RNA and protein expression in PC9 cells under normoxia and hypoxia were analyzed by quantitative RT‐PCR (left) and Western blotting (right). (B) TGFα expression was knocked down in PC9 cells with siRNA. Two specific siRNAs and one non‐specific control were used, and data representative of the siRNA experiment are shown (P = 0.008). (C) Left: PC9 cells were transfected with siTGFα or siControl then incubated under hypoxia (Hy) or normoxia (N) for 48 h, followed by treatment with the indicated concentrations of gefitinib. Each data point is expressed as a percentage of the surviving cells relative to the untreated controls. Right: Bar graphs show the IC 50 values of the cells to gefitinib. The knockdown of TGFα expression significantly reduced the IC 50 value of the hypoxic PC9 cells. P < 0.001. (D) HIF1α expression in PC9 cells under normoxia and hypoxia was analyzed by Western blotting. (E) HIF1α expression was knocked down with siHIF1α in PC9 cells, resulting in the downregulation of TGFα expression (P = 0.004, P = 0.03, respectively). Two specific siRNAs and one non‐specific control were used; data representative of the siRNA experiment is shown. (F) Left: PC9 cells were transfected with siHIF1α or siControl then incubated under hypoxia (Hy) or normoxia (N) for 48 h followed by treatment with the indicated concentrations of gefitinib. Right: Bar graphs show the IC 50 values of cells to gefitinib. The knockdown of HIF1α expression significantly reduced the IC 50 value of the hypoxic PC9 cells. P < 0.001.
Accumulation of HIF1α implicated in hypoxia‐induced TGFα upregulation and gefitinib resistance in PC9 and HCC2935 cells
As a major transcription factor, HIF1α plays a central role in hypoxic cellular responses. Hypoxia upregulated the expression levels of HIF1α in PC9 cells (Fig. 4D) and HCC2935, A549, and HCC827 cells (Fig. S4B). To verify whether the accumulation of HIF1α was a key factor in hypoxia‐induced resistance to gefitinib, we carried out a knockdown of HIF1α using specific siRNAs. The downregulation of HIF1α reduced TGFα expression, which was upregulated by hypoxia in PC9 cells (Fig. 4E). Furthermore, the knockdown of HIF1α expression also significantly reversed the resistance to gefitinib in hypoxic PC9 cells (Fig. 4F). The same result was obtained for hypoxic HCC2935 cells (Fig. S4C). These findings indicate that HIF1α transcriptionally regulates TGFα expression during hypoxia and is implicated in hypoxia‐induced resistance to gefitinib in PC9 and HCC2935 cells.
Introduction of wild‐type EGFR gene into HCC827 cells harboring an activating EGFR mutation
To further confirm the involvement of wild‐type EGFR in hypoxia‐induced resistance to gefitinib in NSCLC cell lines with an activating EGFR mutation, we introduced the wild‐type EGFR gene into HCC827 cells then evaluated their sensitivity to gefitinib under normoxia and hypoxia. Transfection of the wild‐type EGFR vector increased the proportion of wild‐type EGFR in HCC827 cells, which primarily expressed deletion mutant EGFR (Fig. 5A). The relative proportions of wild‐type and mutant EGFR expression became 29.27% and 70.73% for the transfected HCC827 cells. As shown in Figure 5(B), introduction of the wild‐type EGFR gene did not induce the HCC827 cells to show gefitinib resistance under normoxia (P = 0.116). However, HCC827 cells that were transfected with wild‐type EGFR were significantly resistant compared to the mock transfected cells under hypoxia (P = 0.014) (Fig. 5B). In addition, TGFα or HIF1α expression was upregulated by hypoxic exposure in the control and wild‐type EGFR transfected HCC827 cells (Fig. 5C), and hypoxia‐induced gefitinib resistance in wild‐type EGFR transfected HCC827 cells was reversed by the knockdown of TGFα or HIF1α (Fig. 5D). These findings indicate that hypoxia causes resistance to gefitinib in NSCLC cells with both mutant and wild‐type EGFR through the activation of wild‐type EGFR mediated by TGFα and HIF1α.
Figure 5.
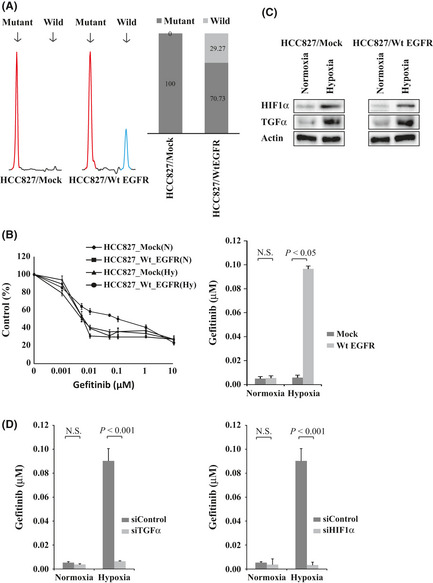
Transfection of wild‐type epidermal growth factor receptor (EGFR) gene into HCC827 cells harboring the activating EGFR mutation. (A) Transfectants of the empty vector and wild‐type EGFR were designated HCC827/Mock and HCC827/Wt EGFR cells, respectively. Left: Electropherograms of the fragment analysis for the HCC827/Mock and HCC827/Wt EGFR cells. Red peak, amplified PCR fragment of exon 19 deletion mutation EGFR; blue peak, wild‐type EGFR. Right: Relative proportions of mutant and wild‐type EGFR expression by fragment analysis. Black bar, mutant EGFR; gray bar, wild‐type EGFR. (B) HCC827/Mock and HCC827/Wt EGFR cells were incubated under hypoxia or normoxia for 48 h followed by treatment with gefitinib. The IC 50 value of the HCC827/Wt EGFR cells was significantly higher than that of the HCC827/Mock cells under hypoxia. P = 0.014. (C) HIF1α and TGFα expression was upregulated by hypoxia in the HCC827/Mock cells (left) and the HCC827/Wt EGFR cells (right) as indicated by Western blotting. (D) HCC827/Wt EGFR cells were transfected with siTGFα or siControl (left) or with siHIF1α or siControl (right) then incubated under hypoxia or normoxia for 48 h, followed by treatment with gefitinib. The IC 50 values of the HCC827/Wt EGFR cells were significantly reduced by the knockdown of TGFα or HIF1α under hypoxia (P < 0.001, P < 0.001, respectively).
Discussion
Recent phase III studies have indicated that first‐line gefitinib for patients with advanced NSCLC who were selected on the basis of EGFR mutations significantly improved their progression‐free survival compared with patients undergoing standard chemotherapy.25, 26 However, even among patients with EGFR‐sensitive mutations, not all responded to treatment with gefitinib.25, 26 Therefore, it is clinically important to elucidate the biological mechanisms responsible for the intrinsic resistance to gefitinib in NSCLC.
In this study, we focused on micro‐environmental factors such as tumor hypoxia with respect to resistance to gefitinib in NSCLC cell lines with different genetic statuses relating to EGFR. HCC827 and PC9 cells that harbored an activating EGFR mutation were highly sensitive to gefitinib under normoxia. However, we found that their sensitivities to gefitinib following hypoxia were different. Hypoxia was involved in resistance to gefitinib in the PC9 cells that expressed not only mutant EGFR but also wild‐type EGFR. In contrast, hypoxia did not induce resistance to gefitinib in the HCC827 cells that harbored only the mutant EGFR. However, introduction of the wild‐type EGFR gene into the HCC827 cells caused resistance to gefitinib under hypoxia, while it did not under normoxia. These findings strongly suggest that hypoxia induces gefitinib resistance in NSCLC with both mutant and wild‐type EGFR, and that wild‐type EGFR might contribute to resistance to gefitinib in NSCLC with an activating EGFR mutation under hypoxia.
Several growth factors, notably TGFα, are HIF1α target genes.16 Krishnamachary et al.27 showed that TGFα expression is induced by hypoxia in cell line HCT116, which comprises human colon carcinoma cells, and that transfection of siRNA against HIF1α reduces TGFα expression under hypoxia. Expression of TGFα is transcriptionally upregulated by HIF1α, and the binding of TGFα to its cognate receptor EGFR also activates a signal transduction pathway that leads to increased HIF1α transcriptional activity of target genes, including TGFα.16 Thus, HIF1α and TGFα contribute to autocrine signaling pathways. In our study, hypoxia markedly upregulated TGFα expression, and HIF1α knockdown reduced TGFα expression in the hypoxic PC9 cells. Furthermore, hypoxia‐induced gefitinib resistance was reversed by the knockdown of HIF1α or TGFα in the PC9 cells and wild‐type EGFR‐transfected HCC827 cells. These findings strongly imply that the accumulation of HIF1α upregulates TGFα expression and that it plays a key role in the development of hypoxia‐induced resistance to gefitinib in NSCLC cells through the activation of wild‐type EGFR mediated by TGFα.
It has been reported that mutant EGFR still requires ligand stimulation for receptor activation.6 However, phosphorylation of mutant EGFR and downstream AKT and ERK are inhibited at concentrations of gefitinib that are 10‐ to 100‐fold lower than those that are necessary to inhibit wild‐type EGFR.7 We previously transfected HEK293 cells with wild‐type EGFR or mutant EGFR expression vectors.28 The stable transfectants of wild‐type EGFR and its deletion mutant were designated 293(W) and 293(D) cells, respectively. Increased phosphorylation and dimerization of the mutant EGFR was detected without ligand stimulation in the 293(D) cells. Stimulation with the ligand of EGFR increased phosphorylation, and EGFR dimers were observed in the 293(W) cells. However, phosphorylation and dimerization were not increased in the 293(D) cells by stimulation with the ligand. We also showed that ERK phosphorylation was not inhibited with 0.2 μM gefitinib treatment in the 293(W) cells, whereas it was completely suppressed with the same concentration of gefitinib in the 293(D) cells.
In this study, we focused on the wild‐type EGFR and its ligand TGFα under hypoxia in the resistance to gefitinib in NSCLC cell lines with EGFR mutation. The PC9 cells expressed wild‐type EGFR, and TGFα upregulated by HIF1α stimulated wild‐type EGFR, resulting in increased phosphorylation of EGFR and ERK in addition to mutant EGFR signaling under hypoxia (Fig. S5). To inhibit the wild‐type EGFR‐mediated activation of signaling pathways, especially the ERK pathway, a higher concentration of gefitinib was required compared to that for mutant EGFR‐mediated signaling. We also previously revealed that treatment with gefitinib inhibited the AKT signaling pathway more strongly than the ERK signaling pathway.28 In our study, regardless of treatment with gefitinib, the activation of ERK persisted, although the phosphorylation of AKT was almost completely suppressed in the hypoxic PC9 cells (Fig. 3). Our results were consistent with a previous finding that gefitinib suppresses the AKT signaling pathway more strongly than the ERK signaling pathway.28
Hepatocyte growth factor also induces gefitinib resistance for NSCLC with EGFR mutations by restoring AKT phosphorylation through the activation of MET.12 In our study, HGF mRNA expression was also upregulated in hypoxic PC9 cells by qPCR (data not shown). However, gefitinib did not restore the AKT phosphorylation (Fig. 3), and treatment with MET kinase inhibitor (PHA‐665752) failed to reverse the resistance to gefitinib in hypoxic PC9 cells, although the MEK inhibitor (U0126) reversed this (data not shown). These findings suggest that the HGF‐MET pathway does not contribute to the hypoxia‐induced resistance to gefitinib in PC9 cells.
A recent clinical study suggests that the genetic heterogeneity of EGFR is important for gefitinib resistance in NSCLC.13 Among tumor tissues from 21 NSCLC patients having activating EGFR mutations, six patients were found to have not only EGFR mutation‐positive tumor cells but also wild‐type EGFR cells. After treatment with gefitinib, the prognosis of these patients was worse than that in patients with only EGFR‐mutant cells.13 These clinical findings are consistent with our hypothesis that the intratumoral genetic heterogeneity of EGFR, especially the presence of sensitive mutations and wild‐type EGFR, is an important factor for determining the therapeutic response to gefitinib in hypoxic NSCLC.
To our knowledge, our study is the first to reveal that hypoxia induces gefitinib resistance in NSCLC with both mutant and wild‐type EGFR through the activation of wild‐type EGFR mediated by the upregulation of TGFα. We suggest that a combination of EGFR‐TKI treatment with HIF1α‐targeting approaches would be more effective than either one alone and will provide new insight into understanding the mechanisms responsible for the intrinsic resistance to EGFR‐TKIs in NSCLC.
Disclosure Statement
The authors declare no conflict of interest.
Supporting information
Fig. S1. Left: Mutant and wild‐type epidermal growth factor receptor (EGFR) plasmids were mixed at various ratios, then the mixed plasmids were amplified as a PCR template for fragment analyses. Calibration curve between mixture ratio of the plasmids and proportion ratio of amplified fragments. Right: Electropherograms of the fragment analysis of the mixtures of mutant and wild‐type EGFR plasmids.
Fig. S2. Data for A549 cells are shown. (A) Left: Electropherograms of the fragment analyses. Right: Relative proportions of wild‐type epidermal growth factor receptor (EGFR) expression. (B) Left: Gefitinib sensitivity curves. Right: IC 50 values of cells to gefitinib. (C) Western blot analysis for total and phosphorylated EGFR, AKT, and ERK.
Fig. S3. (A) Direct sequencing of amplified fragment of exon 19 in epidermal growth factor receptor (EGFR) gene from PC9 cell‐derived genomic DNA. (B) Copy numbers of EGFR gene were determined by quantitative PCR.
Fig. S4. (A) mRNA expression level of epidermal growth factor receptor (EGFR) ligands including EGF, epiregulin, amphiregulin, and TGFα in PC9 cells by quantitative PCR. (B) Hypoxia‐inducible factor‐1α (HIF1α) or transforming growth factor‐α (TGFα) expression in HCC2935, A549, and HCC827 cells by Western blotting. (C) TGFα or HIF1α knockdown by siRNA reversed hypoxia‐induced resistance to gefitinib in HCC2935 cells.
Fig. S5. Proposed model depicting the involvement of wild‐type epidermal growth factor receptor (EGFR), hypoxia‐inducible factor‐1α (HIF1α), and transforming growth factor‐α (TGFα) in hypoxia‐induced resistance to gefitinib in PC9 cells.
Acknowledgments
This study was supported by Grant‐in‐Aids for Scientific Research No. 21591003 (Kazuhisa Takahashi) and No. 23591906 (Fumiyuki Takahashi) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
(Cancer Sci 2012; 103: 1946–1954)
References
- 1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60: 277–300. [DOI] [PubMed] [Google Scholar]
- 2. Ohsaki Y, Tanno S, Fujita Y et al Epidermal growth factor receptor expression correlates with poor prognosis in non‐small cell lung cancer patients with p53 overexpression. Oncol Rep 2000; 7: 603–7. [DOI] [PubMed] [Google Scholar]
- 3. Hirsch FR, Varella‐Garcia M, Bunn PA Jr et al Epidermal growth factor receptor in non‐small‐cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 2003; 21: 3798–807. [DOI] [PubMed] [Google Scholar]
- 4. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–81. [DOI] [PubMed] [Google Scholar]
- 5. Rusch V, Baselga J, Cordon‐Cardo C et al Differential expression of the epidermal growth factor receptor and its ligands in primary non‐small cell lung cancers and adjacent benign lung. Cancer Res 1993; 53: 2379–85. [PubMed] [Google Scholar]
- 6. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 7. Paez JG, Janne PA, Lee JC et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 8. Costa DB, Kobayashi S, Tenen DG, Huberman MS. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR‐mutant non‐small cell lung cancers. Lung Cancer 2007; 58: 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inoue A, Suzuki T, Fukuhara T et al Prospective phase II study of gefitinib for chemotherapy‐naive patients with advanced non‐small‐cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006; 24: 3340–6. [DOI] [PubMed] [Google Scholar]
- 10. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007; 98: 1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small‐cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 2009; 10: 281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 13. Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci 2008; 99: 929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004; 4: 437–47. [DOI] [PubMed] [Google Scholar]
- 15. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2: 38–47. [DOI] [PubMed] [Google Scholar]
- 16. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 2003; 3: 721–32. [DOI] [PubMed] [Google Scholar]
- 17. Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z. Requirement of hypoxia‐inducible factor‐1alpha down‐regulation in mediating the antitumor activity of the anti‐epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther 2008; 7: 1207–17. [DOI] [PubMed] [Google Scholar]
- 18. Nishio K, Arioka H, Ishida T et al Enhanced interaction between tubulin and microtubule‐associated protein 2 via inhibition of MAP kinase and CDC2 kinase by paclitaxel. Int J Cancer 1995; 63: 688–93. [DOI] [PubMed] [Google Scholar]
- 19. Kawamura‐Akiyama Y, Kusaba H, Kanzawa F, Tamura T, Saijo N, Nishio K. Non‐cross resistance of ZD0473 in acquired cisplatin‐resistant lung cancer cell lines. Lung Cancer 2002; 38: 43–50. [DOI] [PubMed] [Google Scholar]
- 20. Ohashi R, Takahashi F, Cui R et al Interaction between CD44 and hyaluronate induces chemoresistance in non‐small cell lung cancer cell. Cancer Lett 2007; 252: 225–34. [DOI] [PubMed] [Google Scholar]
- 21. Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non‐small cell lung cancer cell line resistant to gefitinib. Int J Cancer 2005; 116: 36–44. [DOI] [PubMed] [Google Scholar]
- 22. Tajima K, Ohashi R, Sekido Y et al Osteopontin‐mediated enhanced hyaluronan binding induces multidrug resistance in mesothelioma cells. Oncogene 2010; 29: 1941–51. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi F, Takahashi K, Okazaki T et al Role of osteopontin in the pathogenesis of bleomycin‐induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2001; 24: 264–71. [DOI] [PubMed] [Google Scholar]
- 24. Maegawa M, Arao T, Yokote H et al EGFR mutation up‐regulates EGR1 expression through the ERK pathway. Anticancer Res 2009; 29: 1111–7. [PubMed] [Google Scholar]
- 25. Mok TS, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–57. [DOI] [PubMed] [Google Scholar]
- 26. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–8. [DOI] [PubMed] [Google Scholar]
- 27. Krishnamachary B, Berg‐Dixon S, Kelly B et al Regulation of colon carcinoma cell invasion by hypoxia‐inducible factor 1. Cancer Res 2003; 63: 1138–43. [PubMed] [Google Scholar]
- 28. Sakai K, Arao T, Shimoyama T et al Dimerization and the signal transduction pathway of a small in‐frame deletion in the epidermal growth factor receptor. FASEB J 2006; 20: 311–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Left: Mutant and wild‐type epidermal growth factor receptor (EGFR) plasmids were mixed at various ratios, then the mixed plasmids were amplified as a PCR template for fragment analyses. Calibration curve between mixture ratio of the plasmids and proportion ratio of amplified fragments. Right: Electropherograms of the fragment analysis of the mixtures of mutant and wild‐type EGFR plasmids.
Fig. S2. Data for A549 cells are shown. (A) Left: Electropherograms of the fragment analyses. Right: Relative proportions of wild‐type epidermal growth factor receptor (EGFR) expression. (B) Left: Gefitinib sensitivity curves. Right: IC 50 values of cells to gefitinib. (C) Western blot analysis for total and phosphorylated EGFR, AKT, and ERK.
Fig. S3. (A) Direct sequencing of amplified fragment of exon 19 in epidermal growth factor receptor (EGFR) gene from PC9 cell‐derived genomic DNA. (B) Copy numbers of EGFR gene were determined by quantitative PCR.
Fig. S4. (A) mRNA expression level of epidermal growth factor receptor (EGFR) ligands including EGF, epiregulin, amphiregulin, and TGFα in PC9 cells by quantitative PCR. (B) Hypoxia‐inducible factor‐1α (HIF1α) or transforming growth factor‐α (TGFα) expression in HCC2935, A549, and HCC827 cells by Western blotting. (C) TGFα or HIF1α knockdown by siRNA reversed hypoxia‐induced resistance to gefitinib in HCC2935 cells.
Fig. S5. Proposed model depicting the involvement of wild‐type epidermal growth factor receptor (EGFR), hypoxia‐inducible factor‐1α (HIF1α), and transforming growth factor‐α (TGFα) in hypoxia‐induced resistance to gefitinib in PC9 cells.