

Abstract
Failure of chemotherapy in breast cancer presents a major problem and is often due to elevated expression of ATP binding cassette (ABC)‐type transporters, such as MDR1 protein. It has been shown that MDR1/ABCB1 gene expression is regulated at the chromatin level by DNA methylation and histone acetylation. However, the modified histone residues have not been identified and the role of various histone acetyl transferases (HATs) is not fully understood. By studying a breast carcinoma model cell line and its MDR1‐overexpressing derivative, we show that the histone 3 lysine 9 (H3K9) acetylation level is elevated 100‐fold in the promoter and first exon of the MDR1 gene in the drug‐resistant cell line compared to the drug‐sensitive cell line. The acetylation level of the other examined lysine residues (H3K4, H3K14, H4K8, and H4K12) is weakly or not at all elevated in the MDR1 locus, although their acetylation is generally increased genome‐wide in the drug‐resistant cell. Downregulation of the expression of HATs PCAF and GCN5 by RNAi effectively reduces the expression of MDR1. Unexpectedly, treatment with a p300‐selective inhibitor (HAT inhibitor II) further increases MDR1 expression and drug efflux in the drug‐resistant cells. Our data suggest that repeated exposure to chemotherapy may result in deregulated histone acetylation genome‐wide and in the MDR1 promoter. (Cancer Sci 2012; 103: 659–669)
The main reason for the failure of chemotherapy is the development of multidrug resistance in cancer cells, which is often caused by the elevated level of ATP binding cassette (ABC)‐type transporter proteins that eliminate the chemotherapeutic drugs from the cell. The best characterized ABC transporter in cancer cells is P‐glycoprotein (PgP), which is encoded by the MDR1/ABCB1 gene. The two possible mechanisms that can contribute to the elevated expression of MDR1 are: (i) genetic changes, meaning amplification or translocation of the MDR1 gene;1, 2, 3 or (ii) epigenetic changes, meaning increased expression of MDR1 due to chromatin remodeling.4, 5 Epigenetic changes are preserved through cell division, thus the gene remains in active form after the cessation of chemotherapy.6
Breast cancer is usually initially responsive to chemotherapy, but acquisition of multidrug resistance is associated with worse prognosis. Numerous clinical studies detected PgP expression in a significant percentage of untreated breast cancers, showed that MDR1 mRNA expression increased after exposure to chemotherapy and correlated increased expression with a worse response to treatment.7
Expression of MDR1 is induced rapidly by doxorubicin.8 It can also be activated by drugs that are not PgP substrates, as well as by heat shock and genotoxic stress. The MDR1 gene is transcribed from two promoters.9 The majority of the transcripts originate from the downstream promoter (DSP),10 whereas the upstream promoter (USP), located 112 kb upstream of the former, in the neighboring RPIB9/RUNDC3B gene, is active in some but not all multidrug‐resistant cells.11, 12, 13 The DSP is well characterized; several cis‐regulatory elements and binding factors acting in positive or negative regulation of MDR1 have been described.14, 15 An enhanceosome can form on an inverted CCAAT box binding NF‐Y and recruiting p300/CREB binding protein (CBP)‐associated factor (PCAF), a histone acetyltransferase (HAT).16 The role of histone acetylation and deacetylation in the transcriptional regulation of MDR1 has been established. Co‐transfection of HATs p300, CBP, or PCAF co‐activates the downstream MDR1 promoter–luciferase construct.16, 17 Binding of these HATs to the MDR1 promoter has been determined in vivo.17, 18, 19
Elevated levels of histone acetylation in the chromatin of the MDR1 promoter in drug‐resistant cells have been described before,18, 20, 21 however, the acetylation of specific lysine residues has not been investigated. We examined the acetylation of various histone H3 and H4 residues in a breast carcinoma cell line and its MDR1 overexpressing derivative. We found that H3K9 acetylation is elevated by two orders of magnitude in the major promoter and first exon of the MDR1 gene in a drug‐resistant cell line compared to a drug‐sensitive cell line. Acetylation of the other examined lysine residues was weakly or not at all elevated in the MDR1 locus, although their acetylation was globally increased in the drug‐resistant cells. Simultaneous knockdown of PCAF and GCN5 decreased the expression of MDR1. Unexpectedly, treatment with HAT inhibitor II further increased MDR1 expression and drug efflux in the drug‐resistant cells. Our data suggest that repeated exposure to chemotherapy may result in altered regulation of histone acetylation, and that malignant cells accumulate multiple mechanisms to ensure elevated expression of MDR1 and evasion of therapy.
Materials and Methods
Cell culture and drug treatment.
Breast carcinoma cell line MCF7 was from ATCC (Manassas, VA, USA). The doxorubicin‐resistant MCF7‐KCR cell line had been developed from MCF‐7 by stepwise selection, increasing the dose of doxorubicin from 10 nM to 1 μM.22, 23 Cells were maintained in RPMI medium supplemented with 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA), 4 mM glutamine, and antibiotic–antimycotic solution (Gibco/Life Technologies, Carlsbad, CA, USA) at 37°C in a 5% CO2 atmosphere. MCF7‐KCR cells were maintained in cycles of 1 week in medium containing 1 μM doxorubicin followed by 1 week in drug‐free medium. Before each experiment, cells were grown in drug‐free medium for approximately 1 week. Doxorubicin (Sigma‐Aldrich) was dissolved in PBS at 1.72 mM stock concentration (Fig.1). For drug accumulation assay, cells were incubated with 25 μM doxorubicin for 4 h. After gentle trypsinization and washing, fluorescence of the cells was analysed by FACS on FL‐2 channel using FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA), 10 000 cells were counted and analyzed using CellQuestPro (Becton Dickinson). Trichostatin A (TSA) was dissolved in DMSO to 2 mg/mL; cells were treated for 24 h at the indicated concentrations (2, 3).
Figure 1.
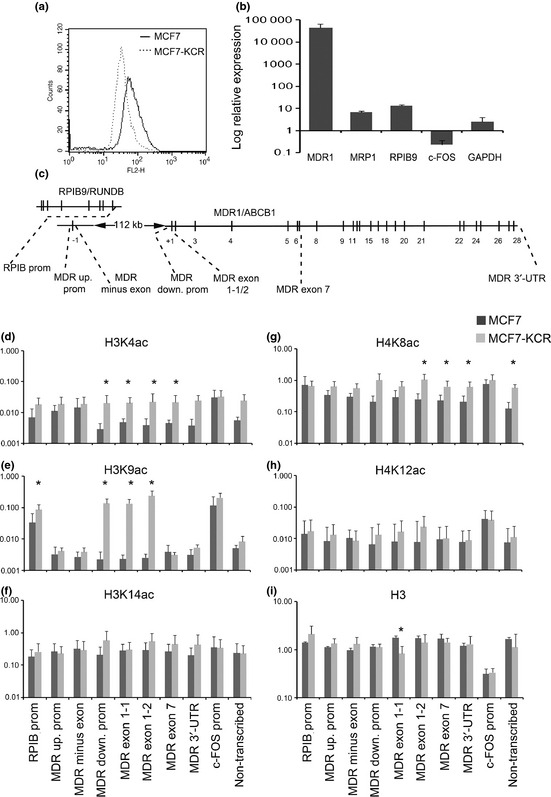
Comparison of MCF7 and MCF7‐KCR breast cancer cell lines for drug accumulation, drug transporter gene expression, and histone acetylation at the MDR1/ABCB1 locus. (a) Cells were treated with 25 μM doxorubicin for 4 h and fluorescence was determined by FACS. MCF7‐KCR cells accumulated less drug, indicating highly elevated ATP binding cassette (ABC) transporter activity. (b) The mRNA levels of MDR1, MRP1, RBIP9, c‐FOS, GAPDH, and 18S for internal control, were quantified by quantitative PCR (qPCR) and are shown in the MCF7‐KCR cell line relative to that in MCF7 cells. (c) Physical map of the MDR1 locus. Exons are indicated as crossing lines, some exons are numbered. Upper line indicates the RPIB9/RUNDC3B gene transcribed in the opposite direction. The positions of qPCR primers used in ChIP experiments are indicated. c‐FOS‐specific primers on chromosome 14 and primers for a non‐transcribed region on chromosome 6 are not shown. The ChIP experiments were carried out using anti‐H3K4ac (d), anti‐H3K9ac (e), anti‐H3K14ac (f), anti‐H4K8ac (g), anti‐H4K12ac (h), and anti‐H3 pan (i) Abs. The quantities of precipitated DNA corresponding to the indicated regions were determined by qPCR, and are expressed as the percent of input. The scales are logarithmic and different in different experiments. Values represent the means of 2–5 experiments. Error bars indicate SD. Down., downstream; prom, promoter; up., upstream; UTR, untranslated region. *P < 0.05, significantly different in MCF7‐KCR versus MCF7 cells.
Figure 2.
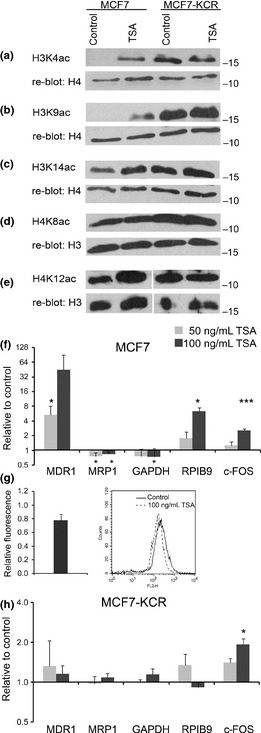
Analysis of changes in genome‐wide histone acetylation, mRNA expression level, and drug accumulation after treatment with trichostatin A (TSA). (a–e) Analysis of genome‐wide acetylation levels of specific histone lysine residues in drug‐sensitive and drug‐resistant cells. Histones were extracted from mock‐treated cells (control) or cells treated with 50 ng/mL TSA for 24 h. Western blot analysis was carried out with the indicated acetyl‐lysine‐specific Abs, and each membrane was reblotted with a control Ab. (f,h) MCF7 and MCF7‐KCR cells were mock‐treated or treated with 50 ng/mL (gray bars) or 100 ng/mL (black bars) TSA for 24 h. The level of the indicated transcripts and 18S RNA (as internal control) was quantified by RT‐quantitative PCR. Expression of each gene is shown relative to that in the mock‐treated control cells. *P < 0.05; ***P < 0.005. (g) MCF7 cell were treated with 100 ng/mL TSA and drug accumulation was determined by FACS. Bar graph shows average and SD of three independent experiments, curves are from one representative experiment.
Figure 3.
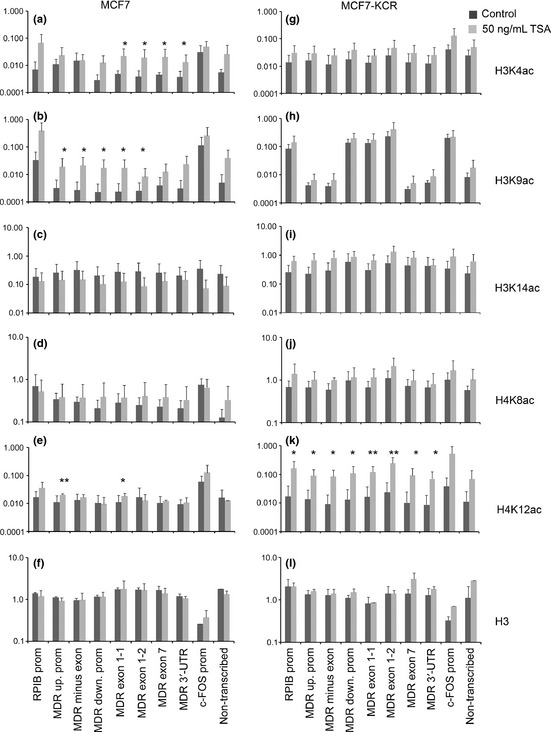
Changes in histone acetylation pattern in the MDR1/ABCB1 locus after treatment with trichostatin A (TSA) were analyzed by ChIP. MCF7 and MCF7‐KCR breast cancer cells were untreated (black) or treated with 50 ng/mL TSA (gray) for 24 h. The ChIP assays were carried out with anti‐H3K4ac (a,g), anti‐H3K9ac (b,h), anti‐H3K14ac (c,i), anti‐H4K8ac (d,j), anti‐H4K12ac (e,k), and anti‐H3 pan Ab for control (f,l). Quantities determined by qPCR are expressed as percent of input. Scales are logarithmic and values represent means of 2–5 experiments. Error bars are SD. *P < 0.05 and **P < 0.01 are significantly different in treated versus untreated cells. Down., downstream; prom, promoter; up., upstream; UTR, untranslated region.
RNA interference, analysis of mRNA expression, and quantitative PCR (qPCR).
For RNA interference, siRNA targeting PCAF, GCN5, ADA2B, or non‐targeting siRNA for control (Dharmacon/Thermo Fisher Scientific, Waltham, MA, USA, or Ambion/Life Technologies) was transfected into cells using DharmaFECT 1 (Dharmacon) transfection reagent following the manufacturer's protocol (Fig.4Fig.). Expression of mRNAs and proteins was examined 48 and 72 h after transfection, respectively. Total RNA was purified from the cells using the RNeasy Plus kit (Qiagen, Hilden, Germany). Reverse transcription was carried out using TaqMan Reverse Transcription Reagents (ABI/Life Technologies) and random hexamers from 1 to 2 μg RNA. Real‐time PCR was carried out with the SYBR‐Green detection method on StepOne Plus Real‐Time PCR System (ABI/Life Technologies). Primers used are summarized in Table S1. Thermal cycling was done for 10 min at 95°C followed by 50 cycles of 95°C for 15 s and 60°C for 45 s. Dissociation (melt) curves were analyzed after each run to confirm primer specificity. The 18S rRNA gene was used for normalization. HAT inhibitor II [2,6 bis‐(3‐bromo‐4‐hydroxybenzylidene), Calbiochem/Merck KGaA, Darmstadt, Germany] was dissolved in ethanol to 21.5 mM concentration; cells were treated for 24 h at the indicated concentrations (5, 6).
Figure 4.
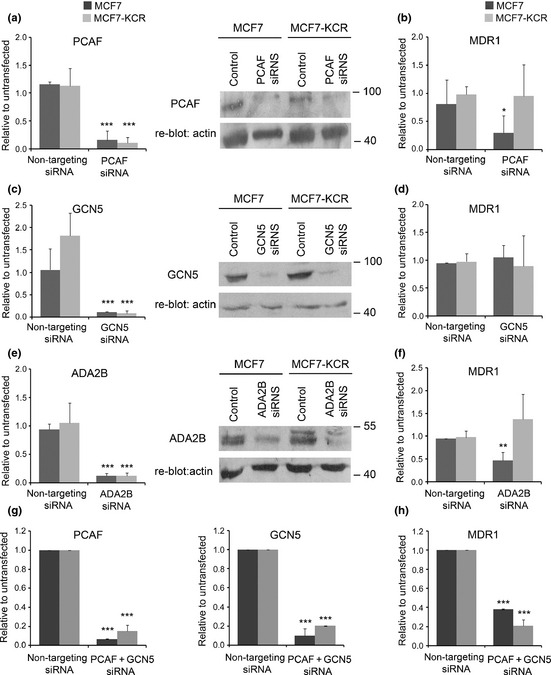
Effect of downregulation of individual subunits of SAGA‐type HAT complexes on the expression level of MDR1. MCF7 (black) and MCF7‐KCR (gray) breast cancer cells were untransfected, or transfected with non‐targeting siRNA for control, or siRNA for the indicated gene. The expression levels of PCAF, GCN5, or ADA2B (left panels of a,c,e, respectively, and g) were determined by RT‐quantitative PCR. Levels of PCAF, GCN5, or ADA2B proteins (right panels of a,c,e, respectively) were determined by Western blotting. Each blot was re‐probed for β‐actin for internal control. The level of MDR1 mRNA was determined by RT‐quantitative PCR (b,d,f,h). Expression of each gene is shown normalized to that in the untransfected control cells. *P < 0.05; **P < 0.01; ***P < 0.005.
Figure 5.
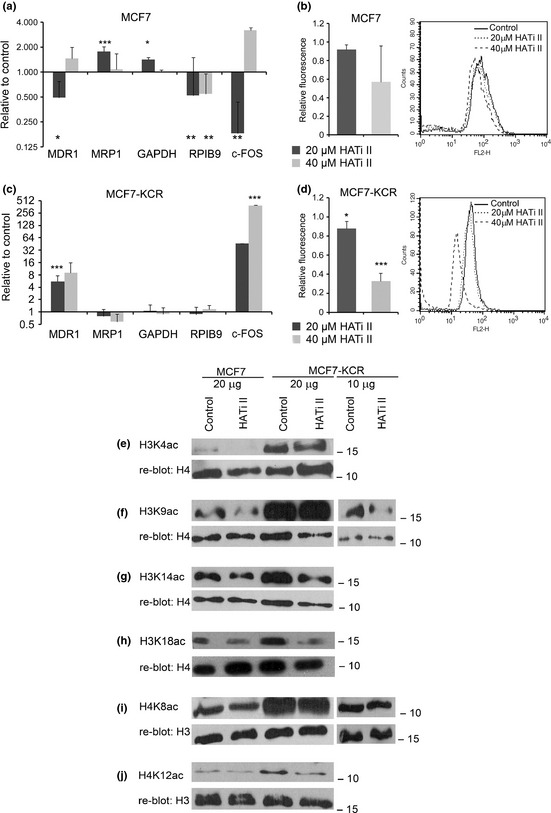
Analysis of the changes in mRNA expression, drug accumulation and genome‐wide histone acetylation levels in MCF7 and MCF7‐KCR breast cancer cells after treatment with histone acetyl transferase (HAT) inhibitor II (HATi II). MCF7 (a,b) and MCF7‐KCR (c,d) cells were treated with 20 or 40 μM HATi II for 24 h. (a,c) The level of the indicated transcripts and 18S RNA (as internal control) was quantified by RT‐quantitative PCR. Expression of each gene is shown relative to that in the mock‐treated control cells. *P < 0.05; **P < 0.01; ***P < 0.005. (b,d) Drug accumulation was determined by FACS. Bar graphs show the average and SD of three independent experiments; curves are from one representative experiment. (e–j) Analysis of the acetylation level of specific histone lysine residues. MCF7 and MCF7‐KCR cells were mock‐treated (control) or treated with 20 μM HATi II for 24 h. Histones were extracted, 20 μg (tracks 1–4) was analyzed by Western blotting with the indicated acetyl‐lysine‐specific Abs, and each membrane was re‐blotted with a control Ab. In order to see the change and not to overexpose the film we also analyzed 10 μg histone extract from MCF7‐KCR cells (f,i, tracks 5,6).
Figure 6.
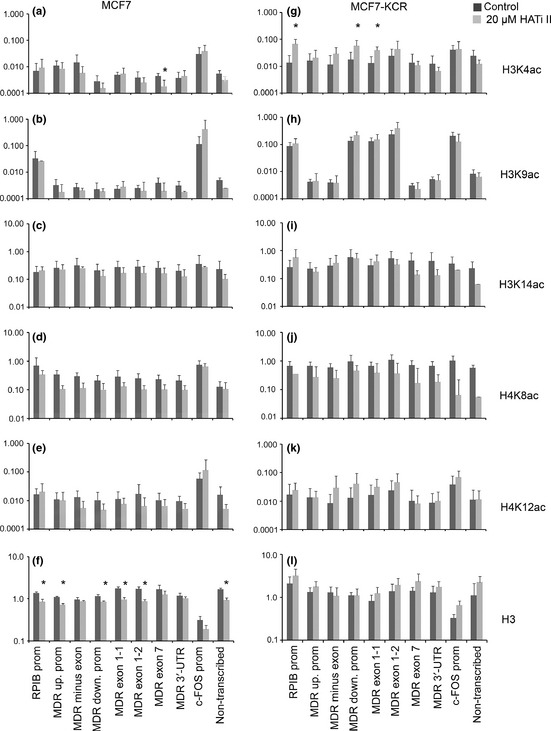
Changes in histone acetylation pattern in the MDR1/ABCB1 locus after treatment with histone acetyl transferase (HAT) inhibitor II (HATi II), analyzed by ChIP assay. MCF7 (left) and MCF7‐KCR breast cancer cells (right) were untreated or treated with 20 μM HATi II for 24 h. Quantities determined by qPCR are expressed as percent of input. Scales are logarithmic and values represent means of 2–5 experiments. Error bars are SD. *P < 0.05 is significantly different in treated versus untreated cells. The ChIP assay was carried out using anti‐H3K4ac (a,g), anti‐H3K9ac (b,h), anti‐H3K14ac (c,i), anti‐H4K8ac (d,j), anti‐H4K12ac (e,k), and anti‐H3 pan Abs for control (f,l). Down., downstream; prom, promoter; up., upstream; UTR, untranslated region.
Western blot analysis and antibodies.
Histone proteins were acid extracted according to the Abcam histone extraction protocol. Equal amounts of proteins were size‐fractionated by 15% SDS‐PAGE and transferred to nitrocellulose membrane. Antibodies were used at the following dilutions for immunoblotting: anti‐H3 (Abcam, Cambridge, UK) 1:10 000; anti‐H3K4acetyl (Millipore, Billerica, MA, USA) 1:5000; anti‐H3K9acetyl (Millipore) 1:5000; anti‐H3K14acetyl (Millipore) 1:5000; anti‐H3K18acetyl (Abcam) 1:10 000; anti‐H4K8acetyl (Abcam) 1:10 000; and anti‐H4K12acetyl (Abcam) 1:500. Antibodies used for ChIP were the same, expect for the acetyl‐H3K9‐specific Ab (Diagenode, Liege, Belgium). Specific Abs for PCAF (#2760)24 and GCN5 (2GC2c11)25 have been described. The ADA2B Ab (#2459) was a generous gift from D. Devys (IGBMC, Illkirch, France).26
Chromatin immunoprecipitation.
The ChIP analysis was done using the Acetyl‐Histone H3 Immunoprecipitation Assay Kit (Millipore) according to the manufacturer's instructions. Lysis buffer was supplemented with protease inhibitor cocktail (Sigma‐Aldrich) and 150 ng/mL TSA to preserve acetylation. One percent of each chromatin sample without immunoprecipitation provided the input DNA control. Immunoprecipitated DNA was analyzed by qPCR, percent of input was calculated using the ΔC t method. Independent experiments were done between two and six times. Statistical analyses between groups was carried out by anova and the Holm‐Sidak post hoc test using SigmaStat (Systat Software Inc, Chicago, IL, USA) and Student's t‐test using Excel (Microsoft, Redmond, WA, USA).
Results
Multiple mechanisms contribute to increased expression of MDR1 in the MCF7‐KCR cell line.
The MCF7 breast carcinoma cell line is sensitive to chemotherapeutic agents, but several drug‐resistant sublines have been established from it.27 We chose the MCF7‐KCR cell line that had been established through stepwise treatment of MCF7 with doxorubicin.22, 23 Fluorescent assay showed lower intracellular doxorubicin accumulation in the MCF7‐KCR cells than in the parental cells (Fig. 1a); the doxorubicin ID50 was 0.5 μM for MCF7 and 50 μM for MCF7‐KCR cells (data not shown). To determine if drug resistance was caused by increased expression of drug transporters, we studied the mRNA expression of MDR1 and MRP1 by RT‐qPCR. We found that the mRNA level of MDR1/ABCB1 was elevated 72 000‐fold in the MCF7‐KCR cell line compared to the parental cell line, whereas that of MRP1/ABCC1 was only elevated sevenfold (Fig. 1b). Expression of RPIB9, the gene partially overlapping MDR1, was also increased 10‐fold, whereas the expression of control genes GAPDH and c‐FOS was not significantly changed. The RT‐PCR with primers specific to the −1 exon of MDR1 showed that transcription was not initiated from the USP (data not shown). To determine if gene amplification was responsible for the enormous increase in the MDR1 expression, we carried out qPCR on genomic DNA. The copy number of the MDR1/ABCB1 locus in the MCF7‐KCR cells was 40‐fold that in the MCF7 cells (Fig. S1). These data suggest that, from each MDR1 gene copy, approximately 2000‐fold more mRNA is expressed in the drug‐resistant cells than in the sensitive cells. Treatment of MCF7‐KCR cells with MDR1‐specific siRNA resulted in a strong reduction of MDR1 message and protein levels (data not shown). However, neither drug accumulation assay nor cell viability assay showed significant functional difference between non‐targeting siRNA‐treated control and MDR1 siRNA‐treated cells, probably due to an effect of the transfection reagent on the cell membrane (data not shown). Thus, we conclude that in the MCF7‐KCR cell line drug resistance is likely caused by a high level of expression of MDR1, and gene amplification is partially but not solely responsible for this. Other mechanisms, possibly epigenetic changes, might also contribute to the high MDR1 mRNA level in the resistant cells.
H3K9ac is highly enriched at downstream promoter and first exon of MDR1 in drug‐resistant cell line.
In order to study the epigenetic regulation of MDR1 expression in the two cell lines, we carried out ChIP using Abs specific to acetylated histone lysine residues H3K4, H3K9, H3K14, H4K8, and H4K12. We analyzed the immunoprecipitated DNA by qPCR over the entire MDR1 locus: using primer pairs specific to the USP, upstream first exon (minus exon); DSP, two regions of the downstream first exon (exon1‐1 and 1‐2); gene body (exon 7); and 3′‐untranslated region (Fig. 1c). We also examined histone acetylation on the promoter of the RPIB9/RUNDC3B gene, which is transcribed from the other strand partially overlapping with MDR1. The c‐FOS promoter and an intergenic region were used as controls for high and low levels of acetylated histones, respectively (see Table S1). In MCF7 cells, acetylation of H3K9 is very low in every studied region of the MDR1 locus, 25‐fold lower than that in the c‐FOS promoter. Importantly, H3K9 acetylation is increased 60‐fold in the DSP region and 110‐fold in the downstream first exon in the drug‐resistant cells compared to the level in the sensitive cells (Fig. 1e). The acetylation level of H3K4 over the MDR1 gene locus in MCF7 cells is approximately sixfold lower than that in the c‐FOS promoter (Fig. 1d). In MCF7‐KCR cells, H3K4 acetylation is slightly increased in the MDR1 locus, in particular at the DSP, first exon and gene body region. Similarly, acetylation values of H4K8 in MCF7 cells are approximately threefold lower at the studied regions of MDR1 than at the c‐FOS promoter; in MCF7‐KCR cells, in the MDR1 DSP and along the gene body, they reach the level of that at the c‐FOS promoter (Fig. 1g). Furthermore, acetylation values of H4K12 at the MDR1 locus are approximately eightfold lower than at the c‐FOS promoter in both cells (Fig. 1h). In contrast, acetylation of H3K14 is uniform in both cells (Fig. 1f). The striking differences between the levels of particular modified histone forms in drug‐resistant and drug‐sensitive cells are unlikely to have resulted from differences in nucleosome densities in the MDR1 locus, because immunoprecipitation carried out with the H3 pan Ab resulted in similar values in the two cell lines (Fig. 1i). These data reveal that highly elevated levels of acetylated H3K9 at the DSP and first exon of the MDR1 gene correlate best with increased MDR1 expression in the drug‐resistant MCF7‐KCR line.
Global histone acetylation levels higher in drug‐resistant than in drug‐sensitive cell lines.
The observed alteration in the histone acetylation levels at the MDR1 locus in the MCF7‐KCR cells prompted us to study the global acetylation of specific histone lysine residues in the two cell lines. Western blot analysis showed that, although to different extent, each examined acetylated forms of histone H3 and H4 were present in higher levels in the drug‐resistant cell line than in the drug‐sensitive counterpart (Fig. 2a–e). Most strikingly, although K4 and K9 acetylated forms of H3 were hardly detectable in the parental cells, they were present at highly elevated levels in the drug‐resistant cells (Fig. 2a,b, tracks 1,3). A less dramatic, but still noticeable, increase in the levels of H3K14, H4K8, and H4K12 was detectable in the drug‐resistant cells compared to the drug‐sensitive line (Fig. 2c–e, tracks 1,3). In order to further characterize the regulation of histone acetylation, we treated the two cell lines with TSA, a histone deacetylase (HDAC) inhibitor, for 24 h. The TSA treatment induced a large increase of H3K4 and H3K9 acetylation and a moderate increase of H3K14, H4K8, and H4K12 acetylation in MCF7 cells (Fig. 2a–e, track 2), but the acetylation level of the studied residues did not increase any further in MCF7‐KCR cells (Fig. 2a–e, track 4). These data show that global increases in the levels of the studied acetylated forms of H3 and H4 are characteristic features of the drug‐resistant cell line, and HDAC inhibition causes no further increase.
Inhibition of HDACs upregulates MDR1 expression and increases H3K9 acetylation at the first exon of MDR1 in drug‐sensitive but not drug‐resistant cells.
In order to obtain further proof of the role of histone acetylation in MDR1 expression, we treated the two studied cell lines with TSA and examined changes in mRNA expression. MDR1 expression increased fivefold or 45‐fold after treatment with 50 or 100 ng/mL TSA, respectively, in the drug‐sensitive line (Fig. 2f), however, it did not change in the drug‐resistant line compared to untreated cells (Fig. 2h). Fluorescent assay showed decreased drug accumulation in MCF7 cells (Fig. 2g) but not in MCF7‐KCR cells (data not shown), indicating that increased MDR1 expression results in drug efflux. Expression of RPIB9 increased sixfold in MCF7 cells, but did not change in MCF7‐KCR cells. However, the expression of MRP1 and GAPDH did not change in either cell line, supporting the model of Wang et al.28 that HDAC inhibitor treatment induces expression of only certain “primed” genes. Therefore, we were interested in whether TSA had any local effect on the chromatin structure of the MDR1 gene, and carried out ChIP analysis. In the MCF7 cells we detected large increases in the level of H3K9 acetylation: 3–10‐fold at the RPIB9 promoter, USP, exon −1, DSP and exon 1 of MDR1 (Fig. 3b). Furthermore, acetylation of H3K4 increased threefold at the gene body (Fig. 3a), and that of H4K12 increased twofold at the USP and exon 1 (Fig. 3e). However, acetylation levels of H3K14 and H4K8 did not significantly change in the studied regions (Fig. 3c,d). After treatment of the drug‐resistant cells with TSA, neither acetylation of H3K9, nor that of H3K4, H3K14, or H4K8 changed at any of the studied regions regardless of whether they were low or high in the untreated cells (Fig. 3a–j). In contrast, acetylation of H4K12 (Fig. 3k) increased 8–16‐fold in MCF7‐KCR cells. The amount of H3 histone did not change significantly in the studied regions in either cell line (Fig. 3f,l). These data suggest that elevated H3K9 acetylation at both promoters and first exons of the MDR1 gene correlates with its increased expression in MCF7 after treatment with TSA.
Simultaneous downregulation of PCAF and GCN5 reduces the level of MDR1 mRNA.
The highly increased acetylation level of H3K9 observed around the transcription start site (TSS) of MDR1 and its elevated expression in MCF7‐KCR cells prompted us to investigate which HAT(s) might contribute to them. It has been shown previously that PCAF participates in the regulation of MDR1 expression.16 Initial RT‐qPCR and Western blot analysis showed that PCAF expression is not elevated, but in fact slightly decreased in the MCF7‐KCR cells compared to the drug‐sensitive cells (data not shown; Fig. 4a, right). We knocked down the expression of PCAF; as well as GCN5, a HAT closely related to PCAF; and ADA2B, an essential subunit of the SAGA‐type HAT complexes;24 in order to test their role in the regulation of MDR1 expression. Treatment of the cells with PCAF‐specific siRNA resulted in a strong reduction of PCAF message and protein levels in both cell lines (Fig. 4a), and a concomitant 70% reduction of MDR1 mRNA level in MCF7 cells, but no change in MCF7‐KCR cells (Fig. 4b). GCN5‐specific siRNA treatment resulted in a 90% reduction of GCN5 mRNA and a similarly strong reduction of GCN5 protein level (Fig. 4c), but the MDR1 mRNA level did not significantly change in either cell line (Fig. 4d). Treatment of the cells with ADA2B‐specific siRNA resulted in an 88% reduction of ADA2B message level in both cell lines (Fig. 4e), and a concomitant 50% reduction of MDR1 mRNA level in MCF7 cells, but no change in MCF7‐KCR cells (Fig. 4f). However, simultaneous knockdown of the expression of PCAF and GCN5 (Fig. 4g) resulted in a strong reduction of MDR1 mRNA levels in both cell lines (Fig. 4h). These data suggest that a complex containing PCAF and ADA2B plays a role in the regulation of the basal expression of MDR1 in the drug‐sensitive cell line. The finding that downregulation of PCAF or GCN5 individually is not able to downregulate MDR1 expression in the drug‐resistant cells, but their simultaneous downregulation is, suggests that these HATs substitute each other in maintaining the elevated expression of MDR1.
Inhibition of p300 HAT activity increases MDR1 expression in drug‐resistant but not drug‐sensitive cells.
P300 was also shown to participate in the activation of MDR1 expression.17 Therefore, we treated the cells with a novel p300 selective inhibitor, HATi II [2,6‐bis‐(3‐bromo‐4‐hydroxybenzylidene)],29 and examined changes in gene expression. After treatment of MCF7 cells for 24 h with 20 μM HATi II, MDR1 expression slightly decreased, whereas that of MRP1 increased twofold (Fig. 5a). Fluorescent assay showed no significant change in doxorubicin accumulation in these cells (Fig. 5b). Unexpectedly, treatment of the MCF7‐KCR cells with HATi II resulted in a sevenfold increase in the expression of MDR1 (Fig. 5c). Furthermore, fluorescent drug accumulation assay also showed that the intracellular doxorubicin level in the HATi II‐treated cells was lower than in the untreated MCF7‐KCR cells, suggesting that the MDR1 protein produced is functional (Fig. 5d). Interestingly, although expression of the other genes did not change, that of c‐FOS increased 300‐fold. This suggests that the increased expression after treatment with HATi II is not unique to MDR1 in the drug‐resistant cells, supporting the hypothesis that the functioning of HATs is altered there. Thus, like PCAF inhibition, inhibition of p300 does not downregulate expression of MDR1 in drug‐resistant cells; on the contrary, it results in an increase in MDR1 expression and drug efflux.
Global acetylation in drug‐resistant cells effectively decreased by treatment with HATi II.
The surprising finding of increased MDR1 and c‐FOS mRNA levels after HAT inhibitor treatment made us wonder how HATi II affected acetylation of specific lysine residues genome‐wide and at specific regions in the MDR1 locus. Although global acetylation of H3K4 and H3K9 were almost undetectably low in MCF7 cells (Fig. 2a,b), we could still see a reduction in their levels after HATi II treatment (Fig. 5e,f, track 2). A less dramatic reduction in the amount of H3K14, H4K8, and H4K12 acetylated histones was clearly detectable in the drug‐sensitive cells (Fig. 5g,j, track 2). A recent study showed that p300 specifically acetylates H3K18.30 Therefore, we examined the acetylation level of that residue and found it also decreased (Fig. 5h). Similarly, acetylation of all six examined histone lysine residues was reduced to various extents in the drug‐resistant cell line (Fig. 5e–j, tracks 3–6). These data show that HATi II treatment was effective in reducing genome‐wide acetylation in both cell lines, and failure to reduce the expression of MDR1 in MCF7‐KCR cells was not due to inactivation of the drug. Next, we examined the specific changes in histone acetylation in the MDR1 locus. Interestingly, the level of H3K4 acetylation increased fivefold at the RPIB9 promoter, DSP, and exon 1 of MDR1 in the drug‐resistant but not drug‐sensitive cells (Fig. 6a,g). There was no statistically significant change in the acetylation levels of H3K9, H3K14, H4K8, or H4K12 in either cell line (Fig. 6b–e,h–k). There was some decrease in histone occupancy around the TSS in MCF7 cells (Fig. 6f,l). These data show that although HATi II effectively reduces histone acetylation genome‐wide, it does not affect or even increase the acetylation level of certain histone residues at the MDR1 locus. Specifically, the increase in H3K4 acetylation coincides with the increase of MDR1 expression in the drug‐resistant cell line after treatment with HATi II.
Discussion
Histones are subject to various post‐translational modifications that can affect transcription. Histone lysine acetylation was proposed to create a more amenable chromatin structure for transcription by neutralizing the positive charge of histones. Moreover, occurring on various residues, acetylation also represents “letters” of the histone code that are interpreted by the transcriptional machinery. Altered genome‐wide acetylation levels of H3K9, H3K18, H4K12, and H4K16 have been found in prostate, lung, kidney, breast, pancreatic, and gastric cancers.31, 32, 33, 34 In particular, H3K18 acetylation was shown to be a prognostic factor in several studies, with high histone acetylation correlating with better prognosis.34 In contrast, a study involving non‐small‐cell lung cancer patients found that higher levels of H3K9 acetylation correlated with shorter survival.35 These contrasting findings suggest that acetylation on various lysine residues may have different functions in different types of cancer. We investigated whether altered genome‐wide histone acetylation might accompany drug resistance. We found that global acetylation levels of H3K4, H3K9, H3K14, H4K8, and H4K12 are higher in drug‐resistant MCF7‐KCR cells than in drug‐sensitive MCF7 cells. Similar to our findings, increased global acetylation has been reported for the OV1/VCR multidrug‐resistant ovarian carcinoma cell line.36
Increased histone H3 and H4 acetylation in the chromatin of the MDR1 promoter, has been found in drug‐resistant cell lines expressing elevated levels of MDR1.18 When compared to a T‐cell leukemia line, two cell lines expressing increased levels of MDR1 mRNA showed 3–30‐fold levels of acetylated H3 in the MDR1 promoter, first exon and transcribed region.18, 20, 21 Interestingly, of doxorubicin‐resistant sarcoma cell lines generated from the same parental cell, some showed elevated H3 acetylation at the DSP, but others at the USP only.11 The individual acetyl‐lysine‐specific Abs were not available at the time of those studies, so the exact residues acetylated have not been identified. We revealed that acetylation of H3 lysine 9 is elevated by two orders of magnitude in the promoter and first exon of the MDR1 gene in a drug‐resistant cell line, whereas H3K4, H3K14, H4K8, and H4K12 acetylation increased only mildly or not at all, compared to the drug‐sensitive parental cell line.
Although most studies correlate H3K4 methylation with transcriptional activity of a given gene, Wang et al.37 reported enrichment of H3K4 acetylation around the TSS of genes with an intermediate level of expression. We found elevated genome‐wide acetylation of H3K4 and a slight increase of H3K4ac in the first exon of MDR1 in MCF7‐KCR cells. Furthermore, Wang et al. characterized active genes with a combinatorial pattern of modifications, including H3K4ac, H3K9ac, H4K8ac, and, for genes with the highest expression, H4K12ac along the gene body. Interestingly, we found H3K9ac alone extremely highly elevated around the TSS, and H4K12ac unchanged along the gene body of MDR1 in the drug‐resistant cells.
Trichostatin A is a potent inhibitor of HDACs and it induces the expression of several genes.28 It is a promising therapeutic candidate against cancer because it induces differentiation, cell cycle arrest, and apoptosis.38, 39 However, several studies have shown that TSA may induce drug resistance, limiting its therapeutic potential.16, 40 In acute myeloid leukemia cells, TSA treatment increased the expression not only of MDR1 but also of genes encoding other drug transporters, BCRP/ABCG2 and MRP8/ABCC11, thereby inducing a very broad drug resistance phenotype.41 In contrast, other reports showed that TSA did not induce MDR1 transcription, although it resulted in a significant increase in the level of acetylated histones at the MDR1 promoter.18, 42 Demethylation of promoter DNA was required for TSA‐inducibility of MDR1 expression in these cells.42, 43 Here, we report that TSA induces MDR1 expression in drug‐sensitive MCF7 cells, but not in drug‐resistant MCF7‐KCR cells. Three studies reported findings similar to ours.21, 44 In a drug‐sensitive small‐cell lung carcinoma line, TSA induced MDR1 expression, but decreased it in the drug‐resistant derivative line. The extent and temporal kinetics of H4 and H3 acetylation at the MDR1 promoter was different in the two cell lines.18 These data suggest that TSA can be a therapeutic candidate for multidrug‐resistant cancers or those with altered histone acetylation.
The finding that H3K9 acetylation around the TSS of MDR1 is very high in our drug‐resistant cell line prompted us to investigate which HAT might be responsible for generating this epigenetic mark. The HATs that may participate in the regulation of MDR1 expression have not been extensively studied. One candidate, PCAF, has been shown to co‐activate an MDR1 promoter–luciferase reporter construct,16 and to bind to the endogenous promoter.18 Our RNA knockdown studies suggest that a SAGA‐type complex involving PCAF and ADA2B,24 but not GCN5, participates in the basal expression of MDR1 in MCF7 cells. Individual downregulation of PCAF, GCN5, or ADA2B did not reduce the very high expression of MDR1 in MCF7‐KCR cells, however, simultaneous downregulation successfully reduced it. This suggests that PCAF and GCN5 substitute each other in maintaining the elevated expression of MDR1 and they may be responsible for generating the elevated H3K9 acetylation pattern at the MDR1 TSS. Both PCAF and p300 can acetylate H3K9 in vitro, although this is not their primary substrate.45, 46 Inhibition of p300 by a specific inhibitor, HATi II,29 resulted in genome‐wide reduction of acetylation on all six studied histone lysine residues in both cell lines. Nevertheless, expression of MDR1 further increased in MCF7‐KCR but not MCF7 cells. The acetylation level of H3K4 increased fivefold at the RPIB9 promoter and around the TSS of MDR1 in the drug‐resistant but not drug‐sensitive cells, whereas acetylation of the other examined residues remained relatively unchanged. There could be several possible explanations for the unexpected induction of MDR1 expression in the drug‐resistant cells. One possibility is that HATi II is sensed by the cell as a xenobiotic, and HATi II activates the pathway that normally induces MDR1 expression.47 However, the fact that HATi II does not induce MDR1 expression in MCF7 argues against this hypothesis. It is more likely that the various HATs and HDACs co‐regulate each other and that these interactions are deregulated in MCF7‐KCR. Inhibition of p300 may result in activation of (an) unidentified HAT(s) and these in turn acetylate H3K4 and further induce MDR1 expression. However, it is questionable whether increased expression of MDR1 is really due to increased acetylation of H3K4. As p300 acetylates transcription factors besides histones, the possibility exists that inhibition of p300 results in a reduced level of acetylation of another factor that regulates MDR1 expression in the drug‐resistant cell line. The role of HDACs in the altered histone acetylation in MCF7‐KCR remains to be explored.
In summary, the drug‐resistant MCF7‐KCR cell line, developed by treatment of MCF7 cells with increasing concentrations of doxorubicin, seems to use multiple mechanisms that ensure elevated expression of MDR1. These mechanisms include gene amplification and epigenetic alterations, including highly elevated H3K9 acetylation around the TSS of MDR1. Both PCAF and GCN5 seem to be responsible for the increased H3K9 acetylation and elevated expression of MDR1. Unexpectedly, inhibition of p300 activity results in a further increase of MDR1 mRNA levels in the drug‐resistant cells, suggesting that there might be additional mechanisms deregulated. Our findings are valuable for the understanding of the role and deregulation of histone modifications in cancer.
Disclosure Statement
The authors declare no conflict of interest.
Supporting information
Fig. S1. MDR1 gene is amplified over 40‐fold in drug‐resistant cells.
Table S1. Primers used for quantitative PCR.
Acknowledgments
This work was supported by the Hungarian National Office for Research and Technology (NKTH, grant number OMFB‐00441/2007 Teller Ede Program NAP‐BIO 06/1) to Professor Eva Kondorosi (BAYGEN Institute, Szeged, Hungary) and by the Hungarian Scientific Research Fund (OTKA 77443) to Imre M. Boros. We thank Professor Joseph Molnar (Department of Medical Microbiology and Immunobiology, University of Szeged, Szeged, Hungary) and the Szeged Foundation for Cancer Research for the MCF7 and MCF7‐KCR cell lines. We are grateful to Professor Laszlo Tora (IGBMC, Illkirch, France) for the GCN5 and PCAF Abs, Didier Devys (IGBMC) for the ADA2B Ab, and Gergely Szakacs (Semmelweis University, Budapest, Hungary) for the MDR1 Ab. We thank Eniko Nagy (University of Szeged) for valuable comments on the manuscript and Professor Eva Kondorosi (BAYGEN Institute) for support.
References
- 1. Fojo AT, Whang‐Peng J, Gottesman MM, Pastan I. Amplification of DNA sequences in human multidrug‐resistant KB carcinoma cells. Proc Natl Acad Sci USA 1985; 82: 7661–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scotto KW, Biedler JL, Melera PW. Amplification and expression of genes associated with multidrug resistance in mammalian cells. Science 1986; 232: 751–5. [DOI] [PubMed] [Google Scholar]
- 3. Mickley LA, Spengler BA, Knutsen TA, Biedler JL, Fojo T. Gene rearrangement: a novel mechanism for MDR‐1 gene activation. J Clin Invest 1997; 99: 1947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen DW, Fojo A, Chin JE et al Human multidrug‐resistant cell lines: increased mdr1 expression can precede gene amplification. Science 1986; 232: 643–5. [DOI] [PubMed] [Google Scholar]
- 5. Nakayama M, Wada M, Harada T et al Hypomethylation status of CpG sites at the promoter region and overexpression of the human MDR1 gene in acute myeloid leukemias. Blood 1998; 92: 4296–307. [PubMed] [Google Scholar]
- 6. Chaudhary PM, Roninson IB. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J Natl Cancer Inst 1993; 85: 632–9. [DOI] [PubMed] [Google Scholar]
- 7. Leonessa F, Clarke R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr Relat Cancer 2003; 10: 43–73. [DOI] [PubMed] [Google Scholar]
- 8. Abolhoda A, Wilson AE, Ross H, Danenberg PV, Burt M, Scotto KW. Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clin Cancer Res 1999; 5: 3352–6. [PubMed] [Google Scholar]
- 9. Ueda K, Clark DP, Chen CJ, Roninson IB, Gottesman MM, Pastan I. The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J Biol Chem 1987; 262: 505–8. [PubMed] [Google Scholar]
- 10. Chin JE, Soffir R, Noonan KE, Choi K, Roninson IB. Structure and expression of the human MDR (P‐glycoprotein) gene family. Mol Cell Biol 1989; 9: 3808–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen KG, Wang YC, Schaner ME et al Genetic and epigenetic modeling of the origins of multidrug‐resistant cells in a human sarcoma cell line. Cancer Res 2005; 65: 9388–97. [DOI] [PubMed] [Google Scholar]
- 12. Raguz S, Randle RA, Sharpe ER et al Production of P‐glycoprotein from the MDR1 upstream promoter is insufficient to affect the response to first‐line chemotherapy in advanced breast cancer. Int J Cancer 2008; 122: 1058–67. [DOI] [PubMed] [Google Scholar]
- 13. Raguz S, Tamburo De Bella M, Tripuraneni G et al Activation of the MDR1 upstream promoter in breast carcinoma as a surrogate for metastatic invasion. Clin Cancer Res 2004; 10: 2776–83. [DOI] [PubMed] [Google Scholar]
- 14. Labialle S, Gayet L, Marthinet E, Rigal D, Baggetto LG. Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochem Pharmacol 2002; 64: 943–8. [DOI] [PubMed] [Google Scholar]
- 15. Scotto KW. Transcriptional regulation of ABC drug transporters. Oncogene 2003; 22: 7496–511. [DOI] [PubMed] [Google Scholar]
- 16. Jin S, Scotto KW. Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF‐Y. Mol Cell Biol 1998; 18: 4377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin W, Liu Y, Xu SG et al UHRF1 inhibits MDR1 gene transcription and sensitizes breast cancer cells to anticancer drugs. Breast Cancer Res Treat 2010; 124: 39–48. [DOI] [PubMed] [Google Scholar]
- 18. El‐Khoury V, Breuzard G, Fourre N, Dufer J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription‐dependent mechanism in a drug‐resistant small cell lung carcinoma cell line model. Br J Cancer 2007; 97: 562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim SN, Kim NH, Lee W, Seo DW, Kim YK. Histone deacetylase inhibitor induction of P‐glycoprotein transcription requires both histone deacetylase 1 dissociation and recruitment of CAAT/enhancer binding protein beta and pCAF to the promoter region. Mol Cancer Res 2009; 7: 735–44. [DOI] [PubMed] [Google Scholar]
- 20. Baker EK, Johnstone RW, Zalcberg JR, El‐Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene 2005; 24: 8061–75. [DOI] [PubMed] [Google Scholar]
- 21. Yatouji S, El‐Khoury V, Trentesaux C et al Differential modulation of nuclear texture, histone acetylation, and MDR1 gene expression in human drug‐sensitive and ‐resistant OV1 cell lines. Int J Oncol 2007; 30: 1003–9. [PubMed] [Google Scholar]
- 22. Kars MD, Iseri OD, Gunduz U, Ural AU, Arpaci F, Molnar J. Development of rational in vitro models for drug resistance in breast cancer and modulation of MDR by selected compounds. Anticancer Res 2006; 26: 4559–68. [PubMed] [Google Scholar]
- 23. Molnár J, Engi H, Gyémánt N et al Multidrug resistance reversal on cancer cells by selected carotenoids, flavonoids and anthocyanins In: Motohashi N, ed. Bioactive Heterocycles VI. Berlin/Heidelberg: Springer, 2008; 133–59. [Google Scholar]
- 24. Nagy Z, Riss A, Fujiyama S et al The metazoan ATAC and SAGA coactivator HAT complexes regulate different sets of inducible target genes. Cell Mol Life Sci 2010; 67: 611–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brand M, Moggs JG, Oulad‐Abdelghani M et al UV‐damaged DNA‐binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 2001; 20: 3187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helmlinger D, Hardy S, Eberlin A, Devys D, Tora L. Both normal and polyglutamine‐ expanded ataxin‐7 are components of TFTC‐type GCN5 histone acetyltransferase‐ containing complexes. Biochem Soc Symp 2006; 73: 155–63. [DOI] [PubMed] [Google Scholar]
- 27. Fairchild CR, Ivy SP, Kao‐Shan CS et al Isolation of amplified and overexpressed DNA sequences from adriamycin‐resistant human breast cancer cells. Cancer Res 1987; 47: 5141–8. [PubMed] [Google Scholar]
- 28. Wang Z, Zang C, Cui K et al Genome‐wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009; 138: 1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Costi R, Di Santo R, Artico M et al Cinnamoyl compounds as simple molecules that inhibit p300 histone acetyltransferase. J Med Chem 2007; 50: 1973–7. [DOI] [PubMed] [Google Scholar]
- 30. Jin Q, Yu LR, Wang L et al Distinct roles of GCN5/PCAF‐mediated H3K9ac and CBP/p300‐mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 2011; 30: 249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seligson DB, Horvath S, Shi T et al Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005; 435: 1262–6. [DOI] [PubMed] [Google Scholar]
- 32. Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol 2008; 15: 1968–76. [DOI] [PubMed] [Google Scholar]
- 33. Elsheikh SE, Green AR, Rakha EA et al Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 2009; 69: 3802–9. [DOI] [PubMed] [Google Scholar]
- 34. Manuyakorn A, Paulus R, Farrell J et al Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol 2010; 28: 1358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barlesi F, Giaccone G, Gallegos‐Ruiz MI et al Global histone modifications predict prognosis of resected non small‐cell lung cancer. J Clin Oncol 2007; 25: 4358–64. [DOI] [PubMed] [Google Scholar]
- 36. Yatouji S, Trussardi‐Regnier A, Trentesaux C, Liautaud‐Roger F, Dufer J. Nuclear texture and chromatin structure in OV1/VCR human multidrug‐resistant cell line. Int J Oncol 2003; 23: 1225–30. [PubMed] [Google Scholar]
- 37. Wang Z, Zang C, Rosenfeld JA et al Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 2006; 5: 37–50. [DOI] [PubMed] [Google Scholar]
- 39. Lee E, Furukubo T, Miyabe T, Yamauchi A, Kariya K. Involvement of histone hyperacetylation in triggering DNA fragmentation of rat thymocytes undergoing apoptosis. FEBS Lett 1996; 395: 183–7. [DOI] [PubMed] [Google Scholar]
- 40. Lee TB, Park JH, Min YD, Kim KJ, Choi CH. Epigenetic mechanisms involved in differential MDR1 mRNA expression between gastric and colon cancer cell lines and rationales for clinical chemotherapy. BMC Gastroenterol 2008; 8: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hauswald S, Duque‐Afonso J, Wagner MM et al Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin Cancer Res 2009; 15: 3705–15. [DOI] [PubMed] [Google Scholar]
- 42. El‐Osta A, Kantharidis P, Zalcberg JR, Wolffe AP. Precipitous release of methyl‐CpG binding protein 2 and histone deacetylase 1 from the methylated human multidrug resistance gene (MDR1) on activation. Mol Cell Biol 2002; 22: 1844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. David GL, Yegnasubramanian S, Kumar A et al MDR1 promoter hypermethylation in MCF‐7 human breast cancer cells: changes in chromatin structure induced by treatment with 5‐Aza‐cytidine. Cancer Biol Ther 2004; 3: 540–8. [DOI] [PubMed] [Google Scholar]
- 44. Tian K, Jurukovski V, Wang XP, Kaplan MH, Xu H. Epigenetic regulation of WTH3 in primary and cultured drug‐resistant breast cancer cells. Cancer Res 2005; 65: 10024–31. [DOI] [PubMed] [Google Scholar]
- 45. Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem 1999; 274: 1189–92. [DOI] [PubMed] [Google Scholar]
- 46. Liu X, Wang L, Zhao K et al The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008; 451: 846–50. [DOI] [PubMed] [Google Scholar]
- 47. Shtil AA, Azare J. Redundancy of biological regulation as the basis of emergence of multidrug resistance. Int Rev Cytol 2005; 246: 1–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. MDR1 gene is amplified over 40‐fold in drug‐resistant cells.
Table S1. Primers used for quantitative PCR.