

Abstract
The gene, collagen triple helix repeat containing 1 (CTHRC1), has been reported to increase in several kinds of human solid cancers and is associated with tumor invasion and metastasis. To date, the expression and function of CTHRC1 in gastric cancer (GC) have not been reported. The aim of this study was to investigate the expression levels and regulatory transcription mechanisms of CTHRC1 in GC. Immunohistochemical analysis revealed that CTHRC1 expression was markedly increased in carcinoma compared with normal gastric mucosa, chronic atrophic gastritis, and intestinal metaplasia (P < 0.05 for all), and this overexpression in tumor was related to depth of tumor invasion. Moreover, RNA interference‐mediated knockdown and ectopic expression of CTHRC1 showed that CTHRC1 promoted tumor cell invasion in vitro. We then investigated the mechanisms underlying the aberrant expression of CTHRC1 in GC and found that CTHRC1 expression was restored after GC cell lines were treated with the demethylating agent, 5‐aza‐2'‐deoxycytidine. Transforming growth factor‐β1 led to an increase in levels of CTHRC1 mRNA and protein. Overall, our data revealed that the upregulated expression of CTHRC1 in gastric carcinogenesis contributes to tumor cell invasion and metastasis, and promoter demethylation and transforming growth factor‐β1 may co‐regulate the expression of CTHRC1. (Cancer Sci 2012; 103: 1327–1333)
Gastric cancer (GC) is one of most common cancers worldwide.1 It has been reported that multiple genetic and epigenetic alterations are implicated in the development of GC,2, 3 but the molecular mechanisms of its pathogenesis remain unclear. More recently, a growing body of published reports has defined a class of genes that function, positively or negatively, in regulating the invasion and metastasis of malignant cancer cells.4, 5, 6 As invasion and metastasis is the leading cause of GC‐related death, identifying novel metastasis‐related molecules may be useful for the prediction of prognosis and the treatment of advanced GC.
Collagen triple helix repeat containing 1 (CTHRC1) was first found in a screen for differentially expressed sequences in balloon‐injured versus normal rat arteries.7 Tang et al.8 reported that CTHRC1 was increased in several kinds of human solid cancers and associated with invasion and metastasis. However, to date, no investigation has addressed the expression of CTHRC1 and the mechanisms that underlie the aberrant expression of CTHRC1 in GC.
In this study, we have found that CTHRC1 expression was increased in gastric carcinogenesis by immunohistochemical (IHC) analysis. This increase was associated with depth of tumor invasion. Moreover, RNA interference‐mediated knockdown and re‐expression of CTHRC1 showed that CTHRC1 promoted tumor cell invasion and metastasis in vitro. In addition, we investigated the mechanisms by which CTHRC1 expression was regulated.
Materials and Methods
Patients and tissue specimens
A total of 116 GC tissue samples were collected from two centers: 51 patients at Renji Hospital (Shanghai, China) from January 2008 to December 2009, and another 65 patients at the No. 3 People's Hospital (Shanghai, China) between August 1992 and December 1999. Gastroscopic paraffin‐embedded specimens from the same period were randomly selected, including 10 specimens of normal gastric mucosa, 46 of chronic atrophic gastritis (CAG), 53 of intestinal metaplasia and 19 of dysplasia. The visual analogue scale of the updated Sydney System was used as the pathological diagnostic criteria for chronic gastritis,9 and a Chinese visual analogue scale was also used.10 The pathomorphological diagnosis of dysplasia and GC was based on the Padova international classification.11 All the paraffin blocks in this study were resliced, restained, and rechecked by two senior pathologists.
All patients and controls gave informed consent, and the study protocol was approved by the Clinical Research Ethics Committee of the Shanghai Jiaotong University School of Medicine (Shanghai, China).
Immunohistochemistry
Rabbit anti‐human CTHRC1 polyclonal antibody was purchased from Abcam (1:250; Cambridge, USA). The IHC streptavidin–peroxidase assay was carried out according to the protocol of the manufacturer. The CTHRC1‐positive gastric cancer tissue samples were used as the positive control; the positive tissue slices were incubated with PBS instead of the primary antibody as the blank control; the gastritis tissue samples served as the negative control. The intensity of staining was graded on a scale from 0 to 3 and the extent of positive immunoreactivity was scored according to the percentage of stained cells (+1, <20% of cells stained; +2, 20–50% stained; and +3, >50% of cells stained). The total score was obtained as the product of intensity and extent of staining. Negative cases had a score of 0 ± 3, positive had a score >4. All the samples were analyzed by two senior pathologists.
Real‐time RT‐PCR
Total RNA was extracted from gastric cancer cells by TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Overall, 1 μg total RNA was transcribed using RT‐Reagents (TaKaRa, Dalian, China), and preserved at −20°C. The mRNA expression level of CTHRC1 was determined by real‐time PCR, as described previously.12 The primer sequences are shown in Table 1.
Table 1.
Primers for each PCR
| Genes | PCR | Primer F (5′–3′) Primer R (5′–3′) | Product size (bp) Temp. (°C) |
|---|---|---|---|
|
CTHRC1 (upstream region of TSS) |
PCR |
TTAGCCGGGCGTGGTGGCG AGGAGACCGAGGAGAGGAACG |
580 60 |
|
CTHRC1 (exon 1) |
PCR |
CCTATTACGGGATGGAAGCTC GCAAGACACACGACACACAGC |
470 60 |
|
CTHRC1 (exon 2) |
PCR |
ATTCTTGACTCCAAGGGCTCA CGCAATTTTCCCAAGATCTATG |
324 60 |
|
CTHRC1 (exon 3) |
PCR |
GAGTGTACATTTACAAAGATG TACAGTAGGCTAGTGAACCAACAT |
310 60 |
|
CTHRC1 (exon 4) |
PCR |
AGAATTACAAACTAGCTTTCTGAAGT GTTGGAAATAATTTTTATTTAACAGATAT |
626 60 |
| CTHRC1 | BSP |
TAGGTTGTATTAATGYGTTTTTTA ACCACRTATCTCTAAAATATTCATT |
322 55 |
| CTHRC1 | RT‐PCR |
CCAACTACAAGCAGTGTTCATGGAG TTGAATGTGAAATACCAACGCTGAC |
175 60 |
| CTHRC1 | Real‐time PCR |
TCATCGCACTTCTTCTGTGGA GCCAACCCAGATAGCAACATC |
76 60 |
| GAPDH |
RT‐PCR Real‐time PCR |
GCACCGTCAAGGCTGAGAAC ATGGTGGTGAAGACGCCAGT |
142 60 |
BSP, bisulfite sequencing PCR; CTHRC1, collagen triple helix repeat containing 1.
Bisulfite modification of DNA and bisulfite genomic sequencing
Genomic DNA samples were extracted from gastric tissues strictly according to the instructions of the manufacturer (Qiagen, Hilden, Germany). Briefly, 1 μg DNA was treated with sodium bisulfite using the EpiTect Bisulfite Kit (Qiagen). Bisulfite induces deamination of unmethylated cytosines, which converts unmethylated CpG sites to uracil. This allows their differentiation by bisulfite genomic sequencing, as described previously.13 The primer sequences are shown in Table 1.
Cancer cell lines
Six human GC cell lines (AGS, MGC803, SNU‐1, SGC7901, MKN28, and MKN45) were routinely maintained in RPMI‐1640 medium (Gibco, Carlsbad, CA, USA), supplemented with 10% FBS at 37°C in a humidified air atmosphere containing 5% CO2. The AGS cell line was obtained from ATCC (Manassas, VA, USA). MKN45, MGC803, SNU‐1, SGC7901, and MKN28 were obtained from Shanghai Cellular Institute of Chinese Academy of Sciences (Shanghai, China). Stable transfected MKN45 cell lines were generated with the pLV‐CHTRC1 vector using standard techniques, and empty controls were generated with the pLV vector.
Mutations analysis of CTHRC1 by DNA sequencing
The primers (shown in Table 1) were designed according to the exons and region upstream of the CTHRC1 transcriptional start site by Primer Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA). Products were purified and sequenced by Generay Biotechnology Shanghai (Shanghai, China).
Treatment with 5‐aza‐2'‐deoxycytidine (5‐aza‐dC) and transforming growth factor (TGF)‐β1
Gastric cancer cells (AGS, MKN45, and SGC7901) were seeded onto a plate at a density of 1 × 106 cells/mL. After 24 h, cells were treated with 5 μmol/L 5‐aza‐dC, a DNA demethylating agent (Sigma‐Aldrich, St. Louis, MO, USA), for 72 h. Cells were then harvested and underwent DNA and RNA extraction.
Before cells were stimulated with 10 ng/mL TGF‐β1 (R&D Systems, Minneapolis, MN, USA), SGC7901 cells and MKN45 cells were placed in serum‐free medium for 24 h. Cells were harvested for real‐time RT‐PCR and Western blotting at the indicated times.
RNA interference
The target siRNA sequences for CTHRC1 were: 5′‐CGGAGUGUACAUUUACAAATT‐3′ (target 1) and 5′‐CGCAUCAUUAUUGAAGAACUATT‐3′ (target 2). Both the RNA duplexes and the control siRNA were synthesized by GenePharma (Shanghai GenePharma, Shanghai, China). For siRNA transfection, GC cells were seeded 24 h prior to the transfection in RPMI‐1640 medium containing 10% FBS. The cells were transfected with 50 nmol/L siRNA using Lipofectamine 2000 (Invitrogen) for 6 h in serum‐free medium. The medium was then replaced with 10% FBS.
Western blot analyses
Western blot analyses were carried out using standard techniques as described previously.14 Briefly, the cells were lysed in RIPA lysate (Beyotime, Beijing, China). Equal amounts of protein (60 μg/lane) from whole cell lysates were subjected to SDS‐PAGE. The proteins were then transferred to nitrocellulose gels (Amersham, Buckinghamshire, UK) and were probed initially with specific primary antibodies, then with the appropriate HRP‐conjugated secondary antibodies (Pierce, Rockford, IL, USA). Proteins were detected using a enhanced chemiluminescence detection kit (SuperSignal West Femto Substrate; Pierce). For the loading control, the membrane was probed with a mAb against GAPDH. The antibodies used in this study were CTHRC1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Smad2‐3 (Cell Signaling Technology, Danvers, MA, USA), pho‐Smad2 (Ser465‐467; Cell Signaling Technology), and GAPDH (Kangchen, Shanghai, China). All primary antibodies were used at a 1:1000 dilution.
Cell migration assay
The cell invasion assay was carried out as described by Hecht et al.15 In brief, chambers with 8‐μm pore polycarbonate membranes, coated with Matrigel on the upper side, were used (Becton Dickinson, San Diego, CA, USA). Cells were transfected with CTHRC1 siRNA (50 nM) for 24 h. Then 1 × 105 transfected cells were harvested and seeded in 0.1% FBS in medium into the upper chamber, whereas medium supplemented with 20% FBS was applied to the lower chamber as a chemoattractant to induce invasion. Cells transfected with non‐specific siRNA were used as the negative controls. After incubation for 48 h, non‐migrated cells on the upper chamber of the filter were removed with a cotton swab. Migrated cells on the bottom surface of the filter were fixed, stained, and counted. To confirm that the cellular invasion was due to the overexpression of CTHRC1, we also filled the upper chamber with either overexpressed levels of CTHRC1 or controls at equivalent volumes.
Cell viability assay
Cell viability was assessed by a tetrazolium salt (WST‐8)‐based colorimetric assay in the Cell Counting Kit 8 (CCK‐8; Dojindo, Kumamoto, Japan).16 Briefly, control and treated cells were seeded onto 96‐well plates at an initial density of 5 × 103 cells/well. At specified time points, 10 μL CCK‐8 solution was added to each well of the plate. The plate was then incubated for 1 h. Cell viability was determined by scanning with a microplate reader at 450 nm. Data were expressed as the percentage of viable cells as follows: relative viability (%) = [A450(treated)−A450(blank)]/[A450(control)−450(blank)] × 100%.
Flow cytometry for the detection of cell cycle progression
Cell cycle analysis was carried out by flow cytometry. Approximately 1 × 106 cells were removed and washed twice with PBS and fixed in ice‐cold ethanol for 1 h. The samples were then concentrated by the removal of ethanol and exposure to 1% (V/V) Triton X‐100 (Sigma‐Aldrich) and 0.01% RNase (Sigma‐Aldrich) for 10 min at 37°C. Cellular DNA was stained in the dark with 0.05% propidium iodide (PI) for 20 min at 4°C. Cell cycle distributions were determined using a flow cytometer (FACSCalibur; Becton Dickinson, San Jose, CA, USA). The data obtained from 10 000 cells were analyzed using the MultiCycle software package (Phoenix Flow Systems, San Diego, CA, USA).
Apoptosis detection
Apoptosis was determined by flow cytometry analysis; an annexin‐V fluorescein isothiocyanate/PI double‐stain assay was carried out in accordance with the protocol of the manufacturer (BioVision, Mountain View, CA, USA). Briefly, after treatment, both floating and trypsinized adherent cells (5 × 105) were collected and resuspended in 500 μL binding buffer containing 5 μL annexin‐V–fluorescein isothiocyanate and 5 μL PI. Samples were then incubated for 5 min in the dark at room temperature. Analysis was carried out immediately afterwards using a flow cytometer.
Statistical analysis
The spss 13.0 software (SPSS, Chicago, IL, USA) was applied in our study. Fisher's exact test and the Pearson's chi‐square ‐test were used to analyze CTHRC1 protein expression in the multistage development of GC. Results in vitro were expressed as the mean ± standard deviation, and the data were analyzed for significance by anova. A value of P < 0.05 was considered to be statistically significant.
Results
Expression of CTHRC1 upregulated in multistage tissues of gastric carcinogenesis
To investigate the change of CTHRC1 expression over the development of GC and to identify the correlations between the expression level of CTHRC1 and the clinicopathological characteristics of GC, we examined CTHRC1 protein expression by IHC in paraffin‐embedded specimens that represented different stages of GC progression.
Normal gastric mucosa, CAG and intestinal metaplasia tissues showed weak staining of the CTHRC1 protein, which was mostly present in the nucleus (Fig. 1A,B). In contrast, there was moderate to strong CTHRC1 expression in GC samples, which was detected mainly in the cytoplasm with partial extracellular space staining (Fig. 1C,D). With the progression of GC, the CTHRC1 expression increased gradually, which was found in 20.0% of normal gastric epithelium samples, 28.3% of CAG samples, 32.1% of intestinal metaplasia samples, 52.6% of dysplasia samples, and 67.2% of GC samples. The differences between normal gastric epithelium, CAG, intestinal metaplasia, and GC were statistically significant (P < 0.01). Overall, 67.2% of GC samples were positive for CTHRC1 staining, and the correlations between CTHRC1 expression and the clinicopathological characteristics of GC are summarized in Table 2. A significant correlation was found between increased CTHRC1 expression and the depth of invasion (P < 0.05, Pearson's chi‐square ‐test), which indicated that CTHRC1 was associated with invasion and metastasis in GC.
Figure 1.
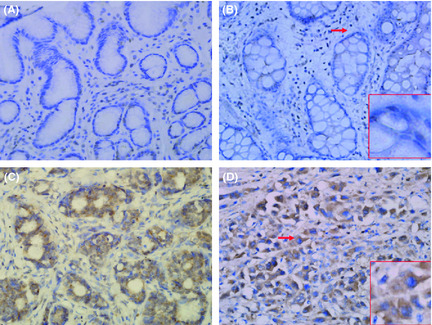
Immunohistochemical staining of gastric tissues with the anti‐collagen triple helix repeat containing 1 (CTHRC1) antibody. The CTHRC1 protein, which was mostly present in the nucleus, was weakly stained in normal gastric mucosa (A) and intestinal metaplasia (B). In contrast, CTHRC1 staining was moderate to strong in gastric cancer samples, and was detected mainly in the cytoplasm with partial extracellular space staining (C,D). Red arrows indicate the enlarged areas in red frames. Original magnification, ×200; ×400 (red frames).
Table 2.
Correlation between collagen triple helix repeat containing 1 (CTHRC1) immunoreactivity and clinical parameters in gastric cancer tissues
| Cases (n = 116) | CTHRC1 | CTHRC1 | P‐value | |
|---|---|---|---|---|
| Positive n (%) | Negative n (%) | |||
| Differentiation | ||||
| Well or moderate | 50 | 36 (72.0) | 14 (28.0) | 0.34 |
| Poor (or no differentiation) | 66 | 42 (78.0) | 24 (38.0) | |
| Lauren type | ||||
| Diffuse | 47 | 27 (57.4) | 20 (42.6) | 0.80 |
| Intestinal | 69 | 38 (55.1) | 31 (44.9) | |
| Depth of invasion | ||||
| T1/T2 | 70 | 42 (60.0) | 28 (40.0) | 0.04 |
| T3/T4 | 46 | 36 (78.3) | 10 (21.7) | |
| Lymph node metastasis | ||||
| − | 63 | 40 (63.5) | 23 (36.5) | 0.35 |
| + | 53 | 38 (71.7) | 15 (28.3) | |
| Size (cm) | ||||
| <5 | 75 | 48 (64.0) | 27 (36.0) | 0.31 |
| ≥5 | 41 | 30 (73.1) | 11 (26.8) | |
P‐value was calculated according to the Pearson's chi‐square‐test. Bold P‐value means that a significant correlation was found between increased CTHRC1 expression and the depth of invasion.
Expression of CTHRC1 in gastric cancer cell lines
We examined the mRNA and protein levels of CTHRC1 in six GC cell lines (AGS, MKN45, MGC803, SNU‐1, SGC7901, and MKN28) by RT‐PCR and Western blotting. These results showed that both CTHRC1 mRNA and protein were detected in four cell lines (shown in Fig. 2). Consistent with the results of the IHC staining in tissues described above, CTHRC1 expression was present in the cytoplasm (Fig. 3).
Figure 2.
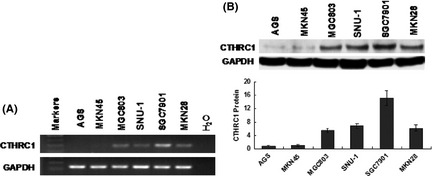
Protein and mRNA expression of collagen triple helix repeat containing 1 (CTHRC1) in cultured gastric cancer cell lines. Both CTHRC1 mRNA and protein were detected by RT‐PCR (A) and Western blot (B) analyses, respectively, in four gastric cancer cell lines (MGC803, SNU‐1, SGC7901, and MKN28).
Figure 3.
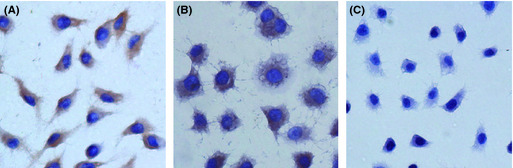
Immunohistochemical staining of gastric cancer cell lines with an anti‐collagen triple helix repeat containing 1 (CTHRC1) antibody. CTHRC1 expression was present in the cytoplasm of MKN28 (A) and SGC7901 (B) gastric cancer cells. No staining was observed in the negative control (C). Original magnification, ×400.
CTHRC1 increases gastric cancer cell migration
Previous studies have showed that CTHRC1 was upregulated in samples of GC, and was associated with increased tumor invasion and metastasis. The effect of the re‐expression of CTHRC1 on the invasiveness of GC cells was tested using the Boyden chamber. The CTHRC1 expression vector was stably transfected into MKN45 cells with silenced CTHRC1. Re‐expression of CTHRC1 in the transfected MKN45/CTHRC1 cells was confirmed by Western blotting (Fig. 4A1). We found that the number of invasive tumor cells was significantly higher in the CTHRC1‐transfected MKN45 cells than in control cells (P < 0.05; Fig. 4A2,3), which suggested that CTHRC1 promoted the invasion of GC cells. To verify the role of CTHRC1 in GC metastasis, we silenced CTHRC1 using siRNA in the SGC7901 cell line, which expresses high levels of CTHRC1, resulting in a decrease in cell invasiveness (P < 0.05; Fig. 4B). These data showed that CTHRC1 increased the invasiveness and metastatic potential of GC cells. In addition, CTHRC1 did not affect cell proliferation or apoptosis in our study (data not shown).
Figure 4.
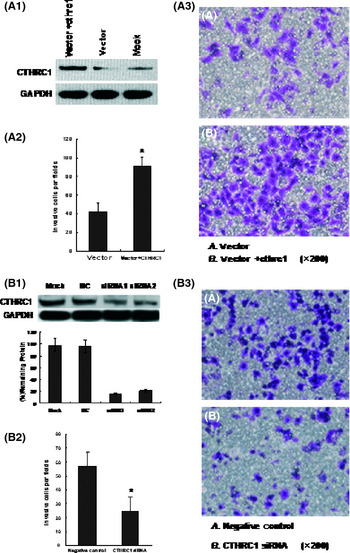
Re‐expression of collagen triple helix repeat containing 1 (CTHRC1) in CTHRC1‐transfected MKN45 gastric cancer cells was confirmed by Western blotting with anti‐CTHRC1 antibody (A1). The number of invasive cells was significantly higher in the CTHRC1‐transfected MKN45 cells than in control cells (P < 0.05; A2,A3). (B) Inhibition of CTHRC1 protein expression by siRNA in SGC7901 cells. The efficiency of inhibition was confirmed by Western blotting (B1). The number of invasive cells was decreased in CTHRC1 siRNA‐transfected cells compared with control cells (P < 0.05; B2,B3).
Aberrant expression of CTHRC1 could not be induced by exonic and regulatory region mutations
The mechanism of increased CTHRC1 with the development of GC remains unknown. Mutation is one of the principal reasons for gene amplification. Using DNA sequencing, we analyzed the exons and region upstream of the CTHRC1 transcriptional start site in three GC cell lines (AGS, MKN45, and SGC7901), three samples of gastritis, and three GC samples. No different bases were found between AGS and MKN45 cell lines with silenced CTHRC1, or in the SGC7901 cell line that expresses high levels of CTHRC1. Furthermore, no mutations were found between samples of gastritis and GC (data not shown). These results revealed that aberrant expression of CTHRC1 was perhaps not caused by mutations of the region described above, so other mechanisms may be involved in regulating the expression of CHTRC1.
Methylation may be associated with transcriptional silencing of CTHRC1 in GC cells
Promoter methylation plays an important role in the development of cancer, and has been reported to be related with transcription. When treated with the demethylating agent 5‐aza‐dC, the expression of the genes regulated by methylation will be upregulated. To test whether promoter methylation regulates CTHRC1 expression, we treated three cell lines (CTHRC1 is silenced in the AGS and MKN45 cell lines; SGC7901 shows high levels of CTHRC1 expression) with 5‐aza‐dC. Treatment with 5‐aza‐dC restored CTHRC1 expression in the AGS and MKN45 cell lines, and markedly elevated CTHRC1 expression in the SGC7901 cell line. There was obvious demethylation observed in the treated AGS and MKN45 cell lines by bisulfite genomic sequencing (Fig. 5), which indicated that methylation was associated with the transcriptional silencing of CTHRC1 in GC cells.
Figure 5.
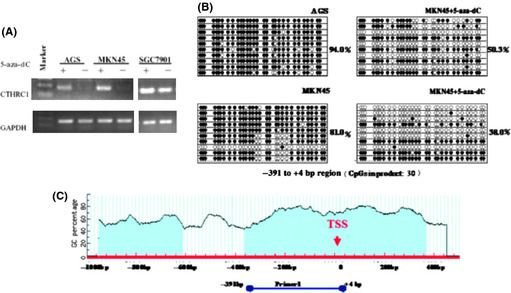
(A) mRNA expression of collagen triple helix repeat containing 1 (CTHRC1) was restored in gastric cancer (GC) cells after treatment with the demethylating agent, 5‐aza‐2'‐deoxycytidine (5‐aza‐dC). (B) Demethylation was observed in the AGS and MKN45 cell lines after treatment with 5‐aza‐dC, as determined by bisulfite genomic sequencing. (C) Cloned bisulfite sequencing was carried out on the CTHRC1 genomic region (−391 to +4 bp) relative to the transcription start site (TSS).
Effect of TGF‐β1 on the regulation of CTHRC1 expression
We treated the SGC7901 cell line with TGF‐β1. As seen in Figure 6, both CTHRC1 mRNA and protein levels increased gradually in response to stimulation with TGF‐β1, with maximal mRNA levels seen at 8 h and maximal protein levels at 24 h after growth factor addition. Meanwhile, pho‐Smad2 increased in a time‐dependent manner, with maximal levels seen between 1 and 8 h after treatment with TGF‐β1. Upregulation of CTHRC1 mRNA levels were also observed in another gastric cancer cell line, MKE45 (data not shown).
Figure 6.
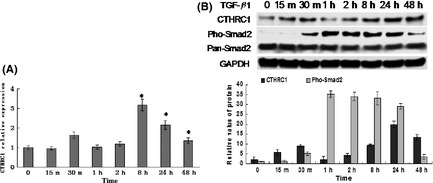
Collagen triple helix repeat containing 1 (CTHRC1) mRNA and protein of SGC7901 cells stimulated with transforming growth factor (TGF)‐β1 (10 ng/mL) for the indicated lengths of time as assessed by real‐time PCR (A) and Western blot (B) analyses. Both CTHRC1 mRNA and protein levels increased gradually in response to stimulation with TGF‐β1. Western blot analyses were also carried out with anti‐pSmad2 and anti‐(pan)Smad2 antibodies, and pho‐Smad2 increased in a time‐dependent manner (B).
Discussion
Gastric carcinogenesis is a multistep and chronic process, where the gastric tissues undergo atrophy, intestinal metaplasia, dysplasia, and ultimately, cancer.2 CTHRC1 is a glycosylated protein that contains a NH2‐terminal signal peptide for extracellular secretion, a short collagen triple helix repeat of 36 amino acids, and a COOH‐terminal globular domain.7, 17, 18 It has been found to be overexpressed in several malignant tumors, including breast cancer,19 GC, and malignant melanoma.8 However, CHTRC1 was simply screened out as being increased in GC, but an initial biochemical and functional characterization of this molecule in GC has not been undertaken.
In this study, using IHC, we observed that the level of CTHRC1 gradually increased with the development of GC. Furthermore, GC samples with deeper depths of invasion showed higher levels of CTHRC1 expression, indicating that CTHRC1 could have important implications in GC invasion and metastasis. Importantly, this result was further confirmed through in vitro experiments. Re‐expression of CTHRC1 in GC cells led to a significant increase in their ability to invade, whereas the knockdown of CTHRC1 gene expression by siRNA had an adverse effect. Therefore, CTHRC1 is a potential metastasis‐related gene in gastric carcinogenesis. In addition, it was interesting to note that CTHRC1 staining of GC was detected mainly in the cytoplasm and extracellular space, but CHTRC1 expression in normal gastric mucosa, CAG, and intestinal metaplasia tissues was mostly present in the nucleus. CTHRC1 is a secreted extracellular protein. Pyagay et al.7 found that CTHRC1 may undergo proteolytic processing and this could have implications for the activity of the molecule. Our ongoing studies showed that there was no difference in mRNA levels of collagen type I and III between control and CTHRC1 overexpressing cells (data not shown). Therefore, it is possible that the secreted form of CTHRC1 overproduced by GC cells may act on the surrounding microenvironment, such as the stromal cells and ECM, and thus promotes tumor invasion.
To date, the regulating mechanism by which CTHRC1 is upregulated in human cancers is not known. There are varied ways to regulate gene expression, including genetic variation, epigenetic variation, and cytokine induction, amongst others. Changes of gene expression are usually caused by mutation in exons and regulatory region (between nucleotides within 1 kb upstream of the transcription start site). In this study, we did not find any mutations in the above regions to be associated with the aberrant expression of CTHRC1. However, the samples are relatively small and further studies are needed to validate this result.
Promoter methylation, which can be a mechanism for the inactivation of tumor suppressor genes, has been associated with cancer‐specific expression differences in human malignancies.20, 21, 22 To date, few examples of promoter hypomethylation of putative oncogenes have been reported.23, 24 Here, our data on 5‐aza‐dC treatment and methylation analyses strongly indicate that CTHRC1 silencing is related to promoter hypermethylation. Hence, promoter demethylation may be one cause of upregulation of CHTRC1 in GC cell lines. Additional studies are needed to investigate the methylation status of CTHRC1 in GC tissues. Thus, CHTRC1 methylation might also function as a prognostic biomarker and therapeutic target.
Sequence analysis of the CTHRC1 promoter region reveals a binding site of Smad, which is responsive to regulation by TGF‐β.8 It has been reported that dysregulation of TGF‐β signaling is implicated in tumor growth, angiogenesis, invasion, and metastasis,25, 26, 27 and the upregulation of TGF‐β1 is present in a variety of human cancers, including GC.28, 29 CTHRC1 is induced by TGF‐β1 and bone morphogenetic protein‐4 in murine NIH3T3 cells.7 Thus, we hypothesized that TGF‐β might also induce CTHRC1 expression during gastric carcinogenesis. Our own in vitro studies showed that both CTHRC1 mRNA and protein levels increased gradually in response to TGF‐β1. In addition, we observed that TGF‐β‐mediated Smad signaling activation was also increased, but this change did not coincide with the upregulation of CTHRC1. These results differed from those of LeClair et al., who reported that Cthrc1 is a cell‐type‐specific inhibitor of TGF‐β, which in turn impacts collagen type I and III deposition, neointimal formation, and the dedifferentiation of smooth muscle cells.30 There are potential confounders for these findings: (i) compared with the function of CTHRC1 in tissue repair, TGF‐β may play a reverse role in tumorigenesis; and (ii) the signaling pathways are very complex. CHTRC1 may participate in the TGF‐β‐mediated Smad signaling pathway, or may have no relation with this pathway and is only regulated by TGF‐β1. Thus, we provide novel mechanistic evidence that TGF‐β1 may upregulate CTHRC1 expression in GC. Further studies will be needed to address the relationship between CTHRC1 and TGF‐β‐mediated Smad signaling or other pathways. In addition, as epigenetic alterations in promoter methylation are associated with downregulation of gene expression, and the CTHRC1 promoter region shows a binding site of Smad, it is possible that promoter demethylation and TGF‐β1 may co‐regulate the expression of CTHRC1. Understanding the mechanism by which CTHRC1 is regulated requires further investigation in GC tissues.
In summary, CTHRC1 is a potential metastasis‐related gene in gastric carcinogenesis. Promoter demethylation and TGF‐β1 may coregulate the expression of CTHRC1. Understanding the mechanism by which CTHRC1 increases cell invasion requires further investigation. Thus, it will not only provide new insights into the metastasis mechanism but provide a new therapeutic strategy for the treatment of GC in the future.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by grants from the National Basic Research Program of China 973 program (Grant No. 2010CB5293), the National High Technology Research and Development Program of China (863 Program) (Grant No. 2006AA02A402), the National Natural Science Foundation of Key Program (Grant No. 30830055), and the Science and Technology Commission of Shanghai Municipality (Grant No. 09DZ1950101) to FJY. Thanks go to Ying‐Chao Wang and Xiao‐Yu Chen for their excellent technical assistance and enthusiastic participation in this study.
References
- 1. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogiatzi P, Vindigni C, Roviello F, Renieri A, Giordano A. Deciphering the underlying genetic and epigenetic events leading to gastric carcinogenesis. J Cell Physiol 2007; 211: 287–95. [DOI] [PubMed] [Google Scholar]
- 3. Jankowski JA, Odze RD. Biomarkers in gastroenterology: between hope and hype comes histopathology. Am J Gastroenterol 2009; 104: 1093–6. [DOI] [PubMed] [Google Scholar]
- 4. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell, 2006; 127: 679–95. [DOI] [PubMed] [Google Scholar]
- 5. Steven CS, Dan T N. Learning therapeutic lessons from metastasis suppressor proteins. Rev Cancer 2009; 9: 253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tie J, Pan Y, Zhao L et al MiR‐218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet 2010; 6: e1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pyagay P, Heroult M, Wang Q et al Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ Res 2005; 96: 261–8. [DOI] [PubMed] [Google Scholar]
- 8. Tang L, Dai DL, Su M, Martinka M, Li G, Zhou Y. Aberrant expression of collagen triple helix repeat containing 1 in human solid cancers. Clin Cancer Res, 2006; 12: 3716–22. [DOI] [PubMed] [Google Scholar]
- 9. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 1996; 20: 1161–81. [DOI] [PubMed] [Google Scholar]
- 10. Chinese Society of Gastroenterology . Consensus on chronic gastritis in China. Chin J Gastroenterol 2006; 11: 674–84. [Google Scholar]
- 11. Rugge M, Correa P, Dixon MF et al Gastric dysplasia, the Padova international classification. Am J Surg Pathol 2000; 24: 167–76. [DOI] [PubMed] [Google Scholar]
- 12. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 2008; 3: 1101–8. [DOI] [PubMed] [Google Scholar]
- 13. Jun HJ, Woolfenden S, Coven S et al Epigenetic regulation of c‐ROS receptor tyrosine kinase expression in malignant gliomas. Cancer Res 2009; 69: 2180–4. [DOI] [PubMed] [Google Scholar]
- 14. Lu R, Wang X, Chen ZF, Sun DF, Tian XQ, Fang JY. Inhibition of the extracellular signal–regulated kinase/mitogen‐activated protein kinase pathway decreases DNA methylation in colon cancer cells. J Biol Chem 2007; 282: 12249–59. [DOI] [PubMed] [Google Scholar]
- 15. Hecht M, Papoutsi M, Tran HD, Wilting J, Schweigerer L. Hepatocyte growth factor/c‐Met signaling promotes the progression of experimental human neuroblastomas. Cancer Res 2004; 64: 6109–18. [DOI] [PubMed] [Google Scholar]
- 16. Wang YY, Zhou GB, Yin T et al AML1‐ETO and C‐KIT mutation/overexpression in t(8;21) leukemia, implication in stepwise leukemogenesis and response to Gleevec. Proc Natl Acad Sci USA 2005; 102: 1104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leclair RJ, Wang Q, Benson MA, Prudovsky I, Lindner V. Intracellular localization of Cthrc1 characterizes differentiated smooth muscle. Arterioscler Thromb Vasc Biol 2008; 28: 1332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. LeClair RJ, Lindner V. The role of collagen triple helix repeat containing 1 in injured arteries, collagen expression, and transforming growth factor beta signaling. Trends Cardiovasc Med 2007; 17: 202–5. [DOI] [PubMed] [Google Scholar]
- 19. Turashvili G, Bouchal J, Baumforth K et al Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 2007; 7: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang JY, Xiao SD. Alteration of DNA methylation in gastrointestinal carcinogenesis. J Gastroenterol Hepatol 2001; 16: 960–8. [DOI] [PubMed] [Google Scholar]
- 21. Dehan P, Kustermans G, Guenin S, Horion J, Boniver J, Delvenne P. DNA methylation and cancer diagnosis: new methods and applications. Expert Rev Mol Diagn 2009; 9: 651–7. [DOI] [PubMed] [Google Scholar]
- 22. Baylin SB, Jones PA. A decade of exploring the cancer epigenome ‐ biological and translational implications. Nat Rev Cancer 2011; 11: 726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Swami M. Epigenetics: demethylation links cell fate and cancer. Nat Rev Cancer 2010; 10: 740. [DOI] [PubMed] [Google Scholar]
- 24. Wild L, Flanagan JM. Genome‐wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta 2010; 1806: 50–7. [DOI] [PubMed] [Google Scholar]
- 25. Oft M, Heider KH, Beug H. TGF‐β signaling is necessary for carcinoma cell invasiveness andmetastasis. CurrBiol 1998; 8: 1243–52. [DOI] [PubMed] [Google Scholar]
- 26. Leivonen SK, VeliMatti K. Transforming growth factor‐β signaling in cancer invasion and metastasis. Int J Cancer 2007; 121: 2119–24. [DOI] [PubMed] [Google Scholar]
- 27. Perera M, Tsang CS, Distel RJ et al TGF‐beta1 interactome: metastasis and beyond. Cancer Genomics Proteomics 2010; 7: 217–29. [PubMed] [Google Scholar]
- 28. Hawinkels LJ, Verspaget HW, van Duijn W et al Tissue level, activation and cellular localisation of TGF‐β1 and association with survival in gastric cancer patients. Br J Cancer 2007; 97: 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu WK, Cho CH, Lee CW et al Dysregulation of cellular signaling in gastric cancer. Cancer Lett 2010; 295: 144–53. [DOI] [PubMed] [Google Scholar]
- 30. LeClair RJ, Durmus T, Wang Q, Pyagay P, Terzic A, Lindner V. Cthrc1 is a novel inhibitor of transforming growth factor‐beta signaling and neointimal lesion formation. Circ Res 2007; 100: 826–33. [DOI] [PubMed] [Google Scholar]