

Abstract
The so‐called “seed and soil” hypothesis proposed by Stephen Paget in 1889 to explain the metastatic behavior of cancer cells and the homing of certain cancers to “selected” sites has been a well‐recognized phenomenon for over a century. What advances have been made to increase our understanding of this phenomenon and what does it really implicate in terms of targets for therapy? (Cancer Sci 2012; 103: 626–631)
The seeds of a plant are carried in all directions; but they can only live and grow if they fall on congenial soil – Stephen Paget, 1889.1
Metastatic cancer arises when cancerous cells from a tumor spread from the primary lesion and form new tumors at another site in the body. Cancers that metastasize are generally more difficult to treat than those that remain at the site of origin, and have a worse prognosis. Most deaths from cancer are a result of metastases that are resistant to conventional therapies, even therapies that might have been successful in the treatment of the primary cancer.2 This might be explained by the metastatic process itself, and the changes a tumor cell must undergo to become a successful initiator of metastases.
The metastatic process consists of several steps, as outlined in Figure 1. Initially, cells of the primary tumor, the so called “seeds” require angiogenesis for increased blood supply and the provision of growth factors. However, this is also necessary for metastasis, as tumor vascularization increases the chances for tumor cells to enter the circulation and become metastatic. For angiogenesis to be involved in metastatic cancer requires an increased vascularization of the primary tumor followed by a breakdown of the extracellular matrix (ECM) to allow tumor cells to intravasate.3
Figure 1.
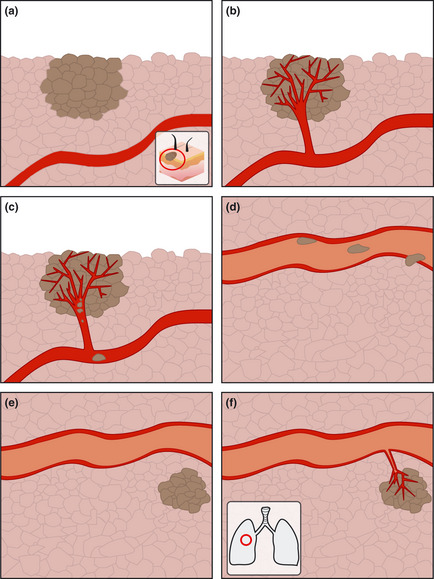
Steps in the metastatic process. (a) primary tumor, (b) angiogenesis in primary tumor, (c) intravasation, (d) migration and extravasation, (e) colonization of new organ and (f) angiogenesis and growth of new tumor.
Following this, the circulating tumor cells must be able to form attachments to their environment; that is, to other cells and the ECM. In this way, they might eventually succeed in extravasation by proteolysis of the ECM and migration into the surrounding tissue. The tumor cell must eventually colonize the site (soil) for metastasis to be successful.3, 4, 5 All of these processes must be completed, and done so successfully, before a metastasis is established.
Of course, these processes are regulated by many pathways, both exogenous and endogenous, and may require that the original cell from the primary tumor undergoes many changes. Interestingly, cells with different metastatic properties have been successfully isolated from the same primary tumor, indicating that the ability of the “seed” of a given tumor to metastasize is complicated even further.6
It is also of note that we are still unaware if the seed that can successfully metastasize is a result of an accumulation of mutations conferring an advantage on the cell and resulting in an ability to thrive elsewhere in the body, or if these metastatic seed cells are an integral part of each primary tumor from the beginning. Perhaps it is even the case that both of these hypotheses are true.
In this way, treatment of the primary tumor and the metastatic tumor are very different due to the possibility that this selection process is occurring. It might be less a matter of a seed simply being “carried in all directions,” but, rather, a complex relationship between the internal changes within the original tumor cell and the influence of the ever changing composition of the soil as it makes its journey.
What is it that decides which organs shall suffer in a case of disseminated cancer?
The importance of the “soil” in metastasis is not to be underestimated in a successful metastatic process. This “soil” may be considered as a host factor, stroma, niche or organ microenvironment.7 It is no coincidence that certain tumors home to characteristic organs. As Stephen Paget emphasized in 1889; “the distribution of the secondary growths is not a matter of chance.”
One cannot dispute the importance of mechanical lodgment of circulating tumor cells and the resulting growth of the arrested cell, in particular when considering metastatic growth in the lung for example.8, 9 This, however, does not explain the many clinical metastases that cannot be accounted for by circulatory patterns and mechanical lodgment of blood borne tumor cells.10
In fact, although the process of metastasis is highly inefficient (and only a few cells in a primary tumor are believed to be capable of forming metastases), the entry of tumor cells to the bloodstream is relatively common in cancer.11, 12 However, it has been documented that <0.01% of circulating tumor cells eventually succeed in forming secondary growths.11 The findings of Tarin et al.13 that the development of secondary cancer was rare even upon direct deposition of millions of tumor cells into the vena cava (to reduce ascites from ovarian cancer) is a good indication of the importance of other factors involved.
The main patterns of seemingly “random” distant metastases are outlined in Table 1. This table does not include regional metastases because they might be explained almost exclusively by mechanical and anatomical processes. These kinds of regional metastases support the hypothesis put forward by Ewing in opposition to Paget's “seed and soil” theory that “the mechanisms of the circulation will doubtless explain most of these peculiarities.”14
Table 1.
Typical sites of metastasis for some common solid tumors
| Cancer type | Primary site | Principal site of metastasis |
|---|---|---|
| Breast adenocarcinoma | Breast | Bone, lung, liver and brain |
| Small cell carcinoma | Lung | Brain, liver and bone |
| Malignant melanoma | Skin | Lung, brain and liver |
| Prostatic adenocarcinoma | Prostate | Bone |
| Testicular carcinoma | Testis | Liver |
| Colorectal adenocarcinoma | Colon/rectum | Liver and lung |
| Neuroblastoma | Mediastinum, abdomen | Liver |
It is of note that Ewing could not, however, explain the strange phenomenon that the spleen escaped from this occurrence with “peculiar frequency,”14 despite its rich blood supply. Studies have shown that the most difficult point in the metastatic process is, in fact, the initiation of growth in the secondary tissue or organ. The tumor cells that have succeeded in travelling to the site of secondary growth might fail to thrive there due to an inability to trigger the angiogenesis necessary for growth, or simply might exit the cell cycle and remain dormant.15, 16
Furthermore, although some cancers share the same principle metastatic sites, the time period between the tumor dissemination, organ infiltration and eventual colonization might differ greatly. This may be described as metastatic latency and is illustrated by such metastases as those from primary breast and lung malignancies. Adenocarcinomas from both breast and lung are known to metastasize to bone, liver and brain. However, while breast metastases may only be detected following years of remission, lung metastases may appear several months following initial detection and diagnosis, even while the primary tumor is a symptomless lesion.17, 18
This raises even more questions about the “seed and soil” relationship of tumor cell and host organ. If it is the case that an accumulation of perfect mutations for colonizing a distant organ is necessary for metastasis, perhaps this can explain the inefficiency of the process. However, is this accumulation less difficult to achieve for colonizing certain organs that regularly host metastatic tumors? What triggers the development of colonizing ability in these tumor cells? What does the “soil” need to provide to allow them to remain in a dormant state for (in some cases) many years? Finally, and, perhaps most importantly, how can we develop strategies to target these mechanisms?
General Features of Metastasis
It is suggested by Nowell that selection pressure results in the acquirement of genetic variability within developing tumors.19 These variations confer increased malignant capabilities on the tumor cell and might result in an increased ability of some tumor cells to metastasize, compared to other non‐metastatic cells within the same tumor. These selected cells might not only have an increased metastatic ability, but might also be more resistant to eradication strategies, such as chemotherapy, and normal homeostatic controls, such as anti‐growth signaling and immune attack by the host.20
Various mechanisms, such as cellular motility and basement membrane degradation, have been implicated in the metastatic process, for example in the entry of cancer cells to the circulation. The aberrant expression of cytoskeletal modifiers, such as RhoC (which has a role in the control of cytoskeletal organization in response to extracellular factors) and Endo180 (a collagen internalization receptor implicated in invasion), can enhance dissemination in metastatic processes in a specific way.21, 22
In addition, the process of epithelial to mesenchymal transition (EMT), where cells lose cell–cell adhesion and dissemination of tumor cells from epithelial cells occurs, has been recognized as an important initiator of metastasis, playing a major role in invasion and intra/extravasation. Several proteins have been identified in this process. For example, Twist, a transcription factor in embryonic development, is identified by Yang et al. as an essential component of EMT induction in metastasis by repression of E‐cadherin and is an attractive protein to target in metastasis.23, 24 Similarly, the process of mesenchymal to epithelial transition, and the mechanisms involved therein, is an equally attractive target.
Aspects of the host vasculature itself are implicated in general metastatic mechanisms: for example, loss of interaction between the Duffy antigen chemokine receptor and CD82 (a known suppressor of metastasis), as well as evasion of immune surveillance through platelet binding.5 This is perhaps also important in the avoidance of shear stress in the vasculature.25
The genetic background of the individual has also been a topic of discussion with regard to susceptibility of that person to metastasis. Hunter suggests that it is not only a pro‐metastatic signature in tumor cells that is important, but also the polymorphisms present in the healthy cells of the individual. These genetic variations increase or decrease the likelihood that a tumor will metastasize.26, 27 Studying the relevant polymorphisms might be a strategy for predicting the likelihood of non‐specific metastasis in an individual.
The means by which the tumor cells and host tissues contribute to metastatic development are outlined in Table 2.
Table 2.
Metastatic mechanisms by tumor cells and host tissue
| Nature of cell | Mechanism promoting metastasis |
|---|---|
| Tumor cell | Stimulation of growth and angiogenesis |
| Movement and invasion | |
| Expression of cell surface receptors for adhesion molecules | |
| Host cell | Vascularization and production of growth factors |
| Expression of chemotactic proteins | |
| Immune response and platelet binding |
Organ Specific Metastasis
Seed: breast carcinoma cell; soil: bone.
Breast and prostate cancer are the most common carcinomas to develop bone metastases. There are characteristics of the bone niche that help to explain this tendency. Biochemical (the presence of cytokines and growth factors) and physical (acidic pH, high extracellular calcium concentration) properties of bone mean that it is a soil for tumor seeds providing a most welcoming microenvironment where they may thrive.28
Looking at breast carcinomas specifically, it has been shown in a number of microarray analyses that there are a small number of genes that differ consistently between the parent tumor and the metastasis. For example, in a study of primary tumor and metastases in 100 breast cancer patients, Gancberg et al.29 found that 6% of metastases had an overexpression of Her2/neu when compared to the primary tumor. In addition, van de Vijver et al.30 used DNA microarray analysis on breast cancer patients to develop a gene expression signature that can predict poor outcome and metastasis. The genes involved in this prognostic signature are involved in regulating cell cycle, invasion, metastasis and angiogenesis.
Kang et al.31 identify that CXCR4 expression in breast cancer cells is a key gene in the expression signature of bone metastasizing tumor cells. Interestingly, CXCR4 is a receptor for CXCL12, a chemotactic protein preferentially expressed by stromal cells in target organs of breast metastases; that is, bone, brain, liver, lymph node and lung.32 CXCR4 also enhances adhesion of cells expressing the protein to endothelial cells through activation of integrin, which further supports the hypothesis that the expression of CXCR4 is essential for metastatic mechanisms.33 Therefore, DPPIV‐dependent depletion of CXCL12 is a potential strategy for development of adjuvant therapy targeting metastatic breast cancer.34
In fact, preclinical studies have shown evidence that blockage of the CXCL12 pathway in breast cancer can be efficacious in a preventative sense; that is, it might delay the metastatic process if the patient is treated near to the time of initial primary tumor establishment.35 However, as this strategy is not practically feasible in a clinical setting, it might not be sufficient as a targeting strategy in breast tumor metastasis.36 However, there is both preclinical and clinical data available to suggest that the use of CXCL12 inhibition in combination with conventional therapies, such as radiotherapy and anti‐angiogenic therapy, might be useful as in many cases the levels of CXCL12 increases following therapy.36 Increased levels also correlate with increased levels of metastatic disease in rectal cancer.37
Therefore, this CXCR4/CXCL12 interaction relationship between both the CXCR4 expressing seed (as in metastatic breast cancer cells) and the soil (stromal cells in bone) remains an intriguing choice of target in metastasis.
Bone remodeling in metastatic breast cancer is also a mechanism to potentially target. Tumor cells release such osteoblastic growth stimulating factors as parathyroid hormone (PTHrP), which results in osteoclastic bone resorption through RANK signaling. This ultimately leads to release of growth factors, including TGFβ, for example, resulting in a pathogenic positive feedback loop with TGFβ stimulating a further increase in the expression of PTHrP from tumor “seed” cells.38 This mechanism might even further promote metastatic development due to the pro‐angiogenic and immunosuppressive properties of TGFβ.39 Although this dual role of TGFβ has been a topic of much debate in recent years, it now appears that although it has a tumor suppressive role early in breast cancer development, overexpression at a later stage might result in anti‐cancer drug resistance, as appears to be the case in patients treated with tamoxifen.40, 41
Indeed, there have been numerous strategies tested in a pre‐clinical setting targeting TGFβ signaling, and some of these strategies are now in clinical trials, with models based on, for example, blockage of synthesis, ligand/receptor binding, antisense oligonucleotides and TGFβ antibodies. As with most anti‐tumor strategies, however, one must bear in mind the suitability of the patient for the treatment, and how much each individual might benefit from a particular strategy.40
Seed: malignant melanoma cell; soil: lung.
The dense vascular surface of the lung makes this organ a suitable site for many primary tumor metastases, including melanoma, breast, bladder, colon, kidney, head and neck.39 Lung metastatic lesions most commonly initiate at the level of small pulmonary arterioles. Here, they must either burst through or breach by another mechanism the endothelial junction of lung‐blood vessels and the basement membrane underneath.42
Initially, studies involving experimental metastasis of malignant melanoma appeared to suggest that an upregulation of adhesion molecule expression (such as VCAM‐1) was necessary in enhancing the number of successful metastases.43 In contrast to this finding, it has more recently been established that there is no upregulation of VCAM‐1 in spontaneous metastasis by melanomas, suggesting that this mechanism is not required for formation of metastasis.44 There is, however, evidence to suggest that, once established, the organ‐specific upregulation of VCAM‐1 in the lungs as stimulated by metastatic tumor cells from the primary melanoma helps to support the growth of the metastases.44
Therefore, targeting strategies for VCAM‐1 expression might be an attractive means to restrict growth and survival of metastatic tumors in the lung. Gosk et al.45 propose one such targeting mechanism using immunoliposomes (IL) directed against VCAM‐1. They demonstrate a selective in vivo targeting of tumor vessels versus non‐affected organ vessels using these specific IL.
Seed: colorectal cancer cell; soil: liver.
The liver is one of the body's most densely vascularized tissues and its unique vascular supply via both the systemic circulation and the portal vein makes it a logical destination for circulating cancer cells. Gastrointestinal malignancies like colorectal carcinomas have a tendency to metastasize to the liver, likely due to the nature of the portal circulation. The vasculature in the liver is unique in comparison with other organs in that the sinusoids are highly permeable and do not represent a major barrier to extravasation.4, 42
It appears that the main barrier to the survival of circulating tumor cells in the liver is the ability to resist the immune response initiated by resident cells. Gene expression profiling of liver metastases from colorectal primary sites have revealed an upregulation of COX‐2, an important player in immune regulation and angiogenesis.46 Inhibition of COX‐2 is now recognized as a potential preventative measure against development of liver metastasis based on studies using metastatic colorectal cancer cell lines.47 The antineoplastic effects of COX‐2 inhibition ex‐vivo using tissue microarrays from liver metastases have also been demonstrated by assessing the number of apoptotic liver cells.48
In addition, it appears that some colorectal cancer cells have been found to express sialyl Lewis‐X (sLex) and sialyl Lewis‐A (sLea) antigens, and this correlates with both their prognosis and metastatic potential.49, 50 Colorectal cancer cells support their adhesion to vascular endothelial cells in the liver through binding of the activated E‐selectin on the vasculature to its ligands, sLex and sLea. It has also been shown that in studies with mice treated with E‐selectin monoclonal antibodies, the treated mice form significantly less metastasis when sialyl Lewis expressing tumor cells are injected.51 More recently, studies by Khatib et al.52 have also supported the hypothesis that the inhibition of tumor‐induced, hepatic microvessel E‐selectin expression might provide a useful strategy for the prevention of hepatic metastasis based on mice studies in vivo.
Seed: small cell carcinoma (lung); soil: brain.
Between 20% and 40% of all cancer patients develop brain metastasis, with 30–50% of patients with lung cancer contributing greatly to this number. The colonization of the brain by a primary tumor results in an extremely poor prognosis for the affected individual, with the median survival time for patients with untreated brain metastasis being just 1–2 months.53 The brain might be colonized in two ways, affecting either the brain parenchyma or the leptomeninges.42
Difficulties in colonizing the brain are mainly as a consequence of its lack of lymphatic drainage and the presence of the blood brain barrier (BBB), restricting even serum proteins unless they are shuttled across by active transport. This barrier limits and regulates molecular exchange at the interface between the blood and neural tissue or its fluid spaces.54, 55
This barrier must, therefore, be overcome by the invading tumor cells. However, mechanisms behind this in human brain metastasis are poorly understood. Focus instead has been on how the cells, once past the BBB, can thrive in the brain. Although glial cells and astrocytes can synthesize a large number of growth factors, cytokines and chemokines to support tumor cells, the metastatic tumor requires an increased vascular supply in order to maintain growth.56
Yano et al. show that highly brain metastatic cells produce a higher level of vascular endothelial growth factor (VEGF) protein than low brain metastatic cells in mice, confirmed both by mRNA expression analysis and immunohistochemistry. They find this expression to be necessary, but not sufficient for the angiogenesis and growth of brain metastasis.57 The inhibition of VEGF in lung to brain metastasis, as in many other malignancies, is a promising target for an adjuvant treatment to radiotherapy. Bevacizumab, a monoclonal antibody for VEGFA has already been shown to have clinical activity in some metastatic cancers when combined with traditional cytotoxic therapy.58, 59, 60
Discussion
Stephen Paget concludes his seminal paper on metastasis in 1888 with the following words: “The best work in the pathology of cancer now is done by those who are studying the nature of the seed. They are like scientific botanists, and he who turns over the records of cases of cancer is only a ploughman, but his observations of the properties of the soil might also be useful.”
This is perhaps a slight understatement with regard to the importance of the soil in many metastases.
As we have seen, many of the mechanisms involved in a successful organ specific metastasis involve not only the specific properties of the metastatic cancer cell, but also an interaction between a specific property of the seed and one of the microenvironment in the soil. Indeed, when considering that some metastases arise up to 20 years after the removal of the primary tumor, tumor cell dormancy in metastatic sites appears to be controlled by a cue from the host tissue determining when the cell might begin to proliferate.
With regard to therapeutic implications, targeting metastatic progression is no mean feat. Table 3 summarizes a small number of current therapeutic targets and therapies, but there are many more. The lack of understanding in progression of primary tumor cells to metastatic cells and whether this occurs early or late in cancer development leads us to question when it is clinically relevant to administer anti‐metastatic targets.56 Recent studies have indicated that it might even be relevant to locally target the primary tumor in order to retard metastatic progression.61 Compounds designed to interrupt metastatic development have not been generally shown to have sufficient efficacy in a clinical setting.
Table 3.
Strategies for development of therapy targeting metastatic cancers
| Primary tumor | Organ of metastasis | Target for therapy | Mechanism(s) targeted | Status |
|---|---|---|---|---|
| Breast carcinoma | Bone | CXCR4/CXCL12 interaction, TGFβ expression | Chemotaxis, angiogenesis, growth | Anti CXCR4 and TGFβ antibodies in clinical trials |
| Malignant melanoma | Lung | VCAM‐1 expression | Adhesion | Methods to downregulate VCAM1 under development |
| Colorectal carcinoma | Liver | Inhibition of COX‐2 and E‐selectin | Inflammation | COX‐2 inhibitor celecoxib shown to help prevent CRC development, E‐selectin inhibitor cimetidine shows modest beneficial effect in CRC |
| Small cell carcinoma | Brain | Inhibition of VEGF | Angiogenesis | VEGF‐A inhibitor bevacizumab currently used for treatment of both lung and brain carcinoma |
CRC, colorectal cancer cell; VEGF, vascular endothelial growth factor.
In conclusion, an increase in our understanding of mechanisms of invasion, tumor cell and stromal interactions has promoted another perspective on Paget's original hypothesis. The genetic background of the individual affecting the non‐malignant cells of the host target organ has meant that the focus of the seed and soil hypothesis has shifted perspective from the seed being the most important factor, as suggested by Paget. It is now recognized that other components of the metastatic tumor's microenvironment are relevant. This increase in our knowledge provides a wealth of possible targets for therapy, many of which might contribute to prolonging life expectancy in patients with metastatic disease, provided they are administered to the right patient at an appropriate time.
Disclosure Statement
The authors have no conflict of interest.
References
- 1. Paget S. The distribution of secondary growths in cancer of the breast. Lancet 1889; 133: 3. [PubMed] [Google Scholar]
- 2. Ribatti D, Mangialardi G, Vacca A. Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin Exp Med 2006; 6: 145–9. [DOI] [PubMed] [Google Scholar]
- 3. Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer 1997; 80: 1529–37. [DOI] [PubMed] [Google Scholar]
- 4. Chambers AF, Groom AC, MacDonald IC. Metastasis: dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2: 563–72. [DOI] [PubMed] [Google Scholar]
- 5. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ‐specific colonization. Nat Rev Cancer 2009; 9: 274–84. [DOI] [PubMed] [Google Scholar]
- 6. Waghorne C, Thomas M, Lagarde A, Kerbel RS, Breitman ML. Genetic evidence for progressive selection and overgrowth of primary tumors by metastatic cell subpopulations. Cancer Res 1988; 48: 6109–14. [PubMed] [Google Scholar]
- 7. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res 2010; 70: 5649–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weiss LA. Pathobiologic overview of metastasis. Semin Oncol 1977; 4: 5–17. [PubMed] [Google Scholar]
- 9. Weiss L, Ward PM. Cell detachment and metastasis. Cancer Metastasis Rev 1983; 2: 111–27. [DOI] [PubMed] [Google Scholar]
- 10. Nicolson GL. Cancer metastasis: tumor cell and host organ properties important in metastasis to specific secondary sites. Biochim Biophys Acta 1988; 948: 175–224. [DOI] [PubMed] [Google Scholar]
- 11. Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I‐5‐iodo‐2′‐deoxyuridine. J Natl Cancer Inst 1970; 45: 773–82. [PubMed] [Google Scholar]
- 12. Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 1975; 35: 218–24. [PubMed] [Google Scholar]
- 13. Tarin D et al Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res 1984; 44: 3584–92. [PubMed] [Google Scholar]
- 14. Ewing J. Neoplastic Diseases: A Treatise on Tumors. Philadelphia & London: WB Saunders Co., 1928; 77–89. [Google Scholar]
- 15. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer 2010; 46: 1181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Cancer Res 2009; 102: 67–101. [DOI] [PubMed] [Google Scholar]
- 17. Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst 1999; 91: 80–5. [DOI] [PubMed] [Google Scholar]
- 18. Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet 2000; 355: 479–85. [DOI] [PubMed] [Google Scholar]
- 19. Nowell PC. The clonal evolution of tumor cell populations. Science 1976; 194: 23–8. [DOI] [PubMed] [Google Scholar]
- 20. Welch DR, Tomasovic SP. Implications of tumor progression on clinical oncology. Clin Exp Metastasis 1985; 3: 151–88. [DOI] [PubMed] [Google Scholar]
- 21. Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000; 406: 532–5. [DOI] [PubMed] [Google Scholar]
- 22. Huijbers IJ et al A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS ONE 2010; 5: e9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang J et al Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–39. [DOI] [PubMed] [Google Scholar]
- 24. Wallerand H et al The epithelial‐mesenchymal transition‐inducing factor TWIST is an attractive target in advanced and/or metastatic bladder and prostate cancers. Urol Oncol 2010; 28: 473–9. [DOI] [PubMed] [Google Scholar]
- 25. Bacac M, Stamenkovic I. Metastatic cancer cell. Annu Rev Pathol 2008; 3: 221–47. [DOI] [PubMed] [Google Scholar]
- 26. Hunter K Welch DR, Liu ET. Genetic background is an important determinant of metastatic potential. Nat Genet 2003; 34: 2–24; author reply 25. [DOI] [PubMed] [Google Scholar]
- 27. Hunter KW. Allelic diversity in the host genetic background may be an important determinant in tumor metastatic dissemination. Cancer Lett 2003; 200: 97–105. [DOI] [PubMed] [Google Scholar]
- 28. Guise T. Examining the metastatic niche: targeting the microenvironment. Semin Oncol 2010; 37(Suppl 2): S2–14. [DOI] [PubMed] [Google Scholar]
- 29. Gancberg D et al Comparison of HER‐2 status between primary breast cancer and corresponding distant metastatic sites. Ann Oncol 2002; 13: 1036–43. [DOI] [PubMed] [Google Scholar]
- 30. van de Vijver MJ et al A gene‐expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347: 1999–2009. [DOI] [PubMed] [Google Scholar]
- 31. Kang Y et al A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–49. [DOI] [PubMed] [Google Scholar]
- 32. Mulle A et al Involvement of chemokine receptors in breast cancer metastasis. Nature 2001; 410: 50–6. [DOI] [PubMed] [Google Scholar]
- 33. Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer Res 2003; 63: 6751–7. [PubMed] [Google Scholar]
- 34. Epstein RJ. The CXCL12‐CXCR4 chemotactic pathway as a target of adjuvant breast cancer therapies. Nat Rev Cancer 2004; 4: 901–9. [DOI] [PubMed] [Google Scholar]
- 35. Huang EH et al A CXCR4 antagonist CTCE‐9908 inhibits primary tumor growth and metastasis of breast cancer. J Surg Res 2009; 155: 231–6. [DOI] [PubMed] [Google Scholar]
- 36. Duda DG et al CXCL12 (SDF1alpha)‐CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res 2011; 17: 2074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu L et al Direct evidence that bevacizumab, an anti‐VEGF antibody, up‐regulates SDF1alpha, CXCR4, CXCL6, and neuropilin 1 in tumors from patients with rectal cancer. Cancer Res 2009; 69: 7905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kozlow W, Guise TA. Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia 2005; 10: 169–80. [DOI] [PubMed] [Google Scholar]
- 39. Langley RR, Fidler IJ. The seed and soil hypothesis revisited–the role of tumor‐stroma interactions in metastasis to different organs. Int J Cancer 2011; 128: 2527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iyer S, Wang ZG, Akhtari M, Zhao W, Seth P. Targeting TGFbeta signaling for cancer therapy. Cancer Biol Ther 2005; 4: 261–6. [DOI] [PubMed] [Google Scholar]
- 41. Barcellos‐Hoff MH, Akhurst RJ. Transforming growth factor‐beta in breast cancer: too much, too late. Breast Cancer Res 2009; 11: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006; 127: 679–95. [DOI] [PubMed] [Google Scholar]
- 43. Okahara H, Yagita H, Miyake K, Okumura K. Involvement of very late activation antigen 4 (VLA‐4) and vascular cell adhesion molecule 1 (VCAM‐1) in tumor necrosis factor alpha enhancement of experimental metastasis. Cancer Res 1994; 54: 3233–6. [PubMed] [Google Scholar]
- 44. Langley RR et al Endothelial expression of vascular cell adhesion molecule‐1 correlates with metastatic pattern in spontaneous melanoma. Microcirculation 2001; 8: 335–45. [DOI] [PubMed] [Google Scholar]
- 45. Gosk S, Moos T, Gottstein C, Bendas G. VCAM‐1 directed immunoliposomes selectively target tumor vasculature in vivo. Biochim Biophys Acta 2008; 1778: 854–63. [DOI] [PubMed] [Google Scholar]
- 46. Pantaleo MA et al Gene expression profiling of liver metastases from colorectal cancer as potential basis for treatment choice. Br J Cancer 2008; 99: 1729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tachimori A, Yamada N, Amano R, Ohira M, Hirakawa K. Combination therapy of S‐1 with selective cyclooxygenase‐2 inhibitor for liver metastasis of colorectal carcinoma. Anticancer Res 2008; 28: 629–38. [PubMed] [Google Scholar]
- 48. Kasper HU et al COX‐2 expression and effects of COX‐2 inhibition in colorectal carcinomas and their liver metastases. Anticancer Res 2010; 30: 2017–23. [PubMed] [Google Scholar]
- 49. Nakamori S et al Increased expression of sialyl Lewisx antigen correlates with poor survival in patients with colorectal carcinoma: clinicopathological and immunohistochemical study. Cancer Res 1993; 53: 3632–7. [PubMed] [Google Scholar]
- 50. Sato M et al The association of sialyl Lewis(a) antigen with the metastatic potential of human colon cancer cells. Anticancer Res 1997; 17: 3505–11. [PubMed] [Google Scholar]
- 51. Brodt P et al Liver endothelial E‐selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer 1997; 71: 612–9. [DOI] [PubMed] [Google Scholar]
- 52. Khatib AM, Fallavollita L, Wancewicz EV, Monia BP, Brodt P. Inhibition of hepatic endothelial E‐selectin expression by C‐raf antisense oligonucleotides blocks colorectal carcinoma liver metastasis. Cancer Res 2002; 62: 5393–8. [PubMed] [Google Scholar]
- 53. Li B et al Comparison of three treatment options for single brain metastasis from lung cancer. Int J Cancer 2000; 90: 37–45. [PubMed] [Google Scholar]
- 54. Abbott NJ. Astrocyte‐endothelial interactions and blood–brain barrier permeability. J Anat 2002; 200: 629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abbott NJ, Ronnback L, Hansson E. Astrocyte‐endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 2006; 7: 41–53. [DOI] [PubMed] [Google Scholar]
- 56. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12: 895–904. [DOI] [PubMed] [Google Scholar]
- 57. Yano S et al Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res 2000; 60: 4959–67. [PubMed] [Google Scholar]
- 58. Yang JC et al A randomized trial of bevacizumab, an anti‐vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003; 349: 427–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hurwitz H et al Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 60. Cobleigh MA et al A phase I/II dose‐escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol 2003; 30: 117–24. [DOI] [PubMed] [Google Scholar]
- 61. Morgan SC, Parker CC. Local treatment of metastatic cancer–killing the seed or disturbing the soil? Nat Rev Clin Oncol 2011; 8: 504–6. [DOI] [PubMed] [Google Scholar]