

Abstract
The cellular response to genotoxic stress is multifaceted in nature. Following DNA damage, the tumor suppressor gene p53 activates and plays critical roles in cell cycle arrest, activation of DNA repair and in the event of irreparable damage, induction of apoptosis. The breakdown of apoptosis causes the accumulation of mutant cells. The elucidation of the mechanism for the p53‐dependent apoptosis will be crucial in applying the strategy for cancer patients. However, the mechanism of p53‐dependent apoptosis remains largely unclear. Here, we carried out ChIP followed by massively parallel DNA sequencing assay (ChIP‐seq) to uncover mechanisms of apoptosis. Using ChIP‐seq, we identified PDCD6 as a novel p53‐responsive gene. We determined putative p53‐binding sites that are important for p53 regulation in response to DNA damage in the promoter region of PDCD6. Knockdown of PDCD6 suppressed p53‐dependent apoptosis. We also observed that cytochrome c release and the cleavage of PARP by caspase‐3 were suppressed by depletion of PDCD6. We further observed that PDCD6 localizes in the nucleus in response to DNA damage. We identified the nuclear localization signal of PDCD6 and, importantly, the nuclear accumulation of PDCD6 significantly induced apoptosis after genotoxic stress. Therefore, we conclude that a novel p53‐responsive gene PDCD6 is accumulated in the nucleus and induces apoptosis in response to DNA damage.
Transcription factor p53 is reported to be mutated frequently in human cancer cells and is the gene responsible for Li‐Fraumeni syndrome.1, 2 Thus, p53 is considered as the most important tumor suppressor gene.3, 4, 5, 6 p53 is activated by DNA damage, including UV and oxidative stress.7, 8, 9, 10 Activated p53 induces the expression of p21 or 14‐3‐3σ, and blocks cell cycle progression at the G1 or G2 phase.11, 12, 13, 14 Irreparable DNA damage results in p53‐dependent apoptosis through the induction or repression of genes such as BAX or Bcl‐2 to avert the accumulation of mutant cells.15, 16, 17, 18 p53 can act in the cytosol and mitochondria to promote apoptosis through a transcription‐independent mechanism.19 Thus, p53 is an important factor in the determination of cell fate; that is, survival or apoptosis in the DNA‐damaged cells.20, 21, 22 Consequently, elucidating the mechanism of the p53‐dependent apoptosis is important for applying the strategy in cancer patients. Some of the apoptosis‐factor induced by p53 pertain to the Bcl‐2 family.23 In unstressed cells, Bcl‐2 binds to Bax/Bak to suppress apoptosis;24 however, upon exposure to DNA damage, the expression of Bax/Bak, tBid,25 Noxa26 and Puma27 is upregulated by p53 to induce apoptosis. Although many p53 target genes that participate in apoptosis have been reported,28 there are still unidentified genes regulated by p53. To elucidate the molecular mechanism underlying apoptosis, it is important to uncover how proteins interact with genes and to regulate its expression. In the present study, we carried out ChIP followed by massively parallel DNA sequencing assay (ChIP‐seq) to identify direct p53‐target genes. Until recently, microarray analysis has been used to identify the p53‐responsive genes; however, there has been little comprehensive analysis using ChIP‐seq. Until recently, the studies using ChIP‐seq have been carried out to predict the genomic loci of transcription factor binding sites. However, conventional analysis has been limited by the scale and resolution. These drawbacks have been resolved by the recent development of the automated sequencer. This technology enables us to obtain high‐resolution genome‐wide mapping and comprehensive analysis. In this context, exploration of the p53‐responsive gene using ChIP‐seq could be valuable.
In this study, we used ChIP‐seq to identify PDCD6 as a novel p53‐responsive gene. PDCD6 (programmed cell death 6) is a calcium‐binding modulator protein involved in cell proliferation and death and is reported as a 22‐kDa protein containing an open reading frame encoding 191 amino acids. It is a member of the penta EF‐hand protein family with an affinity for calcium in the micromolar range, similar to calmodulin.29, 30 PDCD6, also known as apoptosis‐linked gene‐2 (ALG‐2), was identified in a cell death trap assay as a pro‐apoptotic protein in a functional screen of T‐cell hybridoma cells.31 PDCD6 could be involved in calcium‐dependent apoptosis.32 The characterized target of PDCD6 is AIP1/Alix, an adaptor protein, which induces calcium‐dependent apoptosis and reduces tumorigenicity.33, 34 Other work has demonstrated that PDCD6 interacts with Sec31A, a component of the COPII complex involved, in a calcium‐dependent way and regulates its subcellular localization.35 However, there is no report that p53 directly binds to PDCD6 and induces apoptosis in response to DNA damage in cancer cells. In this study, we suggest that a novel function of PDCD6 is involved in p53‐dependent apoptosis.
Materials and Methods
Cell culture
H1299 (non‐small lung carcinoma cell line) cells were cultured in DMEM supplemented with 10% heat‐inactivated FBS, 100 U/mL penicillin, 100 µg/mL streptomycin and 2 mM l‐glutamine. A549 (Human lung adenocarcinoma epithelial cell line) cells were cultured in RPMI 1640 medium supplemented with 10% heat‐inactive FBS, 100 U/mL penicillin and 100 µg/mL streptomycin.
Plasmids
PDCD6 cDNA was amplified by PCR using pfuUltra High‐Fidelity DNA Polymerase (Stratagene, La Jolla, CA, USA) from A549 cDNA cloned into the pEGFP‐C1 vector.36, 37 Primer sequences are: the sense primer, 5′‐GCAGATCTATGGCCGCCTACTCTTACCGC‐3′; and the antisense primer, 5′‐CGCGAATTCTCATACGATACTGAAGACCAT‐3′. The Flag‐p53 was constructed as previously described.38 The PDCD6 p53‐consensus sequence was amplified by PCR from genomic DNA of A549 cells and cloned into the pGL3 basic vector (Promega, Madison, WI, USA), which carries the luciferase gene. Primer sequences are: the sense primer, 5′‐CGACGCGTCGGGAGACCACGCATTTCCTG‐3′; and the antisense primer, 5′‐CCGCTCGAGCGCGCACCTCTGGAAAAC‐3′. A deletion mutant of Luc‐PDCD6D was constructed by deleting the region of PDCD6 from −324812 to −324841. Primer sequences for the GFP‐PDCD6 NLS mutant (GFP‐PDCD6ΔNLS) are: 5′‐TGGAACGTTTTCCAGATATCAGACACCGAG‐3′; and the antisense primer, 5′‐CTCGGTGTCTGATATCTGGAA AACGTTCCA‐3′.
Cell transfection
Plasmid DNA was transfected using FuGENE9 (Roche, Basel, Switzerland), according to the manufacturer's protocol. siRNA were obtained from Invitrogen (Stealth RNAi). siRNA of PDCD6 was designed as previously described.39, 40 Transfection of siRNA was performed with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol.
Immunoblotting and antibodies
Cultured cells were washed twice with chilled PBS and resuspended in lysis buffer (50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothretiol, 10 mg/mL aprotinin, 1 mg/mL leupeptin, 1 mg/mL pepstatin A and 1% NP‐40). Cell lysates were centrifuged for 5 min at 4°C. The supernatants were separated by SDS‐PAGE and transferred to nitrocellulose membranes. The membranes were incubated with anti‐caspase‐3 (BD Transduction Labs, Franklin Lakes, NJ, USA), anti‐PDCD6 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐p53 (Santa Cruz Biotechnology), anti‐p21WAF1 (Oncogene Research Products, Boston, MA, USA), anti‐GFP (Nacalai Tesque, Kyoto, Japan) or anti‐tubulin (Sigma‐Aldrich, St. Louis, MO, USA). Immune complexes were incubated with secondary antibodies and visualized by chemiluminescence (PerkinElmer, Waltham, MA, USA).
Real‐time RT‐PCR analysis
Total RNA was isolated from cells using RNeasy spin column kits (Qiagen, Hilden, Germany), according to the manufacturer's protocol. Total RNA (5 µg) was amplified using a Super Script III First‐Strand Synthesis System for RT‐PCR (Invitrogen), following the manufacturer's protocol. The PCR reaction was performed using Power SYBR Green Master Mix (15 µL Power SYBR Green Master Mix, 0.3 µL each of 5 µM primer, 5 µL cDNA and 9.4 µL water) using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The PCR program was as follows: incubation for 10 min at 95°C, denaturation for 15 s at 95°C, annealing for 60 s at 60°C and extension for 30 s at 72°C. Cumulative fluorescence was measured at the end of the extension phase of each cycle. The results were normalized to GAPDH.
ChIP sequencing assay
Transfected H1299 cells were harvested and washed with chilled PBS, then incubated in 1% formaldehyde for 15 min at room temperature for chromatin cross‐linking. The cells were washed with chilled PBS again. Cell pellets were resuspended in SDS lysis buffer (1% SDS, 10 nM EDTA, 50 mM Tris–HCl, pH 8.0, 1 mM DTT, 1 mM PMSF, 1 mM Na3VO4, 10 µg/mL aprotinin, 1 µg/mL leupeptin and 1 µg/mL pepstatin A). The lysates were sonicated until the length of DNA fragments were between 200 and 500 bp. Then, 50 µL of the each lysate was used as an input. The remainder was diluted twofold in washing buffer (50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 0.1% NP‐40 and protease inhibitors, as described above). The dilution was subjected to immunoprecipitation with Flag‐agarose for 1–2 h at 4°C with rotation. The beads were then pelleted by centrifugation and washed sequentially with 300 µL of the following buffers: low salt buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.0 and 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.0 and 0.5 M NaCl), LiCl buffer (0.25 M LiCl, 1 mM EDTA, 10 mM Tris–HCl, pH 8.0 and 1% deoxycholate) and Tris‐EDTA buffer (twice). Precipitated chromatin complexes were removed from the beads by shaking with 150 µL elution buffer (1% SDS and 0.1 M NaHCO3) for 15 min; this was repeated once. NaCl was added at a final concentration of 200 mM for 6 h at 65°C for cross‐linking reversal. Then, the extraction buffer (50 mM Tris–HCl, pH 6.8, 10 mM EDTA and 40 µg/mL proteinase K) was added for 1 h at 45°C to digest the remaining proteins. DNA was recovered by phenol‐chloroform‐isoamyl alcohol (25:24:1) extraction and precipitated with 0.1 volume of 3 M sodium acetate and 2.5 volumes of ethanol in −80°C. The precipitated DNA was sequenced and analyzed by TaKaRa Bio (Shiga, Japan). PCR amplification was then performed using the chromatin immunoprecipitated fragments with the following oligonucleotide pairs: for PDCD6, 5′‐CCCCGGAAGTGGTGATAAT‐3′ and 5′‐GAAAACGTTCCACAGGAAGC‐3′; and for p21WAF1, 5′‐GGTCTGCTACTGTGTCCTCC‐3′ and 5′‐CATCTGAACAGAAATCCCAC‐3′.
TUNEL assay
Cells cultured in poly‐D‐lysine‐coated four‐well chamber slides were transfected with plasmids or siRNA and then treated with 1 mg/mL adriamycin (ADR) for 24 h. The apoptotic effect was measured using the DeadEnd Fluorometric TUNEL system (Promega). The apoptotic cells expressing GFP plasmids were detected using FluoroLink Cy5‐dUTP (GE Healthcare, Buckinghamshire, UK). Morphological changes in the nuclear condensation of cells undergoing apoptosis were detected by staining with DAPI.
Immunofluorescence assay
A549 cells cultured in chamber slides were fixed with 3% paraformaldehyde for 10 min. Cells were rinsed with PBS and incubated with 0.1% Triton X‐100 for 15 min. After washing and subsequent blocking with 10% goat serum in PBS for 1 h, cells were incubated with anti‐PDCD6 (1:200 dilution) for 2 h to overnight at 4°C. Secondary antibodies were then applied for 1 h. Nuclei were stained with DAPI.
Results
Comprehensive analysis of p53 target genes
ChIP‐seq was performed to identify direct binding genes of p53. H1299 cells, p53 null cell lines, were transfected with Flag vector as a control or Flag‐tagged wild‐type p53 (Flag‐p53). ChIP assay was performed using anti‐Flag antibody. We analyzed the DNA binding sites of p53 by the high throughput sequencing approach and isolated 5208 regions of 2332 genes. Based on the ChIP‐seq data, we identified direct p53 target genes, which were annotated in the vicinity of mapped sequences. Peak regions in sequences obtained from ChIP‐seq were represented by the score (Fig. 1A). This score was calculated from the number of overwrapped sequences. In this study, we focused on the PDCD6 gene because its score was considerably high. More importantly, the ChIP‐seq peak region of PDCD6 in the UCSC genome browser (Fig. 1B) contained p53 binding sites (Fig. 1B; indicated by red in the box), and was located within intron 1 (324762‐324904). To confirm the results from ChIP‐seq, H1299 cells transfected with Flag vector or Flag‐p53 were subjected to ChIP assay with anti‐p53 antibodies (Fig. 2A). The result demonstrated that the p53 was directly bound to promoter regions of PDCD6. We further examined the gene expression of PDCD6 by real‐time RT‐PCR. Ectopic expression of p53 induced transactivation of PDCD6 (Fig. 2B). As a control, ChIP and RT‐PCR for p21WAF1 were analyzed (Fig. S1A and B). Immunoblot analysis showed that the protein levels of PDCD6 were increased in p53‐transduced cells but not in mock cells (Fig. 2C). To determine whether PDCD6 is induced by naturally expressing p53, we investigated the expression of PDCD6 mRNA levels by treating p53 positive cell lines, A549 (human alveolar adenocarcinoma cell line) and U2OS (human osteosarcoma cell line) with various concentrations of ADR. We confirmed that the mRNA levels of PDCD6 are markedly augmented after ADR exposure in A549 cells. By contrast, the mRNA levels of PDCD6 were slightly increased in U2OS cells (Fig. 2D). In addition, the protein levels of PDCD6 were increased by ADR in A549 cells but not in H1299 cells (Fig. 2E). To confirm that PDCD6 expression is controlled by p53, A549 cells were transfected with scramble siRNA or p53 siRNA followed by treatment with ADR. The results of real‐time RT‐PCR and immunoblot analyses indicated that both PDCD6 mRNA and protein increased in A549 cells. In contrast, no upregulation was observed when p53 was knocked down (Fig. 2F).
Figure 1.

Comprehensive analysis of p53 target gene. (A) The ChIP sequencing score datasets. (B) Peak regions of PDCD6 in sequences obtained from the ChIP sequencing were visualized by the UCSC Genome Browser. The sequence of the highest peak is shown in the box. The p53 binding sites within the sequence of the highest peak are indicated in red color.
Figure 2.
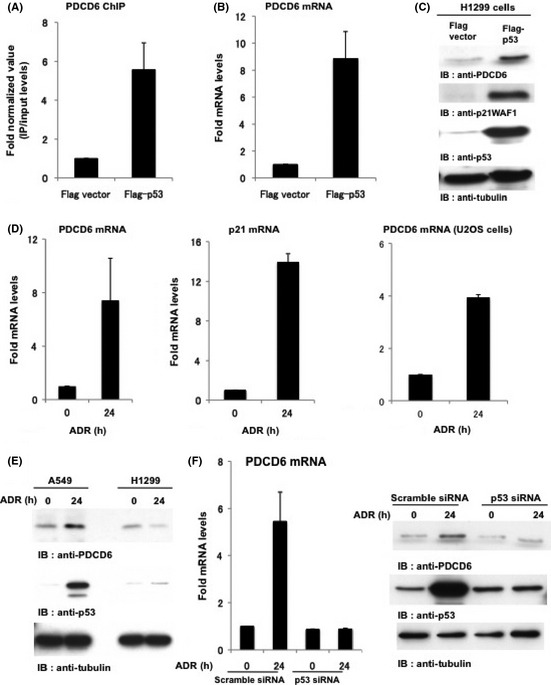
PDCD6 is a novel target of p53. (A) PDCD6 expression is regulated by p53. H1299 cells were transfected with Flag vector or Flag‐p53. To perform ChIP assays, choromatin‐protein complexes were immunoprecipitated with anti Flag‐p53 antibodies. Real‐time PCR amplification was performed to analyze the chromatin immunoprecitated fragments using appropriate primers for PDCD6. (B) H1299 cells were transfected with Flag vector or Flag‐p53. The mRNA level of PDCD6 was quantified by real‐time RT‐PCR. The value is normalized to GAPDH. The data represents the mean ± SD from three independent experiments, each performed in triplicate. (C) H1299 cells were transfected with Flag vector or Flag‐p53 and then analyzed by western blotting. IB, immunoblotting. (D) A549 cells and U2OS cells were treated with 1 µg/mL adriamycin (ADR) and incubated for 24 h. The mRNA level of PDCD6 was quantified by real‐time RT‐PCR. The value was normalized to GAPDH. The data represent the mean ± SD from three independent experiments, each performed in triplicate. (E) A549 cells and H1299 cells were treated with 1 µg/mL ADR for the indicated times and then analyzed by western blotting. (F) A549 cells were transfected with scramble siRNA or p53 siRNA and treated with 1 µg/mL ADR for 24 h. The mRNA level of PDCD was quantified by real‐time RT‐PCR. The value was normalized to GAPDH (left). The data represent the mean ± SD from three independent experiments, each performed in triplicate. Cell lysates were subjected to immunoblot analysis with anti‐PDCD6, anti‐p53 or anti‐tubulin (right).
Identification of putative consensus sequence for the p53‐binding in the PDCD6
The mRNA and protein levels of PDCD6 were increased in accordance with p53 expression. These results indicate that PDCD6 is a downstream target of p53. Thus, we next analyzed a consensus sequence for the p53‐binding in the promoter region of PDCD6. Based on the ChIP‐seq results, we found a putative consensus sequence for the p53‐binding in PDCD6 (Fig. 3A). To determine if p53 binds to this sequence, we constructed reporter vectors with or without this region (pGL3‐PDCD6 or pGL3‐PDCD6D, respectively; also see Fig. 3B), and carried out the luciferase assay. Luciferase activity in A549 cells transfected with the Luc‐PDCD6 was markedly increased (Fig. 3C). By sharp contrast, activity of Luc‐PDCD6Δ was significantly diminished. Importantly, the depletion of p53 completely reduced luciferase activity of Luc‐PDCD6 in A549 cells (Fig. 3C). Taken together, these results suggest that p53 binds to and activates PDCD6 via the consensus sequence of the p53‐binding.
Figure 3.
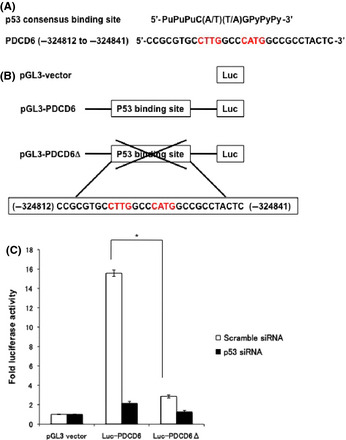
Identification of p53 consensus sequence in PDCD6. (A) The putative p53 response elements are depicted in the upstream of PDCD6. (B) Schematic representation of p53 consensus binding site‐driven luciferase reporter constructs. (C) A549 cells transfected with scramble siRNA or p53 siRNA were transduced with luciferase vector, Luc‐PDCD6 or Luc‐PDCD6Δ, and treated with 1 µg/mL adriamycin for 24 h. Luciferase activities were measured after 48 h post‐transfection. The relative fold increase of activity compared with the control was calculated. The data represent the mean ± SD from three independent experiments, each performed in triplicate.
PDCD6 induces apoptosis in response to DNA damage
The mechanism of cell death is regulated by p53, which upregulates many genes for the p53‐dependent apoptosis after DNA damage. Because our results suggested that PDCD6 is a direct target of p53 and is upregulated in the DNA damage response, we aimed to determine if PDCD6 is involved in apoptosis in a p53‐dependent manner. To investigate this possibility, we performed TUNEL assay to determine whether apoptosis is suppressed by the knockdown of PDCD6 in A549 cells. The result showed that p53‐dependent apoptosis was substantially attenuated by suppressing PDCD6 (Fig. 4A). We further verified whether overexpression of PDCD6 induces apoptosis upon genotoxic stress. The TUNEL assay results demonstrated that apoptotic cells were considerably increased by ectopic expression of PDCD6 (Fig. 4B). These results suggest that PDCD6 contributes to induction of apoptosis. We further confirmed that PDCD6 is involved in the activation of apoptosis‐related factors, such as caspase‐3, which plays an important role as a critical modulator in the apoptosis progression,41 and PARP cleavage is a significant indicator, as well as a useful marker for cellular apoptotic events. Caspase‐3 activity was suppressed by knockdown of PDCD6 following ADR exposure (Fig. 4C). We also observed cytochrome c release from mitochondria to cytoplasm after DNA damage, but it was reduced in cells silenced for PDCD6 (Fig. 4D). These observations collectively indicate that PDCD6 participates in the p53‐dependent apoptosis through the caspase‐3 activation and the cytochrome c release.
Figure 4.
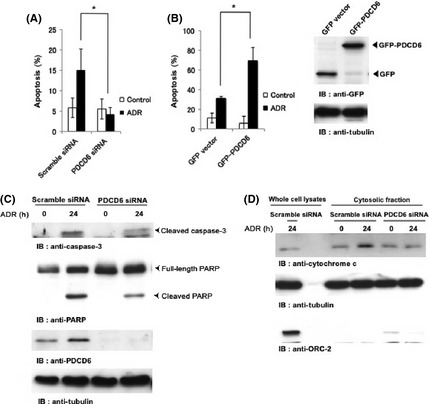
PDCD6 induces apoptosis in response to DNA damage. (A) A549 cells were transfected with scramble siRNA or PDCD6 siRNA. Cells were left untreated (open bar) or treated with 1 µg/mL adriamycin (ADR) (closed bar). The percentage of apoptotic cells was quantified by TUNEL assay. The data represent the mean ± SD from three independent experiments, each performed in triplicate. (B) A549 cells were transfected with GFP vector or GFP‐PDCD6. After 24 h post‐transfection, cells were left untreated (open bar) or treated with 1 µg/mL ADR for 24 h. The percentage of apoptotic cells was quantified by TUNEL assay. The data represent the mean ± SD from three independent experiments, each performed in triplicate. An asterisk indicates P < 0.05. Cell lysates were subjected to western blotting with anti‐GFP or anti‐tubulin (right panels). (C) A549 cells were transfected with scramble siRNA or PDCD6 siRNA and treated with 1 µg/mL ADR for 24 h. Cell lysates were subjected to immunoblot analysis with anti‐caspase‐3, anti‐PARP, anti‐PDCD6 or anti‐tubulin. IB, immunoblotting. (D) A549 cells were transfected with scramble siRNA or PDCD6 siRNA and treated with 1 µg/mL ADR for 24 h. Cell lysates and cytosolic fraction were subjected to immunoblot analysis with anti‐cytochrome c, anti‐tubulin or anti‐ORC‐2.
PDCD6 targets to the nucleus in response to DNA damage
We verified that PDCD6 is involved in the p53‐dependent apoptosis. However, the mechanism by which PDCD6 promotes apoptosis remains to be clarified. To address this issue, we investigated subcellular localization of endogenous PDCD6 by immunostaining assay before and after ADR treatment in A549 cells. The results demonstrated that PDCD6 was localized in the nucleus after ADR exposure. By contrast, in control cells, PDCD6 was localized ubiquitously in cytoplasm (Fig. 5A). Importantly, we found a nucleus localization signal (NLS)42 at the very N‐terminal end of PDCD6. To confirm whether this NLS is functional, we constructed the plasmid in which putative NLS sequence is deleted (Fig. 5B). The results indicated that nuclear localization was abrogated with the mutant PDCD6, suggesting that the signal is required for its nuclear localization (Fig. 5C). We used TUNEL assay to further examine whether the NLS mutant induces apoptosis in response to DNA damage. The results showed that expression of PDCD6ΔNLS reduced induction of apoptosis compared with that of intact PDCD6 (Fig. 5D). Of note, the transfection efficiency with each GFP‐PDCD6 and GFP‐PDCD6ΔNLS was approximately 65% (Fig. S2). These findings suggest the possibility that nuclear localization of PDCD6 is, at least in part, necessary for induction of the p53‐dependent apoptosis.
Figure 5.
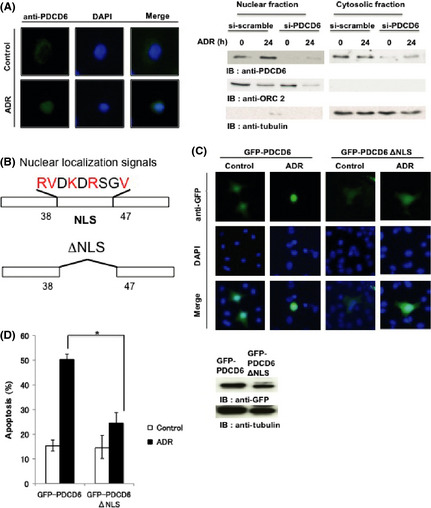
PDCD6 is localized in the nucleus in response to DNA damage. (A) A549 cells were transfected with scramble siRNA or PDCD6 siRNA and left untreated or treated with 1 µg/mL adriamycin (ADR) for 24 h. Immunofluorescence was performed with anti‐PDCD6. The nuclei were stained with DAPI. Western blotting was performed with anti‐PDCD6, anti‐ORC2 and anti‐tubulin. (B) Schematic representation of a nuclear localization signal (NLS) in PDCD6 and the mutant PDCD6 that was deleted in the NLS (PDCD6ΔNLS). (C) A549 cells were transfected with GFP‐PDCD6 or GFP‐PDCD6ΔNLS and left untreated or treated with ADR for 24 h followed by immunofluorecence assay. The nuclei were stained with DAPI. (D) A549 cells were transfected with scramble GFP‐PDCD6 or GFP‐PDCD6ΔNLS. Cells were left untreated (open bar) or treated with 1 µg/mL ADR (closed bar). The percentage of apoptotic cells was quantified by TUNEL assay. The data represent the mean ± SD from three independent experiments, each performed in triplicate. Cell lysates were subjected to immunoblot analysis with anti‐GFP or anti‐tubulin (right panels).
Discussion
In this study, through analysis with ChIP‐seq, we identified PDCD6 as a p53‐responsive gene for the first time. The ChIP‐seq used in this research has three particular features for screening p53‐direct binding genes. First, using the ChIP‐seq analysis novelty advanced our research. Although there have been many explorations using microarray analysis, ChIP‐seq analysis of p53‐responsive genes has seldom been undertaken. PDCD6, which was obtained from this comprehensive analysis, is known as a calcium binding protein of the penta EF‐hand protein family.29, 30 There is no report showing that PDCD6 is a direct transactivation gene of p53. In the current study, we clarified PDCD6 as a p53‐target gene (Fig. 2A–C). Furthermore, we found a consensus sequence of the p53‐binding (Fig. 3A) in the promoter region of PDCD6 and discovered that p53 used this site to promote expression of PDCD6 (Fig. 3C). These findings suggest that ChIP‐seq is a useful tool for exploring direct binding genes for p53. Second, ChIP is useful for isolating specific DNA fragments to which transcription factors and other proteins directly interact. In addition, in comparison with microarray analysis, the ChIP‐seq is not deviated from the probe design or the number of probe sets. This is useful for highly specific and sensitive analysis. Finally, the ChIP‐seq reaches a genome‐wide range of analysis on account of the recent development of the automated sequencer. Indeed, through this comprehensive analysis, we obtained novel p53‐responsive genes (Fig. 1A). The characteristics mentioned above suggest that using ChIP‐seq could be the most valuable method for exploring the p53‐responsive gene.
In a cell death assay, PDCD6 was previously identified as a pro‐apoptotic protein.29, 30 PDCD6 modulates endoplasmic reticulum‐stress‐stimulated stress cell death.43 In this study, we found that PDCD6 is localized in the cytoplasm ubiquitously; however, PDCD6 was translocated to the nucleus in response to DNA damage (Fig. 5A). Importantly, nuclear targeting of PDCD6 is required for induction of apoptosis (Fig. 5D). Based on these findings, our result led us to speculate that nuclear PDCD6 plays a role in the p53‐dependent apoptosis. Moreover, in condition of PDCD6 knockdown, cytochrome c release and caspase‐3 activation, which are involved in downstream of apoptotic cascade, were prohibited. Ectopic expression of PDCD6 significantly reduced the expression levels of the Bcl‐xL and the Bcl‐2 proteins. By contrast, the levels of Bax increased following overexpression of PDCD6.44 Thus, PDCD6 is, at least in part, involved in the signaling cascade of the p53‐responsive apoptotic machinery. These findings suggest that manipulation of nuclear targeting of PDCD6 could be useful for a novel cancer therapy by inducing apoptosis in cancer cells.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. P21WAF1 was also regulated by p53.
{kind=link}
Fig. S2. Evaluation of the transfection efficiency.
{kind=link}
Acknowledgments
This work was supported by grants from the Ministry of Education, Science and Culture of Japan (to K. Y. and Y. M.), the Ichiro Kanehara Foundation (to K. Y.), Terumo Life Science Foundation (to K. Y.) and Kobayashi Foundation for Cancer Research (to K. Y.).
(Cancer Sci, doi: 10.1111/j.1349-7006.2012.02362.x, 2012)
References
- 1. Birch JM, Blair V, Kelsey AM et al Cancer phenotype correlates with constitutional TP53 genotype in families with the Li‐Fraumeni syndrome. Oncogene 1998; 17: 1061–8. [DOI] [PubMed] [Google Scholar]
- 2. Olivier M, Goldgar DE, Sodha N et al Li‐Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003; 63: 6643–50. [PubMed] [Google Scholar]
- 3. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–99. [DOI] [PubMed] [Google Scholar]
- 4. Finlay CA, Hinds PW, Levine AJ. The p53 proto‐oncogene can act as a suppressor of transformation. Cell 1989; 57: 1083–93. [DOI] [PubMed] [Google Scholar]
- 5. Sherr CJ. Principles of tumor suppression. Cell 2004; 116: 235–46. [DOI] [PubMed] [Google Scholar]
- 6. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol 2009; 1: a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408: 307–10. [DOI] [PubMed] [Google Scholar]
- 8. Lane DP, Lain S. Therapeutic exploitation of the p53 pathway. Trends Mol Med 2002; 8: S38–42. [DOI] [PubMed] [Google Scholar]
- 9. Yoshida K, Miki Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci 2010; 101: 831–5. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida K. Nuclear trafficking of pro‐apoptotic kinases in response to DNA damage. Trends Mol Med 2008; 14: 305–13. [DOI] [PubMed] [Google Scholar]
- 11. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk‐interacting protein Cip1 is a potent inhibitor of G1 cyclin‐dependent kinases. Cell 1993; 75: 805–16. [DOI] [PubMed] [Google Scholar]
- 12. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature 1993; 366: 701–4. [DOI] [PubMed] [Google Scholar]
- 13. el‐Deiry WS, Tokino T, Velculescu VE et al WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75: 817–25. [DOI] [PubMed] [Google Scholar]
- 14. Hermeking H, Lengauer C, Polyak K et al 14‐3‐3 sigma is a p53‐regulated inhibitor of G2/M progression. Mol Cell 1997; 1: 3–11. [DOI] [PubMed] [Google Scholar]
- 15. Lindsten T, Ross AJ, King A et al The combined functions of proapoptotic Bcl‐2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–9. [DOI] [PubMed] [Google Scholar]
- 17. Sun Y. p53 and its downstream proteins as molecular targets of cancer. Mol Carcinog 2006; 45: 409–15. [DOI] [PubMed] [Google Scholar]
- 18. Ho J, Benchimol S. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ 2003; 10: 404–8. [DOI] [PubMed] [Google Scholar]
- 19. Speidel D. Transcription‐independent p53 apoptosis: an alternative route to death. Trends Cell Biol 2010; 20: 14–24. [DOI] [PubMed] [Google Scholar]
- 20. Nakamura Y. Isolation of p53‐target genes and their functional analysis. Cancer Sci 2004; 95: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levine AJ. p53 the cellular gatekeeper for growth and division. Cell 1997; 88: 323–31. [DOI] [PubMed] [Google Scholar]
- 22. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 2007; 25: 725–38. [DOI] [PubMed] [Google Scholar]
- 23. Burlacu A. Regulation of apoptosis by Bcl‐2 family proteins. J Cell Mol Med 2003; 7: 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei MC, Zong WX, Cheng EH et al Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El‐Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 2002; 4: 842–9. [DOI] [PubMed] [Google Scholar]
- 26. Oda E, Ohki R, Murasawa H et al Noxa, a BH3‐only member of the Bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000; 288: 1053–8. [DOI] [PubMed] [Google Scholar]
- 27. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–94. [DOI] [PubMed] [Google Scholar]
- 28. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- 29. Maki M, Narayana SV, Hitomi K. A growing family of the Ca2+‐binding proteins with five EF‐hand motifs. Biochem J 1997; 328: 718–20. [PMC free article] [PubMed] [Google Scholar]
- 30. Krebs J, Saremaslani P, Caduff R. ALG‐2: a Ca2+‐binding modulator protein involved in cell proliferation and in cell death. Biochim Biophys Acta 2002; 1600: 68–73. [DOI] [PubMed] [Google Scholar]
- 31. Vito P, Lacana E, D'Adamio L. Interfering with apoptosis: Ca(2+)‐binding protein ALG‐2 and Alzheimer's disease gene ALG‐3. Science 1996; 271: 521–5. [DOI] [PubMed] [Google Scholar]
- 32. Tarabykina S, Moller AL, Durussel I, Cox J, Berchtold MW. Two forms of the apoptosis‐linked protein ALG‐2 with different Ca(2+) affinities and target recognition. J Biol Chem 2000; 275: 10514–8. [DOI] [PubMed] [Google Scholar]
- 33. Chatellard–Causse C, Blot B, Cristina N, Torch S, Missotten M, Sadoul R. Alix (ALG‐2‐interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J Biol Chem 2002; 277: 29108–15. [DOI] [PubMed] [Google Scholar]
- 34. Wu Y, Pan S, Luo W, Lin SH, Kuang J. Hp95 promotes anoikis and inhibits tumorigenicity of HeLa cells. Oncogene 2002; 21: 6801–8. [DOI] [PubMed] [Google Scholar]
- 35. Yamasaki A, Tani K, Yamamoto A, Kitamura N, Komada M. The Ca2+‐binding protein ALG‐2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol Biol Cell 2006; 17: 4876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14‐3‐3 proteins regulates nuclear targeting of c‐Abl in the apoptotic response to DNA damage. Nat Cell Biol 2005; 7: 278–85. [DOI] [PubMed] [Google Scholar]
- 37. Yoshida K, Liu H, Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem 2006; 281: 5734–40. [DOI] [PubMed] [Google Scholar]
- 38. Yoshida K, Wang HG, Miki Y, Kufe D. Protein kinase Cdelta is responsible for constitutive and DNA damage‐induced phosphorylation of Rad9. EMBO J 2003; 22: 1431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoj BR, la Cour JM, Mollerup J, Berchtold MW. ALG‐2 knockdown in HeLa cells results in G2/M cell cycle phase accumulation and cell death. Biochem Biophys Res Commun 2009; 378: 145–8. [DOI] [PubMed] [Google Scholar]
- 40. la Cour JM, Hoj BR, Mollerup J, Simon R, Sauter G, Berchtold MW. The apoptosis linked gene ALG‐2 is dysregulated in tumors of various origin and contributes to cancer cell viability. Mol Oncol 2008; 1: 431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004; 23: 2934–49. [DOI] [PubMed] [Google Scholar]
- 42. Cokol M, Nair R, Rost B. Finding nuclear localization signals. EMBO Rep 2000; 1: 411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rao RV, Poksay KS, Castro‐Obregon S et al Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J Biol Chem 2004; 279: 177–87. [DOI] [PubMed] [Google Scholar]
- 44. Park SH, Lee JH, Lee GB et al PDCD6 additively cooperates with anti‐cancer drugs through activation of NF‐kappaB pathways. Cell Signal 2012; 24: 726–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. P21WAF1 was also regulated by p53.
Fig. S2. Evaluation of the transfection efficiency.