

Abstract
Photodynamic therapy (PDT) is an effective therapeutic regime for lung cancer. Mitochondrial functional failure is considered to be one of the most important factors causing cell death after PDT. However, the detailed mechanisms that are involved are still unclear. We previously reported that apurinic/apyrimidinic endonuclease (APE1) plays a critical role in regulating sensitivity to PDT in the lung cancer A549 cell line. An important mitochondrial regulatory role for APE1 has recently been reported, so therefore we explored the role of APE1 in cell survival after PDT‐induced oxidative stress through regulation of mitochondrial function. We first observed that photoirradiation induced the mitochondrial translocation of APE1. The ability of APE1 to regulate mitochondrial membrane potential and reactive oxygen species (ROS) production after photoirradiation was tested in APE1 knockdown A549 cells. APE1‐deficient A549 cells were characterized as having a lower mitochondrial membrane potential and higher ROS production, which led to increased apoptosis through the mitochondrial pathway after PDT. Additionally, unexpected activity of APE1 was observed in mitochondria: the control of mitochondrial transcriptional activity by redox regulation of mitochondrial transcription factor A (TFAM). Furthermore, two dominant‐negative mutants of APE1 were overexpressed to enhance their individual activities in mitochondria. The results suggest that both these APE1 activities play a role in the regulation of mitochondrial function but through different mechanisms. The present study not only provides possible mechanisms for APE1 in regulating survival after photoirradiation but also uncovers a new activity of APE1 in mitochondria. (Cancer Sci 2012; 103: 882–888)
Photodynamic therapy (PDT) is a promising therapeutic strategy for cancer.1 Reactive oxygen species (ROS) generated by laser in the presence of photosensitizers are the major killing effectors of PDT. Many hydrophobic photosensitizers localize in the mitochondria and exert their primary action there. Therefore, mitochondria are the most likely candidates to initiate apoptosis induced by PDT with respect to both the source of the ROS and the major subcellular target of the photosensitizers.2 Once irradiated by PDT, mitochondrial DNA (mtDNA) will be damaged leading to the downregulation of mitochondrial electron transport chain (ETC.) function and an increase of ROS production. Imbalanced ROS production renders more mtDNA damage, causing a vicious cycle leading to mitochondrial malfunction. Consequently, mitochondrial proapoptotic agents are released to the cytosol to trigger the apoptotic cascade. During this process, the damage to mtDNA, proximal to the source of ROS, plays a crucial role in promoting this cycle.3 However, little is known about which molecules are critical in this process and the detailed mechanisms that are involved. Thus, better understanding of the molecular mechanism involved in initiation of PDT‐induced apoptosis will be beneficial for improving the therapeutic outcome of PDT.4
Most of the mtDNA damage caused by PDT consists of oxidative lesions that are mainly repaired by base excision repair (BER).5 Human apurinic/apyrimidinic endonuclease (APE1) is an essential enzyme in BER, which is the major DNA repair mechanism in mitochondria. APE1 is reported to translocate into mitochondria under oxidative stress.6, 7 Pioneering studies regarding mitochondrial APE1 functions showed that on mitochondrial translocation of APE1, oxidative stress‐induced apoptosis is effectively prevented.8 Our previous study showed that mitochondrial translocation of APE1 is beneficial for mtDNA repair and cell survival following hydrogen peroxide challenge.9 In contrast, APE1 is a redox factor that regulates the DNA binding activity of a number of transcription factors playing a critical role in oxidative stress signaling, such as NF‐κB, AP‐1 and p53.10 Taken together, these data indicate that APE1 is clearly critical for cell survival after oxidative stress.11, 12 Our previous reports showed that knockdown of APE1 by an adenovirus vector expressing APE1 shRNA enhanced PDT‐induced tumor cell killing both in vitro and in vivo.13 However, we did not examine the specific role of mitochondrial‐localized APE1 after PDT.
In the present study, we tested the hypothesis that on translocation into mitochondria, APE1 protects cells from PDT‐induced apoptosis. We further explored the molecular mechanism of the photoirradiation protective effects of mitochondrial APE1. Our results indicate that mitochondrial APE1 plays a protective role in resistance to PDT‐induced apoptosis. The translocation of APE1 from the nucleus to the mitochondria is indicative of the cell killing effector mechanism of PDT. In addition to its repair activity, the redox regulatory function of APE1, which involves the control of mitochondrial transcription factor A (TFAM) transcriptional activity, was also observed in mitochondria.
Materials and Methods
Materials
Lipofectamine 2000 and RNAiMAX Transfection Reagent, TRIzol, ROS Detection reagents H2DCF‐DA and primers were from Invitrogen (Carlsbad, CA, USA). E3330, mitochondria staining kit, synthetic siRNA against APE1 (sequence referred to in Wang et al.14) were from Sigma‐Aldrich (St Louis, MO, USA).
Constructs
Overexpression vectors were constructed using the same strategies as described previously.9 To add the mitochondrial targeting sequence to the 5′ end of the APE1 cDNA sequence, the overlap extension PCR method (or splicing by overlap extension [SOE]) for gene splicing was applied. The downstream primer contained a FLAG tag sequence and a Xho I site. The PCR fragment was then subcloned into the EcoR I and Xho I site of pcDNA3.1(+). The Cys 65 Ser and His 309 Asn mutants were then made using the QuikChange Site‐Directed Mutagenesis Kit (Stratagene Cloning Systems, La Jolla, CA, USA).
Cell culture and PDT
The human non‐small‐cell lung cancer cell line A549 was purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were maintained in α‐minimum essential medium (MEM) supplemented with 10% (v/v) fetal bovine serum, 50 mg/mL penicillin/streptomycin and 2 mM l‐glutamine in 5% CO2 at 37°C and passaged 2–3 times a week. The PDT was applied as mentioned previously.13
Mitochondrial membrane potential assay (MMP)
The MMP was assessed using the JC‐1 mitochondria staining kit (Sigma Aldrich, St Louis, MO, USA) for flow cytometry, following the manufacturer's recommendations. Briefly, cells were treated with various doses of photoirradiation for the indicated time, then incubated in medium containing JC‐1 probe (2.5–5 μg/mL) for 30 min at 37°C. After washing with ice‐cold JC‐1 binding buffer twice, MMP was measured immediately using flow cytometry.
ROS production measurement
The ROS production was measured following the manufacturer's instructions. Briefly, A549 cells were plated in 12‐well plates and then treated with photoirradiation at various doses. After incubation at 37°C for the indicated time, cells were washed twice with PBS and then incubated with 20 μM H2DCF‐DA in 0.2% BSA‐PBS at 37°C for 45 min before immediate analysis using flow cytometry (FACScan, BD Bioscience, San Jose, CA, USA). The mean fluorescent intensity of 10 000 analyzed cells in each treatment group was normalized by the mean fluorescent intensity of the control group of each cell line, and then the ratios were taken as a measurement for the total ROS load.
Cytochrome c release assay
To measure the cytochrome c release from mitochondria to cytoplasm following oxidative stress, mitochondria‐free cytosolic fractions were subjected to western blot analysis. Mitochondria‐free cytosolic fractions were prepared using the protocol described previously.9
Western blot assay and antibodies
An equal amount of whole cell lysate or organelle extracts was electrophoresed using 10% SDS‐PAGE. Western blots were performed as described previously.15 Suppliers of antibodies used for the western blot were as follows: anti‐APE1 (Novus, Littleton, CO, USA), anti‐Flag (M2; Sigma Aldrich), anti‐TFAM (Abcam, Cambridge, MA, USA), anti‐β‐actin (Sigma Aldrich), anti‐Cytochrome c (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐Caspase 9 (Cell Signaling Technology, Boston, MA, USA) and anti‐TFB2M (Abcam).
Quantitative RT‐PCR
Total RNA was extracted using a TRIzol‐based protocol according to the manufacturer's instruction. Subsequently, cDNA was synthesized using 1 μg of total RNA in the ReverTra Ace reverse transcription kit (Toyobo, Osaka, Japan). Quantitative RT‐PCR was performed using SYBR Premix Ex Taq (Takara, Dalian, China) in a LightCycler 480 Real‐Time PCR System (Roche, Indianapolis, IN, USA). Primer pairs for MT‐COX1 and β‐actin were designed to yield 152 and 289 bp separately. Mitochondrial DNA and nuclear DNA lesions were quantified using quantitative extended‐length PCR as described previously.9
Co‐immunoprecipitation assay
Cells were harvested and washed with ice‐cold PBS. Total cell lysates prepared using immunoprecipitation (IP) lysis buffer were pre‐cleared by incubating with protein A/G agarose resin (Pierce, Rockford, IL, USA) for 30 min on ice, then co‐immunoprecipitated for 3 h using primary antibodies. After incubation, protein A/G agarose resin was added for 1 h at 4°C. Both the agarose beads and the binding proteins were then precipitated and mixed with sample buffer and incubated at 100°C for 5 min. The samples were then subjected to western blotting analysis.
Electrophoretic mobility‐shift assay (EMSA)
The EMSA was performed according to the manufacturer's instruction for the LightShift Chemiluminescent EMSA kit (Pierce) with minor modifications. The biotin conjugated probes containing the TFAM binding site in bidirectional promoters for transcribing H‐ and L‐strands, TFAMF: 5′‐GGTGGCCTGACGCATTCCCCAA‐3′ and TFAMR: 5′‐TTGGGGAAT GCGTCAGGCCACC‐3′, were synthesized (Invitrogen, Shanghai, China).
MTT assay
The MTT assay was performed as described previously.9 Briefly, 5 mg/mL MTT (15 μL/well) was added after 24–72 h post PDT treatment. Cells were cultured for 4 h at 37°C. The culture medium was then removed and 150 μL of DMSO was added into each well. The plates were shaken on a swing bed for 10 min and the optical density (OD) value at 492 nm was determined using a microplate reader. Cell viability (%) = OD value of the treatment group/OD value of the control group × 100%.
Results
PDT can induce mitochondrial‐mediated apoptosis
To test the effects of dose and time of PDT on apoptosis of A549 cells, especially the mitochondrial‐mediated pathway, we measured apoptosis by flow cytometry using Annexin V‐FITC/PI staining. The results indicated that apoptosis was initiated within 12 h and reached the peak at 24–36 h after a 5 kJ/m2 PDT treatment (Fig. 1A). Because mitochondria are sensitive to oxidative stress and the photosensitizers are mainly targeted to mitochondria, we then tested the effect on mitochondrial function after PDT using a JC‐1 staining assay. The MMP started to significantly decrease as early as 3 h after PDT and kept decreasing in a time‐dependent manner (Fig. 1B). We then tested mitochondrial‐mediated apoptosis activation through the early event of cytochrome c release and later event of caspase 9 cleavage.16, 17 The results indicated that cytochrome c was released to the cytosol as early as 3 h post‐PDT and the time course of caspase 9 activation was consistent with cytochrome c release suggesting that PDT induces mitochondrial‐mediated apoptosis in a time‐dependent manner (Fig. 1C).
Figure 1.
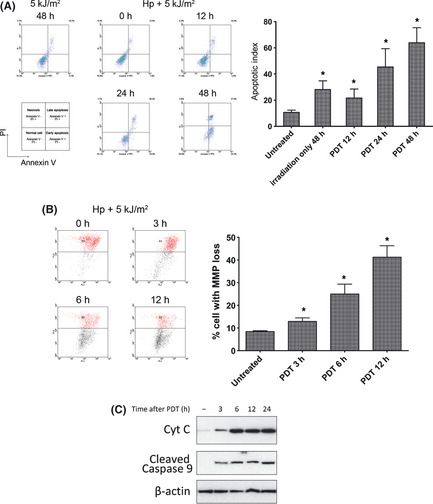
Photodynamic therapy (PDT) induces significant apoptosis of A549 cells through a mitochondrial‐specific pathway. (A) Flow cytometry results of apoptotic assays at different times after photoirradiation with or without photosensitizer (Hp), with the untreated group as a negative control. The x axis is for Annexin‐V‐FITC staining and the y axis is for propidium iodide (PI) staining in all graphs represented. The cells undergoing early apoptosis are Annexin‐V positive and PI negative in the lower right quadrants. The data for the percentage of apoptotic cells (referred to as the apoptosis index) of each group are also shown in the bar graph. (B) Flow cytometry assay for mitochondrial membrane potential (MMP) alteration after photoirradiation. The lower shift of the spots represents the loss of MMP. The percentages of cells with MMP loss are shown in the bar graph. (C) The cytoplasmic fractions were subjected to immunoblotting with anti‐cytochrome C (Cyt C) antibody and anti‐cleaved Caspase 9 antibody. Anti‐β‐actin antibody was used as a cytoplasmic fraction loading control. Cyt C was used as an indicator of pro‐apoptotic factors released from mitochondria, and cleaved Caspase 9 was used as an indicator for apoptotic effects initiated by the mitochondrial pathway. *Statistically significant from the untreated control (P < 0.01).
Alteration of APE1 subcellular distribution after PDT treatment
We then tested if APE1 subcellular localization is altered following PDT treatment using western blot. The results indicated that the mitochondrial APE1 level was elevated after PDT treatment in a dose‐ and time‐dependent manner (Fig. 2). Because the sensitivity of western blotting is higher than that of the microscopy assay, we actually detected a very subtle mitochondrial APE1 level in untreated A549 cells, which began to increase as early as 30 min. These results suggested that APE1 is a quick response protein to mtDNA damage, which is in accord with its main mitochondrial activity of DNA repair.
Figure 2.
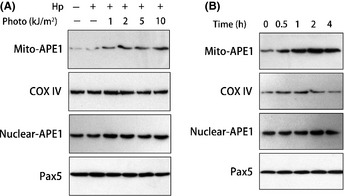
Mitochondrial expression of apurinic/apyrimidinic endonuclease (APE1) in A549 cells was induced by photoirradiation in a time‐ and dose‐dependent manner. (A) Cells received different doses of photoirradiation and were collected 1 h post‐irradiation. The mitochondrial and nuclear extracts were prepared and then used in a western blot assay. Fractions were immunoblotted against APE1, cytochrome c oxidase IV (COX IV) (loading control for mitochondrial fraction) and Pax5 (loading control for nuclear fraction). (B) A time course of APE1 expression in the mitochondria and nuclei post‐5 kJ/m2 photoirradiation. The western blots shown represent three independent experiments.
ROS production and mtDNA damage are increased in APE1‐deficient cells
To investigate the functional meaning of the APE1 mitochondrial translocation in response to PDT‐induced stress, the endogenous ROS production and mtDNA damage were measured in APE1 knockdown A549 cells. The ROS production, which represents mitochondrial electron transport chain (ETC.) function, is measured by the fluorescent probe DCFH‐DA. The results indicated that initial ROS production levels, which were measured at 15 min post‐PDT treatment, were approximately fourfold higher in APE1 knockdown cells (Fig. 3A). This suggested that the intrinsic sensitivity to PDT‐induced oxidative stress differs between APE1‐deficient and APE1‐proficient cells. In addition, ROS production was measured at a later time point of 1–4 h post‐irradiation and the results indicated that tolerance of PDT treatment is impaired in APE1‐deficient cells. Additionally, we observed that APE1‐deficient cells harbor a much weaker mtDNA repair capacity, which is evident by the extended and prolonged mtDNA damage after PDT irradiation (Fig. 3B). The results of the mtDNA lesion quantitation assay are also consistent with the ROS production and further proved the previous hypothesis that the unrepaired ROS‐induced mtDNA lesions promote mitochondrial ETC. malfunction and further increase the ROS production.
Figure 3.
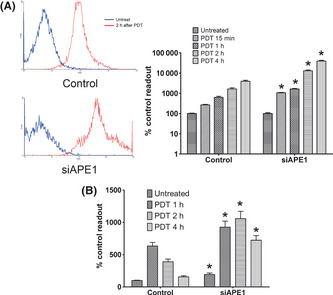
Apurinic/apyrimidinic endonuclease (APE1) deficiency promotes intracellular reactive oxygen species (ROS) production and extensive mitochondrial DNA (mtDNA) damage after photoirradiation. (A) Intracellular ROS production was measured using dichlorofluorescein diacetate (DCFH‐DA) staining and assayed using flow cytometry. A549 cells received 5 kJ/m2 photoirradiation and were collected for ROS assay at 15 min and 1, 2 and 4 h post‐photoirradiation. The ROS production at 2 h post‐photoirradiation versus the HpD only control are shown in the histogram and the data were statistically processed and are shown in the bar graph. (B) Mitochondrial DNA lesions induced by photoirradiation at various times were measured using quantitative extended length PCR. Data were obtained from three independently performed experiments. *Statistically significant from scramble RNA transfected control (P < 0.01). PDT, photodynamic therapy.
Unexpected role of APE1 in mitochondrial transcriptional activation
Since we detected the downregulation of the mitochondrial ETC., we then hypothesized that APE1 might be involved in this process. The previous theory considered it to be due to the loss‐of‐expression of some important ETC. genes caused by the transcriptional blocking effect of oxidative mtDNA lesions. However, we found that the expression of some mitochondrial genome encoded ETC. genes was decreased in unchallenged APE1‐deficient cells and thought it might be caused by impaired mitochondrial transcriptional activity. We then tested the protein–protein interaction between APE1 and the important mitochondrial transcriptional factor, TFAM, by Co‐IP. As shown in Figure 4, when performing the APE1 precipitation, TFAM and TFB2M were detected in the immunocomplex and when performing the TFAM immunoprecipitation, APE1 and TFB2M were also detected in the complex. These results suggest that after entry into mitochondria, APE1 interacts with TFAM and might be involved in mitochondrial transcriptional initiation. We also noticed that the interaction between APE1 and the TFAM/TFB2M transcriptional complex is dependent on the cellular reduction‐oxidation status (data not shown). Additionally, GST pulldown assay showed the direct interaction between APE1 and TFAM (Fig. 4B).
Figure 4.
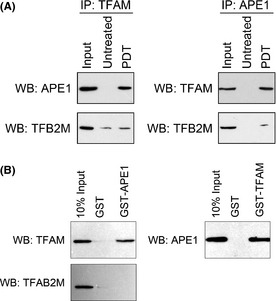
Apurinic/apyrimidinic endonuclease (APE1) interacts with a mitochondrial transcription complex. (A) Co‐immunoprecipitation with either mitochondrial transcription factor A (TFAM) or APE1 antibody in mitochondrial extracts of A549 cells that received 5 kJ/m2 photoirradiation were performed at 2 h post‐treatment. APE1, TFB2M or TFAM antibodies were then used in western blots to detect the presence of these proteins in the immunocomplex. Mitochondrial extracts from untreated A549 cells were also included as a control, and pre‐precipitation mitochondrial extracts from photoirradiated A549 cells were used as an input control. (B) Glutathione S‐transferases (GST)‐tagged APE1 or TFAM proteins were incubated with mitochondrial extracts from photoirradiated A549 cells and then precipitated using GST affinity beads. Antibodies against GST, TFAM or APE1 were applied in subsequent western blots to detect the direct interaction between APE1 and TFAM. IP, immunoprecipitation; WB, western blot.
To determine whether APE1 promotes mitochondrial transcription by interaction with TFAM, we assayed the DNA binding activity of TFAM using EMSA (Fig. 5). The probe contained predicted binding sites of TFAM in bidirectional promoters for transcribing H‐ and L‐strands. First, TFAM DNA binding activity is dependent on the reduction‐oxidation status of the incubation system. As shown in Figure 5(B), the DNA binding activity of TFAM was decreased in a hydrogen peroxide contained system, while it increased in a DTT contained system. To further elucidate the requirement of APE1 for DNA binding activity of TFAM, EMSA was performed in both the A549 cell with and without APE1 knockdown. The results indicated that the affinity of TFAM to the probe dramatically decreased in APE1‐deficient cells. Since previous reports indicated that E3330 might block the mitochondrial transport of APE1,18 we then used isolated mitochondrial crude extracts that were treated with E3330 in vitro during incubation with the probe. The results showed E3330 actually blocked TFAM binding to the probe, which indicated that the redox activity of APE1 might be involved.
Figure 5.
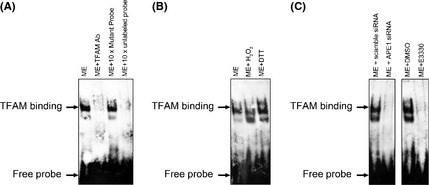
Apurinic/apyrimidinic endonuclease (APE1) regulates mitochondrial transcription factor A (TFAM) transcriptional activity in mitochondria through its redox activity. (A) The DNA binding activity of TFAM to the mitochondrial promoter region was measured using an electrophoretic mobility‐shift assay (EMSA) at 2 h post‐5 kJ/m2 photoirradiation. Mitochondrial extracts from photoirradiated A549 cells were incubated with DTT or hydrogen peroxide, which represents reduced and oxidative conditions, respectively. Reaction with biotin‐labeled mutated probe, TFAM antibody or × 100 unlabeled mutated probe were applied as controls for the specificities of either the probe or the transcription factor. (B) DNA binding activities of TFAM were affected by hydrogen peroxide (oxidation) and DTT (reduction) treatment. (C) DNA binding activities of TFAM were measured using EMSA in both APE1‐deficient or scramble RNA transfected A549 cells. The DNA binding activity of TFAM in the mitochondrial extract was affected by the APE1 redox inhibitor E3330 pretreatment. The EMSA images shown represent three independent experiments. ME, mitochondrial extracts.
Exogenously expressed mitochondrial‐specific APE1 protects cells from cell death induced by PDT
We previously reported specifically expressing APE1 in mitochondrial rendered enhanced cell survival after oxidative stress in human endothelial cells.9 We then tested this using mitochondrial targeting sequence (MTS)‐fused, full‐length, wild‐type APE1 (mt‐flAPE1) with Cys65 mutation vector (mt‐C65SAPE1) or His309 mutation vector (mt‐H309N) to observe its effects on the cell death induced by PDT. A previous study in our laboratory9 indicates that at 48 h post‐transfection, MTS‐fused APE1 protein were mainly located in the mitochondria, whereas wild‐type APE1 was mainly located in the nucleus. We confirmed by both EMSA and AP endonuclease assays that the APE1 activity is intact in mitochondrial fractions of mt‐flAPE1 transfected A549 cells but that the redox activity was reduced in mitochondrial fractions of mt‐C65SAPE1 transfected A549 cells, and repair activity was reduced in mitochondrial fractions of mt‐H309N transfected A549 cells (Fig. 6 A,B). We then performed MTT to measure the overall cell death after PDT. Mitochondrial overexpression of APE1 by both mt‐C65SAPE1 and mt‐H309NAPE1 significantly enhanced cell survival after PDT, when compared with wild‐type APE1 mitochondrial transfection or vector only control. However, mt‐flAPE1 overexpression showed an even higher survival rate than that of mt‐C65SAPE1 or mt‐H309NAPE1, suggesting that the activity of APE1 plays a role in mitochondrial functional maintenance after PDT and is beneficial for cell survival (Fig. 6C,D).
Figure 6.
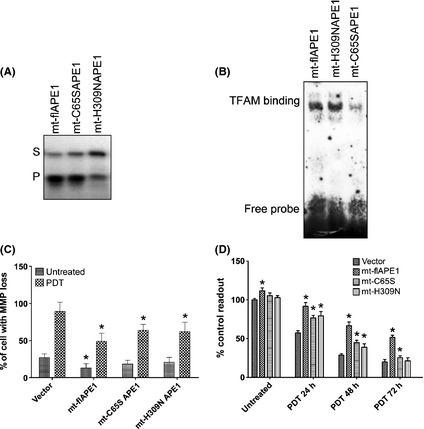
Both DNA repair and redox activities of apurinic/apyrimidinic endonuclease (APE1) are involved in regulation of mitochondrial function and cell survival after photoirradiation. Confirmation of APE1 activity alteration and survival beneficial in the mitochondrially overexpressed wild type (mt‐flAPE1), DNA repair‐deficient mutant (mt‐H309NAPE1) and redox‐deficient mutant (mt‐C65SAPE1) of APE1. (A) The DNA repair activity of wild type and mutants of APE1 was measured using a oligonucleotide incision assay. (B) Redox activity of wild type and mutants of APE1 was measured using electrophoretic mobility‐shift assay (EMSA). (C) Evaluation of MMP post‐photoirradiation was performed using flow cytometry at 6 h after 5 kJ/m2 photoirradiation. Photoirradiation was applied at 48 h post‐transfection. *Statistically significant from vector alone transfected control (P < 0.01). (D) The MTT assays were performed to measure cell survival 24–72 h after 5 kJ/m2 photoirradiation. *Statistically significant from vector alone transfected control (P < 0.01). Data shown in the bar graph are from three independent experiments.
Discussion
In the present study, we found that APE1 translocates to mitochondria in a time‐ and dose‐dependent manner after PDT irradiation in the presence of Hp as photosensitizer. After mitochondrial translocation, APE1 acts as a protective factor to maintain mitochondrial function after PDT. Additionally, we observed that APE1 also exerts its redox activity in mitochondria to regulate mitochondrial transcriptional initiation. Using a mitochondrial‐targeted APE1 expressing vector, we confirmed that mitochondrial APE1 enhances A549 cell survival after PDT irradiation and that both mtDNA repair and redox regulation activities are involved.
Our results indicate that APE1 deficiency increased initial ROS production in response to PDT. As the major effector of cell killing by PDT, the steady state of ROS level is determined by the balance between ROS production and ROS scavenging. The ETC. capacity is mainly responsible for ROS production and the intracellular antioxidative system is responsible for ROS scavenging. We demonstrated here in the present study and our other study (Li et al., Third Military Medical University, Chongqing, manuscript in preparation) that the expression of some ETC.‐related genes, including cytochrome c oxidase VI (COX VI) and COX I, are regulated by redox activity of APE1. In contrast, some antioxidants, especially the thioredoxin system, were also related to the APE1 level. The ROS production increase in APE1 knockdown cells was also observed in a previous study.19 Vascotto et al. performed a global gene expression assay between an APE1 stable knockdown HeLa cell and its control cell. They found a subset of redox signaling genes downregulated in APE1‐deficient cells. In addition to our research, this could be another possible explanation to the intracellular ROS increase caused by APE1 knockdown. As shown in previous reports, mitochondrial function is not only a critical target of but also a sensor for oxidative stress.20 Therefore, manipulation of mitochondrial function after oxidative stress is one of the most effective methodologies to modify the cellular sensitivity to oxidative stress. APE1 has recently been found to be crucial for mitochondrial function. Vascotto et al. developed a series of APE1 stable knockdown and knockin HeLa cell lines, which are powerful tools for APE1 research.21 By using those cell lines, they found that Cys65 is critical for APE1 mitochondrial translocation. An APE1 Cys65 to Ser mutant knockin cell exhibits a lower mitochondrial transmembrane potential and increased apoptosis after hydrogen peroxide or rotenone‐induced oxidative stress. We further observed a loss of transcriptional activity by nuclear respiratory factor 1 (NRF‐1), an important mitochondrion‐related transcription factor, in APE1 deficient cells (Li et al., Third Military Medical University, Chongqing, manuscript in preparation). However, those studies failed to describe the actual role of APE1 in mitochondrial regulation after transport into the mitochondria.
APE1 has a unique role in mitochondria. The present study demonstrates a new regulatory role for APE1 in mitochondria through its redox activity. To the best of our knowledge, this is the first report showing that the redox function of APE1 is exerted in mitochondria. We demonstrated an interaction between APE1 and TFAM, which further proves that TFAM is functionally related to APE1 in mitochondria. TFAM is a protein that resides in mitochondria and protects mtDNA22 and participates in mtDNA damage repair.23 Canugovi et al.23 suggest that TFAM has a competitive role with some BER proteins including OGG1 and APE1. In addition, TFAM also functions as a transcription factor in mitochondria, which makes it a possible target for regulation by APE1. Our current results indicate that TFAM is under redox regulation and that APE1 is present in the TFAM/TFB2M transcription complex. As a result, the mitochondrial genome encoded genes are downregulated in APE1‐deficient cells. Therefore, it is reasonable to conclude that APE1 regulates TFAM transcriptional activity through its redox function.
Moreover, we used a pair of overexpression vectors that targeted wild‐type, repair and redox‐deficient APE1 proteins to mitochondria. We then further mutated the Cys65 residue24 to eliminate the redox activity and the His309 residue25 to eliminate the repair activity. However, we failed to observe comparable enhancement of cell resistance to PDT in the redox‐ or repair‐deficient group when compared with the wild‐type group. This result demonstrated that both the repair and redox activities of APE1 are involved in the regulation of mitochondrial function. Although the repair activity increased in the N terminal truncated form of APE1,26 we failed to get higher or even equal survival enhancement in the mitochondrially expressed N‐terminal deletion APE1 with Cys65 mutation vector (data not shown), even though the repair activity is much higher than the wild‐type APE1. This result further suggested that the redox activity of APE1 plays a considerable role in the regulation of mitochondrial function. Although we observed a redox‐related function of APE1 in mitochondria, further studies are required to characterize the exact mechanism and its functional or molecular connection to APE1 mtDNA repair activity.
Taken together, the current study characterizes the role of mitochondrial APE1 in PDT and provides evidence for the role of APE1 in redox regulation of mitochondrial transcription but not necessarily a detailed mechanism of this regulation. Since oxidative stress induction is the common feature shared by most therapeutic strategies for cancer, the present study might have profound implications for sensitizing cells for cancer treatment, including radiotherapy and some chemotherapy.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The authors thank Mrs Yu‐Xin Yang, Miss Ling Liao and Mr Yi Cheng for their kind and excellent technical assistance. This study was supported by the National Natural Science Foundation of China (NSFC) grants to Z.‐Z. Y. (No. 30970865, No. 30571849) and to M.‐X. L. (No. 30900553).
References
- 1. Simeonov A, Kulkarni A, Dorjsuren D et al Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS ONE 2009; 4: e5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta 2007; 1776: 86–107. [DOI] [PubMed] [Google Scholar]
- 3. Kessel D, Sun HH. Enhanced responsiveness to photodynamic therapy‐induced apoptosis after mitochondrial DNA depletion. Photochem Photobiol 1999; 70: 937–40. [PubMed] [Google Scholar]
- 4. Moor AC. Signaling pathways in cell death and survival after photodynamic therapy. J Photochem Photobiol, B 2000; 57: 1–13. [DOI] [PubMed] [Google Scholar]
- 5. Srivastava DK, Berg BJ, Prasad R et alMammalian abasic site base excision repair. Identification of the reaction sequence and rate‐determining steps. J Biol Chem 1998; 273: 21203–9. [DOI] [PubMed] [Google Scholar]
- 6. Tomkinson AE, Bonk RT, Linn S. Mitochondrial endonuclease activities specific for apurinic/apyrimidinic sites in DNA from mouse cells. J Biol Chem 1988; 263: 12532–7. [PubMed] [Google Scholar]
- 7. Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref‐1: more than a passive phenomenon? Antioxid Redox Signal 2005; 7: 367–84. [DOI] [PubMed] [Google Scholar]
- 8. Frossi B, Tell G, Spessotto P, Colombatti A, Vitale G, Pucillo C. H(2)O(2) induces translocation of APE/Ref‐1 to mitochondria in the Raji B‐cell line. J Cell Physiol 2002; 193: 180–6. [DOI] [PubMed] [Google Scholar]
- 9. Li MX, Wang D, Zhong ZY et al Targeting truncated APE1 in mitochondria enhances cell survival after oxidative stress. Free Radic Biol Med 2008; 45: 592–601. [DOI] [PubMed] [Google Scholar]
- 10. Evans AR, Limp‐Foster M, Kelley MR. Going APE over ref‐1. Mutat Res 2000; 461: 83–108. [DOI] [PubMed] [Google Scholar]
- 11. Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell Mol Life Sci 2010; 67: 3589–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aditi Bapat MF, Kelley MR. Going ape as an approach to cancer therapeutics. Antioxid Redox Signal 2009; 11: 651–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang ZZ, Li MX, Zhang YS et al Knock down of the dual functional protein apurinic/apyrimidinic endonuclease 1 enhances the killing effect of hematoporphrphyrin derivative‐mediated photodynamic therapy on non‐small cell lung cancer cells in vitro and in a xenograft model. Cancer Sci 2010; 101: 180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther 2004; 3: 679–86. [PubMed] [Google Scholar]
- 15. Li M, Zhong Z, Zhu J et al Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J Biol Chem 2010; 285: 14871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perkins CL, Fang G, Kim CN, Bhalla KN. The role of Apaf‐1, caspase‐9, and bid proteins in etoposide‐ or paclitaxel‐induced mitochondrial events during apoptosis. Cancer Res 2000; 60: 1645–53. [PubMed] [Google Scholar]
- 17. Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 2001; 153: 319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vascotto C, Bisetto E, Li M et al Knock‐in reconstitution studies reveal an unexpected role of Cys‐65 in regulating APE1/Ref‐1 subcellular trafficking and function. Mol Biol Cell 2011; 22: 3887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vascotto C, Cesaratto L, Zeef LA et al Genome‐wide analysis and proteomic studies reveal APE1/Ref‐1 multifunctional role in mammalian cells. Proteomics 2009; 9: 1058–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii N. Role of oxidative stress from mitochondria on aging and cancer. Cornea 2007; 26: S3–9. [DOI] [PubMed] [Google Scholar]
- 21. Vascotto C, Fantini D, Romanello M et al APE1/Ref‐1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol Cell Biol 2009; 29: 1834–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007; 7: 39–44. [DOI] [PubMed] [Google Scholar]
- 23. Canugovi C, Maynard S, Bayne AC et al The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 2010; 9: 1080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res 2008; 643: 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maher RL, Bloom LB. Pre‐steady‐state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J Biol Chem 2007; 282: 30577–85. [DOI] [PubMed] [Google Scholar]
- 26. Chattopadhyay R, Wiederhold L, Szczesny B et al Identification and characterization of mitochondrial abasic (AP)‐endonuclease in mammalian cells. Nucleic Acids Res 2006; 34: 2067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]