

Abstract
DNA‐damaging strategies, such as radiotherapy and the majority of chemotherapeutic therapies, are the most frequently used non‐surgical anti‐cancer therapies for human cancers. These therapies activate DNA damage/replication checkpoints, which induce cell‐cycle arrest to provide the time needed to repair DNA damage. Due to genetic defect(s) in the ATM (ataxia‐telangiectasia mutated)‐Chk2‐p53 pathway, an ATR (ATM‐ and Rad3‐related)‐Chk1‐Cdc25 route is the sole checkpoint pathway in a majority of cancer cells. Chk1 inhibitors are expected to selectively induce the mitotic cell death (mitotic catastrophe) of cancer cells. However, recent new findings have pointed out that Chk1 is essential for the maintenance of genome integrity even during unperturbed cell‐cycle progression, which is controlled by a variety of protein kinases. These observations have raised concerns about a possible risk of Chk1 inhibitors on the clinics. In this review, we summarize recent advances in Chk1 regulation by phosphorylation, and discuss Chk1 as a molecular target for cancer therapeutics. (Cancer Sci 2012; 103: 1195–1200)
Cellular genetic information (DNA) is constantly being attacked by exogenous (e.g. chemicals, ultraviolet [UV], ionizing radiation [IR]) or endogenous (e.g. free radicals or by‐products of intracellular metabolism) genotoxic agents. These DNA alternations activate DNA damage checkpoints, which inhibit cell cycle progression from G1 to S (the G1/S checkpoint), during DNA replication (the intra‐S phase checkpoint), or from G2 to mitosis (the G2/M checkpoint). The cell cycle arrest allows cells to repair damaged DNA, thus preventing the transmission of genetic errors to daughter cells. When a repair is unsuccessful owing to excessive DNA damage or a genetic defect, the checkpoint activation facilitates the elimination of damaged cells from the proliferation pool through the induction of semi‐permanent cell cycle arrest (senescence) or cell death (apoptosis). DNA damage is detected by several signaling pathways that ultimately inhibit cyclin‐dependent kinases (Cdks).1, 2, 3, 4, 5
One important pathway is the ataxia‐telangiectasia mutated (ATM)‐checkpoint kinase 2 (Chk2)‐p53 pathway.1, 2, 3, 4, 5, 6, 7 Ataxia‐telangiectasia mutated is activated mainly in response to a double‐strand break1, 2, 3, 4, 5, 6, 7, 8 or oxidative stress.9 Ataxia‐telangiectasia mutated also induces Chk2‐Thr68 phosphorylation, which triggers the catalytic activation of Chk2.1, 2, 3, 4, 5, 6, 7, 8, 10 The activation of an ATM‐Chk2 kinase cascade stabilizes p53 (a transcription factor) through the phosphorylation.1, 2, 3, 4, 5, 6, 7, 8, 10, 11 Stabilized p53 induces cell cycle arrest (including senescence) or apoptosis by regulating the expression of several proteins such as the Cdk inhibitor p21 or the pro‐apoptotic BAX and PUMA proteins.1, 2, 3, 4, 5, 6, 7, 8, 12 This signaling pathway not only prevents cell cycle‐progression from the G1 to the S phase but also works as tumor suppressors.1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 12
On the other hand, an ATM‐ and Rad3‐related (ATR)‐Chk1‐Cdc25 pathway plays critical roles mainly in the intra‐S phase or G2/M checkpoint.1, 2, 3, 4, 5, 6, 7, 8, 13, 14 In response to stalled replication or damaged DNA (induced by UV, IR, DNA‐damaging reagents, etc.), ATR is activated at a replication protein A (RPA)‐coated single strand DNA (ssDNA; Fig. 1).1, 2, 3, 4, 5, 6, 13, 14, 15 The activated ATR phosphorylates Chk1 at Ser317 and Ser345.1, 2, 3, 4, 5, 16 This phosphorylation induces both catalytic activation16, 17, 18 and nuclear accumulation19 of Chk1 (Table 1). Chk1 then phosphorylates and thereby inactivates Cdc25 proteins, a family of dual‐specificity protein phosphatases (Fig. 2a).1, 2, 3, 4, 5, 6, 7, 8, 13, 14, 15, 20, 21 Since Cdc25 phosphatases activate Cdks (Fig. 2a),20, 21, 22, 23, 24 Cdc25 inhibition by Chk1 induces cell‐cycle arrest,1, 2, 3, 4, 5, 6, 7, 8, 13, 14, 15, 25 which provides time to repair damaged DNA lesions.26
Figure 1.
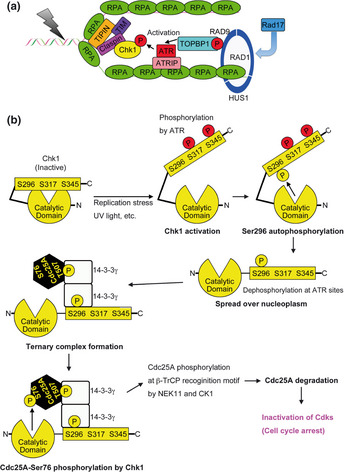
Model for ATR‐Chk1‐Cdc25A pathway in DNA damage/replication checkpoint. (a) ATR‐Chk1 pathway is activated in ssDNA lesions. (b) Chk1 phosphorylation shift from Ser317 and Ser345 (ATR sites) to Ser296 plays critical roles in Cdc25A degradation. See text for details.
Table 1.
Chk1 phosphorylation sitesa and physiological roles of these phosphorylations
| Phosphorylation site | Responsible kinase | Binding partner(s) | Stimuli | Possible function |
|---|---|---|---|---|
| Ser280 | p90 RSK | Growth factor stimulation | Chk1 transport from cytoplasm to nucleus | |
| p90 RSK | UV irradiation | Acceleration of Chk1 activation processes | ||
| Ser286, Ser301 | Cdk1 | Crm‐1/exportin 1b | G2/M transition | Chk1 transport from nucleus to cytoplasm |
| Ser296 | Chk1 | 14‐3‐3γ | Checkpoint responses |
Chk1 translocation to nucleoplasm Cdc25A phosphorylation/degradation |
| Ser317, Ser345 | ATR | Checkpoint responses | Chk1 catalytic activation | |
| Ser345 | ATR | 14‐3‐3 β and ζζ | Checkpoint responses | Nuclear accumulation of Chk1 |
All phosphorylation sites indicated above are located in C‐terminal, regulatory region (outside of the catalytic domain) of Chk1.
Ser286/Ser301 phosphorylation may induce conformational change of Chk1, which promotes Chk1 binding to Crm‐1/exportin 1.
Figure 2.
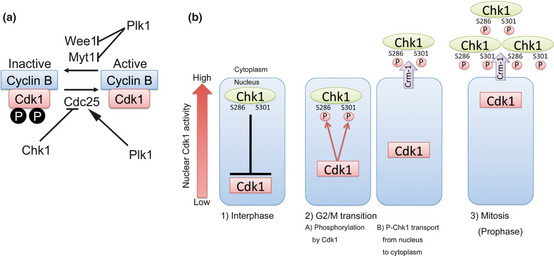
A positive feedback loop between Chk1 and Cyclin‐dependent kinase 1 (Cdk1) at G2/M transition. (a) The control of Cyclin B/Cyclin‐dependent kinase 1 (Cdk1) activity. Myt1 and Wee1 kinases phosphorylate Cyclin‐dependent kinase 1 (Cdk1) at Thr14 and Tyr15, inhibiting Cyclin B/Cyclin‐dependent kinase 1 (Cdk1) activity. The Thr14 and Tyr15 phosphorylations are antagonized by Cdc25 family phosphatases. The activities of other Cyclin‐dependent kinases (Cdks) are also controlled by the balance of Myt1/Wee1 and Cdc25 activities. Chk1 phosphorylates and inhibits Cdc25A, Cdc25B, and Cdc25C. Thus, Chk1 works as a negative regulator of Cdks including Cyclin B/Cyclin‐dependent kinase 1 (Cdk1). On the other hand, Plk1 phosphorylates and activates Cdc25C. Plk1 also phosphorylates and inactivates Myt1 and Wee1 kinases. Thus, Plk1 participates in initial activation of Cyclin B/Cyclin‐dependent kinase 1 (Cdk1). (b) Before mitosis (at S or G2 phase), Chk1 (green) has basal activity in unperturbed cells. Since Chk1 (green) is predominantly localized in nucleus (blue) of interphase cells, it would be expected to inhibit Cdc25 phosphatase activity at nucleus (blue) and thus block premature activation of Cyclin‐dependent kinase 1 (Cdk1; red) in nuclei of interphase cells. Once initial activation of Cyclin‐dependent kinase 1 (Cdk1; red) occurs (likely by Plk1), Cyclin‐dependent kinase 1 (Cdk1; red) starts to phosphorylate Chk1 at Ser286 and Ser301. This phosphorylation induces Chk1 translocation from nucleus to cytoplasm by Crm‐1/exportin 1 (purple)‐mediated transport. Elimination of Chk1 kinase activity from nucleus triggers more activation of Cyclin‐dependent kinase 1 (Cdk1; red) in nucleus and thereby promotes mitosis. See text for details.
Only ATR was long believed to functionally regulate Chk1. It has become increasingly evident, however, that a variety of protein kinases phosphorylate and thereby regulate Chk1 (Table 1). Such new evidence displays the Chk1 function not only in checkpoint responses but also during unperturbed cell‐cycle progression (Table 1). This review will focus on the recent advances in our knowledge of Chk1 regulation by phosphorylation. In addition, we will discuss Chk1 as a molecular target for anti‐cancer therapies.
Chk1 Phosphorylations in DNA Replication/Damage Checkpoints: Importance of Chk1 Autophosphorylation at Ser296
In response to ssDNA generated by a broad spectrum of damaged DNA (e.g. stalled replication forks or UV photoproducts), ATR is activated as follows (summarized in Fig. 1a). Replication protein A complexes rapidly coat the ssDNA lesions and then recruit/activate ATR in a stable complex with ATR‐interacting protein (ATRIP).13, 14, 15, 27 Simultaneously, RPA binds to a clamp loader RAD17, which loads the proliferating cell nuclear antigen (PCNA)‐related RAD9‐HUS1‐RAD1 (9‐1‐1) heterotrimer.13, 14, 15, 28, 29 DNA topoisomerase II binding protein 1 (TOPBP1) is then recruited to ssDNA lesions through the molecular interaction of its breast cancer susceptibility gene 1 (BRCA1) carboxyl‐terminal (BRCT) domains with RAD9 phosphorylated by ATR.13, 14, 15, 30, 31, 32 Efficient activation of ATR requires a molecular interaction between ATR and TOPBP1.13, 14, 15, 32 Active ATR phosphorylates numerous substrates including Chk1. For an efficient phosphorylation of Chk1 by ATR, Chk1 needs to form a complex with the mediator protein Claspin.33, 34, 35, 36 The complex of a Timeless (TIM) and TIM‐interacting protein (TIPIN)36 facilitates the accumulation of Chk1 and Claspin to ssDNA lesions through a TIPIN association with RPA2.37
The cell cycle arrest in an ATR signaling cascade is established mainly through the inhibition of Cdc25 activity by Chk1 (Fig. 2a).1, 2, 3, 4, 5, 6, 7, 8, 13, 14, 15, 25 However, there remained two large questions about a signaling from Chk1 to Cdc25. Based on the localization of Chk1 activators (ATR, Claspin etc.), Chk1 is considered to be initially activated at the sites of DNA damage (Fig. 1a). In support of this idea, Ser345‐phosphorylated Chk1 is predominantly observed at the area where ATR is activated.38 On the other hand, Cdc25 is distributed diffusely in the nucleus.39 Thus, the question remains as to how Chk1 spreads over the nucleus to phosphorylate Cdc25. Another question is the role of 14‐3‐3 proteins in a signaling from Chk1 to Cdc25. Studies in fission yeast first suggested that a 14‐3‐3 protein RAD24 participates in a signal from Chk1 to Cdc25.21, 40 In mammalian cells, three isoforms have been identified: Cdc25A, Cdc25B, and Cdc25C.21 From the viewpoint of evolutionary conservation, Cdc25B and Cdc25C have received more attention.21, 40 Chk1 can phosphorylate those isoforms on conserved serine residues, creating phosphoserine‐binding sites for 14‐3‐3.21, 40 Moreover, the binding of 14‐3‐3 functionally inhibits Cdc25B and Cdc25C.21, 40 However, these sites are likely to be phosphorylated mainly by other kinases in mammalian cells, and the level of phosphorylation does not dramatically change after DNA damage.21, 40, 41, 42, 43 Genetic studies have also revealed that normal cell cycle progression and checkpoint responses were observed in mice and cells lacking Cdc25B and/or Cdc25C.44, 45, 46 In sharp contrast, Cdc25A −/− mice47 die early in embryogenesis, like ATR −/−(48, 49 ) or Chk1 −/−(50, 51) mice. Chk1 generates 14‐3‐3 binding sites on Cdc25A,52 suggesting the possibility that 14‐3‐3 proteins may modulate a signaling from Chk1 to Cdc25A. However, the role of such a binding has remained poorly understood.
The answer to these two questions lies in the novel Chk1 phosphorylation during the checkpoint responses (Fig. 1b, Table 1). We found that Chk1‐Ser296 is autophosphorylated after Chk1 phosphorylation at Ser317 and Ser345 by ATR (which implies Chk1 activation).38 After Chk1‐Ser296 autophosphorylation, Ser317 and Ser345 are rapidly dephosphorylated by phosphatases such as protein phosphatase 2A (PP2A),38, 53 or PPM1D (also called Wip1 or PP2Cδ).54 Since Ser296‐phosphorylated Chk1 is diffusely observed in the nucleus (probably throughout the nucleoplasm),38 the phosphorylation shift from ATR sites to Ser296 plays an important role in the spread of Chk1 signals. That shift also induces an ordered binding of Chk1 to specific 14‐3‐3 subtypes. β and ζ subtypes of 14‐3‐3 bind Chk1 in a Ser345 phosphorylation‐dependent manner.19 On the other hand, Ser296‐phosphorylated Chk1 binds to 14‐3‐3 in a γ subtype‐specific manner.38 Dimer formation by 14‐3‐3 proteins facilitates their function as platforms for the generation of complexes between Chk1 and Cdc25A. This complex formation is required for a Chk1‐induced Cdc25A phosphorylation at Ser7638 (a rate‐limiting phosphorylation site for Cdc25A degradation),21, 55, 56 whereas the Cdc25A phosphorylation at Thr507 (a phosphorylation site required for 14‐3‐3 binding)21, 52 occurs in the complex‐formation‐independent manner.38 Ser76 phosphorylation on Cdc25A facilitates its phosphorylation at Ser79, Ser82 and Ser88 by NIMA (never in mitosis gene A)‐related kinase 11 (NEK11)57 and casein kinase 1 (CK1).58 The phosphorylations by NEK11 and CK1 generate a β‐transducin repeated‐containing protein (β‐TrCP; E3 ligase) recognition motif (phosphodegron) on Cdc25A and then lead to Cdc25A degradation in a proteasome‐dependent manner.21 Cdc25A degradation prevents a premature activation of Cdks (Fig. 2a) and induces cell cycle arrest after DNA damage. Therefore, in mammalian checkpoint systems, Chk1‐Ser296 autophosphorylation triggers a signaling from Chk1 to Cdc25A not only through the spread of Chk1 throughout the nucleus, but also through the complex formation between Chk1 and Cdc25A on 14‐3‐3γ (Fig. 1b, Table 1).
Chk1 Phosphorylations in Unperturbed Cell‐Cycle Progression
Accumulating evidence suggests that Chk1 also plays critical roles in normal (unperturbed) cell‐cycle progression. Essential functions of Chk1 during development are well documented by the early embryonic lethality of Chk1‐knockout mice.50, 51 Chk1 is considered to monitor replication forks even during normal S‐phase progression.50, 59, 60 Chk1 activity is especially likely to be necessary in preventing late‐origin firing and then irreversible replication fork collapse.60
Outside the S‐phase control, Chk1 is also known to restrain mitosis at the G2 phase without exogenously introducing DNA damage.61, 62, 63 Since Chk1 has a basal activity in interphase,61, 62, 63, 64, 65 it would be expected to inhibit Cdc25 phosphatase activity. Thus, Chk1 indirectly prevents the unscheduled activation of a mitosis‐inducer kinase, Cyclin‐dependent kinase 1 (Cdk1; we used Cyclin‐dependent kinase 1 to clearly distinguish Chk1 in the text) even under unperturbed condition (Fig. 2a).61, 62, 63, 64, 65 In other words, cells have to activate Cyclin‐dependent kinase 1 to enter into mitosis. Accumulating evidence has suggested that Polo‐like kinase 1 (Plk1) is likely to play critical roles in initial activation of Cyclin‐dependent kinase 1 through Cdc25 activation and Wee1/Myt1 inhibition (Fig. 2a).25, 66, 67 We have recently established that novel Chk1 phosphorylation also participates in the activation process of Cyclin‐dependent kinase 1 at the G2/M transition. Once initial activation of Cyclin‐dependent kinase 1 occurs (likely by Plk1; Fig. 2a), it starts to phosphorylate Ser286 and Ser301 on Chk1 at the G2/M transition (Fig. 2b, Table 1).68 This phosphorylation facilitates Chk1 translocation from the nucleus to the cytoplasm by Crm‐1/exportin 1.62 Initial activation of Cyclin‐dependent kinase 1 triggers Chk1 phosphorylation at Ser286 and Ser301, which promotes the cytoplasmic sequestration of Chk1. This elimination of Chk1 kinase activity from nucleus triggers more activation of Cyclin‐dependent kinase 1 in nucleus. This positive feedback loop promotes cell cycle progression from G2 to the M phase (Fig. 2b, Table 1). Chk1 activity may also be also modulated by Plk1, which generates β‐TrCP phosphodegron on Claspin (an essential activator of Chk133, 34, 35, 36), thereby inducing Claspin degradation.4, 25, 69, 70
Chk1 is known to constantly shuttle between cytoplasm and nucleus (Fig. 3).17, 19 Since Chk1 is localized predominantly in the nucleus from G1 to G2 phase (during an interphase),62 there may exist a mechanism by which Chk1 prefers to localize in the nucleus during the cell‐cycle progression. We have recently demonstrated that Chk1 is phosphorylated at Ser280 by p90 ribosomal S6 kinase (p90 RSK) downstream of Ras‐mitogen‐activated protein kinase (MAPK) cascade in response to growth‐factor stimulation (Fig. 3, Table 1).71 Chk1‐Ser280 phosphorylation promotes Chk1 translocation from the cytoplasm to the nucleus.71 Since Chk1 is activated in the nucleus,19, 63, 72 such nuclear accumulation is likely to be of great use in the preparation for DNA damage response. In support of this hypothesis, Ser280 phosphorylation accelerates Chk1 activation processes (the phosphorylation by ATR and the autophosphorylation; see Table 1) after UV irradiation.71
Figure 3.
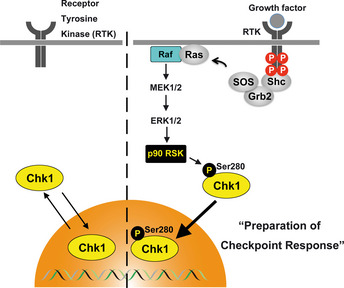
Potential role of Chk1 phosphorylation at Ser280 by p90 RSK. Chk1 constantly shuttles between cytoplasm and nucleus. Without stimulation of receptor tyrosine kinase (RTK) with growth factor, Chk1 is diffusely localized both in the cytoplasm and in the nucleus. Following stimulation of RTK with growth factor, p90 RSK is activated downstream of mitogen‐activated protein kinase (MAPK) cascade and then phosphorylates Chk1 specifically at Ser280. Ser280 phosphorylation promotes nuclear retention of Chk1. Thus, Chk1 is localized predominantly in the nucleus during the cell proliferation. Since Chk1 is activated in nucleus, such nuclear accumulation is likely to be of great use in preparation for the DNA damage response.
In the conditional Chk1 haploinsufficiency mouse model, Chk1 +/− mammary epithelia exhibit an inappropriate entry into S phase (Chk1 failure is likely in the G1 phase), an accumulation of spontaneous DNA damage during DNA replication (a failure in S phase progression), and a failure to restrain mitotic entry (a failure at the G2/M transition).73 Therefore, Chk1 activities in unperturbed cell‐cycle progression are also essential for the maintenance of genome integrity during cell proliferation.
Targeting Chk1 in Cancer Therapy: Pros and Cons of Chk1 Inhibitors
Cancer cell death induced by excessive DNA damage is a general strategy of various cancer therapies, such as radiotherapy and the majority of chemotherapeutic therapies. Such treatments target genetically instable cancer cells, but also damage normal cells, especially highly proliferative cells (such as epithelia in the gastrointestinal tract, hair follicles, and bone marrow). Those undesirable injuries are a major problem in clinics and limit effectiveness in curing cancer. Therefore, it is of great importance to understand the differences of DNA damage/replication checkpoint responses between normal and cancerous cells.
Normal cells have two major DNA damage response pathways, ATM‐Chk2‐p53 and ATR‐Chk1‐Cdc25 (see Introduction). Once the ATM‐Chk2‐p53 pathway is impaired, chromosomal instability (CIN; also called genomic instability) results, which can foster the evolution of cancer cells. In support of this notion, hereditary ATM, CHEK2, or TP53 mutations have been identified in cancer predisposition syndromes.3, 74 In sporadic human cancers, defects in the ATM‐Chk2‐p53 pathway frequently occur due to mutations in or deletions of ATM, CHEK2, or TP53.1, 2, 3, 4, 5, 6, 7, 8, 10, 11, 12 The ATR‐Chk1‐Cdc25 pathway is therefore the sole DNA damage checkpoint route in a majority of cancer cells.3, 74 In fact, Chk1 inhibition induces a catastrophic CIN in cancer cells, which results in mitotic cell death (mitotic catastrophe).3, 74 Since Chk1 inhibitors have been demonstrated to sensitize tumors to radiation or other existing chemotherapeutic agents (various DNA‐damaging agents or anti‐metabolites) in preclinical models, a number of Chk1 inhibitors are now being evaluated in Phase I or II clinical trials.3, 74
In this strategy, whether the ATM‐Chk2‐p53 pathway can compensate for the Chk1 function in normal cells, especially under unperturbed conditions is an important consideration in using Chk1 inhibitors in clinics. Chk1 −/− mice exhibit early embryonic lethality,50, 51 suggesting that Chk1 inhibition can not be completely compensated at least for its development. However, it is more important to understand the effects of Chk1 inhibition in adult mice. In this respect, the mouse model of RNA interference (RNAi)‐mediated gene knockdown may recapitulate the clinical effects of anti‐cancer drugs more clearly.75, 76, 77 In the case of Plk1 (a potent molecular target for cancer), gene‐knockout and ‐knockdown mice exhibit different phenotypes; Plk1 null‐mice are embryonically lethal,78 whereas adverse effects are very rare in the Plk1‐silencing adult mice.75 Thus, information about the effects of Chk1‐ or Cdc25A‐silencing in a mouse model as well as about the clinical trials of Chk1 or Cdc25A inhibitors will be required to evaluate Chk1 or Cdc25A as a molecular target for cancer.
Conclusions
Recent progress discussed in this review illustrates that the Chk1 function is regulated more dynamically than was previously thought. In a variety of cellular events, Chk1 is phosphorylated by several kinases, which change the subcellular localization and binding partners of Chk1. These recent findings provide new insights into the biological function of Chk1 not only in the checkpoint responses but also in normal cell cycle progression, raising concerns about undesirable effects of Chk1 inhibitors on normal cells. At present, the underlying mechanisms of Chk1 function in normal (unperturbed) cell‐cycle progression remain poorly understood. For example, it is largely unknown whether the pathways upstream/downstream of Chk1 differ between perturbed and unperturbed conditions. If those are different molecules between two conditions, it may be beneficial to pharmacologically target the protein(s) acting specifically in the checkpoint pathway (under a perturbed condition). Therefore, the development of therapies that exploit the cancer‐specific checkpoint defects calls for a more advanced molecular understanding of the Chk1‐mediated pathway (especially in normal cell‐cycle progression), which will open additional avenues for individualized cancer therapies.
Disclosure Statement
Ichiro Izawa and Masaki Inagaki were supported by the Takeda Research Foundation in 2011. The rest of the authors have no conflict of interest.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, and from the Ministry of Education, Science, Technology, Sports and Culture of Japan; by a Grant‐in‐Aid for the Third‐Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health and Welfare, Japan; by the Uehara Memorial Foundation; by the Astellas Foundation for Research on Metabolic Disorders; by the Naito Foundation; by the Daiichi‐Sankyo Foundation of Life Science and by the Takeda Science Foundation. We sincerely apologize to all our colleagues whose work we were unable to cite due to space limitations.
References
- 1. Jackson SP, Bartek J. The DNA‐damage response in human biology and disease. Nature 2009; 461: 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Medema RH, Macurek L. Checkpoint control and cancer. Oncogene 2011; doi: 10.1038/onc.2011.451. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 4. Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19: 238–45. [DOI] [PubMed] [Google Scholar]
- 5. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25: 409–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double‐strand break repair. Philos Trans R Soc Lond B Biol Sci 2011; 366: 3562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis 2006; 21: 3–9. [DOI] [PubMed] [Google Scholar]
- 8. McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol 2004; 16: 629–33. [DOI] [PubMed] [Google Scholar]
- 9. Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330: 517–21. [DOI] [PubMed] [Google Scholar]
- 10. McGowan CH. Checking in on Cds1 (Chk2): a checkpoint kinase and tumor suppressor. BioEssays 2002; 24: 502–11. [DOI] [PubMed] [Google Scholar]
- 11. Appella E, Anderson CW. Post‐translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001; 268: 2764–72. [DOI] [PubMed] [Google Scholar]
- 12. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–12. [DOI] [PubMed] [Google Scholar]
- 13. Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 2011; 36: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9: 616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J 2011; 436: 527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao H, Piwnica‐Worms H. ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21: 4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katsuragi Y, Sagata N. Regulation of Chk1 kinase by autoinhibition and ATR‐mediated phosphorylation. Mol Biol Cell 2004; 15: 1680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walker M, Black EJ, Oehler V, Gillespie DA, Scott MT. Chk1 C‐terminal regulatory phosphorylation mediates checkpoint activation by de‐repression of Chk1 catalytic activity. Oncogene 2009; 28: 2314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14‐3‐3 binding following phosphorylation on Ser‐345. J Biol Chem 2003; 278: 25207–17. [DOI] [PubMed] [Google Scholar]
- 20. Neely KE, Piwnica‐Worms H. Cdc25A regulation: to destroy or not to destroy–is that the only question? Cell Cycle 2003; 2: 455–7. [PubMed] [Google Scholar]
- 21. Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 2007; 7: 495–507. [DOI] [PubMed] [Google Scholar]
- 22. Nurse P. Cyclin dependent kinases and cell cycle control (nobel lecture). ChemBioChem 2002; 3: 596–603. [DOI] [PubMed] [Google Scholar]
- 23. Doree M, Hunt T. From Cdc2 to Cdk1: when did the cell cycle kinase join its cyclin partner? J Cell Sci 2002; 115: 2461–4. [DOI] [PubMed] [Google Scholar]
- 24. Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2: 21–32. [DOI] [PubMed] [Google Scholar]
- 25. Lindqvist A, Rodriguez‐Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009; 185: 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9: 297–308. [DOI] [PubMed] [Google Scholar]
- 27. Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 2003; 300: 1542–8. [DOI] [PubMed] [Google Scholar]
- 28. Zou L, Liu D, Elledge SJ. Replication protein A‐mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA 2003; 100: 13827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 2003; 1: E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee J, Kumagai A, Dunphy WG. The Rad9‐Hus1‐Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 2007; 282: 28036–44. [DOI] [PubMed] [Google Scholar]
- 31. Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9‐Hus1‐Rad1 (9‐1‐1) clamp activates checkpoint signaling via TopBP1. Genes Dev 2007; 21: 1472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR‐ATRIP complex. Cell 2006; 124: 943–55. [DOI] [PubMed] [Google Scholar]
- 33. Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 2000; 6: 839–49. [DOI] [PubMed] [Google Scholar]
- 34. Chini CC, Chen J. Claspin, a regulator of Chk1 in DNA replication stress pathway. DNA Repair 2004; 3: 1033–7. [DOI] [PubMed] [Google Scholar]
- 35. Kumagai A, Kim SM, Dunphy WG. Claspin and the activated form of ATR‐ATRIP collaborate in the activation of Chk1. J Biol Chem 2004; 279: 49599–608. [DOI] [PubMed] [Google Scholar]
- 36. Errico A, Costanzo V. Differences in the DNA replication of unicellular eukaryotes and metazoans: known unknowns. EMBO Rep 2010; 11: 270–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kemp MG, Akan Z, Yilmaz S et al Tipin‐replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem 2010; 285: 16562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kasahara K, Goto H, Enomoto M, Tomono Y, Kiyono T, Inagaki M. 14‐3‐3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J 2010; 29: 2802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bekker‐Jensen S, Lukas C, Kitagawa R et al Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 2006; 173: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mohammad DH, Yaffe MB. 14‐3‐3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair 2009; 8: 1009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng CY, Graves PR, Ogg S et al C‐TAK1 protein kinase phosphorylates human Cdc25C on serine 216 and promotes 14‐3‐3 protein binding. Cell Growth Differ 1998; 9: 197–208. [PubMed] [Google Scholar]
- 42. Bulavin DV, Higashimoto Y, Popoff IJ et al Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001; 411: 102–7. [DOI] [PubMed] [Google Scholar]
- 43. Schmitt E, Boutros R, Froment C, Monsarrat B, Ducommun B, Dozier C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J Cell Sci 2006; 119: 4269–75. [DOI] [PubMed] [Google Scholar]
- 44. Ferguson AM, White LS, Donovan PJ, Piwnica‐Worms H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol 2005; 25: 2853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lincoln AJ, Wickramasinghe D, Stein P et al Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat Genet 2002; 30: 446–9. [DOI] [PubMed] [Google Scholar]
- 46. Chen MS, Hurov J, White LS, Woodford‐Thomas T, Piwnica‐Worms H. Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol Cell Biol 2001; 21: 3853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ray D, Terao Y, Nimbalkar D et al Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res 2007; 67: 6605–11. [DOI] [PubMed] [Google Scholar]
- 48. Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- 49. de Klein A, Muijtjens M, van Os R et al Targeted disruption of the cell‐cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10: 479–82. [DOI] [PubMed] [Google Scholar]
- 50. Takai H, Tominaga K, Motoyama N et al Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev 2000; 14: 1439–47. [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Q, Guntuku S, Cui XS et al Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14: 1448–59. [PMC free article] [PubMed] [Google Scholar]
- 52. Chen MS, Ryan CE, Piwnica‐Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14‐3‐3 binding. Mol Cell Biol 2003; 23: 7488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leung‐Pineda V, Ryan CE, Piwnica‐Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1‐regulated protein phosphatase 2A circuit. Mol Cell Biol 2006; 26: 7529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 2005; 19: 1162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jin J, Shirogane T, Xu L et al SCFbeta‐TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev 2003; 17: 3062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 2004; 23: 2050–6. [DOI] [PubMed] [Google Scholar]
- 57. Melixetian M, Klein DK, Sorensen CS, Helin K. NEK11 regulates CDC25A degradation and the IR‐induced G2/M checkpoint. Nat Cell Biol 2009; 11: 1247–53. [DOI] [PubMed] [Google Scholar]
- 58. Honaker Y, Piwnica‐Worms H. Casein kinase 1 functions as both penultimate and ultimate kinase in regulating Cdc25A destruction. Oncogene 2010; 29: 3324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Syljuasen RG, Sorensen CS, Hansen LT et al Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25: 3553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Maya‐Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 2007; 26: 2719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kramer A, Mailand N, Lukas C et al Centrosome‐associated Chk1 prevents premature activation of cyclin‐B‐Cdk1 kinase. Nat Cell Biol 2004; 6: 884–91. [DOI] [PubMed] [Google Scholar]
- 62. Enomoto M, Goto H, Tomono Y et al Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol Chem 2009; 284: 34223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsuyama M, Goto H, Kasahara K et al Nuclear Chk1 prevents premature mitotic entry. J Cell Sci 2011; 124: 2113–9. [DOI] [PubMed] [Google Scholar]
- 64. Sorensen CS, Syljuasen RG, Falck J et al Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation‐induced accelerated proteolysis of Cdc25A. Cancer Cell 2003; 3: 247–58. [DOI] [PubMed] [Google Scholar]
- 65. Shimada M, Niida H, Zineldeen DH et al Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage‐induced transcriptional repression. Cell 2008; 132: 221–32. [DOI] [PubMed] [Google Scholar]
- 66. Macurek L, Lindqvist A, Lim D et al Polo‐like kinase‐1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455: 119–23. [DOI] [PubMed] [Google Scholar]
- 67. Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320: 1655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shiromizu T, Goto H, Tomono Y et al Regulation of mitotic function of Chk1 through phosphorylation at novel sites by cyclin‐dependent kinase 1 (Cdk1). Genes Cells 2006; 11: 477–85. [DOI] [PubMed] [Google Scholar]
- 69. Mailand N, Bekker‐Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 2006; 23: 307–18. [DOI] [PubMed] [Google Scholar]
- 70. Peschiaroli A, Dorrello NV, Guardavaccaro D et al SCFbetaTrCP‐mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 2006; 23: 319–29. [DOI] [PubMed] [Google Scholar]
- 71. Li P, Goto H, Kasahara K et al P90 RSK arranges Chk1 in the nucleus for monitoring of genomic integrity during cell proliferation. Mol Biol Cell 2012; doi: 10.1091/mbc.E11-10-0883. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sanchez Y, Wong C, Thoma RS et al Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997; 277: 1497–501. [DOI] [PubMed] [Google Scholar]
- 73. Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004; 6: 45–59. [DOI] [PubMed] [Google Scholar]
- 74. Ma CX, Janetka JW, Piwnica‐Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011; 17: 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Raab M, Kappel S, Kramer A et al Toxicity modelling of Plk1‐targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat Commun 2011; 2: 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov 2006; 5: 741–54. [DOI] [PubMed] [Google Scholar]
- 77. Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell 2002; 108: 135–44. [DOI] [PubMed] [Google Scholar]
- 78. Lu LY, Wood JL, Minter‐Dykhouse K et al Polo‐like kinase 1 is essential for early embryonic development and tumor suppression. Mol Cell Biol 2008; 28: 6870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]