

Abstract
Thromboxane A 2 (TXA 2) is a prostanoid formed by thromboxane synthase using the cyclooxygenase product, prostaglandin H(2), as the substrate. TXA 2 was shown to enhance tumor metastasis, but the underlying mechanism remains unclear. B16F1 melanoma cells were intravenously injected into TXA 2 receptor (TP) knockout mice (TP −/−) and wild‐type littermates (WT). TP −/− showed a reduction in B16F1 lung colonization and mortality rate, which were associated with a decreased number of platelets. Platelet activation as assessed by P‐selectin expression was suppressed in TP −/−. A selective P‐selectin neutralizing antibody decreased the lung colonization in WT mice, but not in TP −/−. The expression of P‐selectin glycoprotein ligand‐1 in B16F1 and HUVEC were enhanced by treatment with U46619, a thromboxane analog. The plasma levels of vascular endothelial growth factor (VEGF) and stromal‐derived factor (SDF)‐1 were lower in TP −/−. In TP −/−, the mobilization of progenitor cells expressing CXCR4+ VEGFR1+ from bone marrow and the recruitment of those cells to lung tissues were suppressed. These results suggest that TP signaling plays a critical role in tumor colonization through P‐selectin‐mediated interactions between platelets‐tumor cells and tumor cells‐endothelial cells through the TP signaling‐dependent production of VEGF and SDF‐1, which might be involved in the mobilization of VEGFR1+ CXCR4+ cells. Blockade of TP signaling might be useful in the treatment of tumor metastasis. (Cancer Sci 2012; 103: 700–707)
Tumor metastasis is responsible for over 90% of cancer‐associated mortality, yet the mechanism of tumor metastasis remains poorly understood. Tumor metastasis to distant tissues depends on interactions between tumor cells and the host microenvironment within the circulation and target tissues. This involves blood cells, components of the coagulation system, stromal cells and the extracellular matrix. Cells within the microvasculature that contribute to metastasis are endothelial cells, platelets, lymphocytes, macrophages, fibroblasts and bone marrow‐derived progenitor cells.1
Platelets have been implicated in the development of tumor metastasis.2, 3, 4, 5, 6 Thrombocytosis has been associated with advanced, often metastatic, stages of cancer and with poor prognosis in a variety of tumors. Suppression of platelet–tumor cell association by antiplatelet agents or anticoagulants potently inhibits experimental metastasis.7, 8 Interactions between platelets and tumor cells facilitate tumor cell arrest at the endothelium and assist tumor cell survival within the bloodstream, with the subsequent formation of experimental metastasis.3, 9 The platelet–tumor cell and tumor cells‐endothelial cells aggregates might supply tumor cells with critical stimulatory growth factors and cytokines, which are released on platelet activation.10, 11
Thromboxane A2 (TXA2) is a potent stimulator of platelet activation and aggregation, and of vascular constriction, and exerts its biological activity by binding to a G protein‐coupled specific receptor, the thromboxane prostanoid receptor (TP). Thromboxane synthase (TXS) and its product, TXA2, have been shown to promote not only tumor proliferation, invasion and angiogenesis in a variety of tumor entities,12, 13, 14 but also tumor metastasis.15, 16, 17 TXS expression is associated with angiogenesis and metastasis in non‐small cell lung cancer patients.18 TXS inhibitors suppress the formation of experimental metastasis of the lung16 and the liver.19 A potential role for the TP in tumor cell invasion and metastasis has also recently been shown. Increased TP receptor expression has been shown in patients with breast cancer,20 lung cancer,21 bladder cancer22 and prostate cancer.23 The levels of TP expression were linked to poor prognosis in patients with breast cancer, showing that TP might be a prognostic factor for this disease.20
These findings shown that the TXA2–TP signaling pathway in tumor cells is crucial for metastasis. However, the contribution of endogenous TXA2 in platelets as an important part of the tumor metastasis microenvironment remains to be clarified. In the present study, the possible involvement of TP signaling in platelet–tumor cell interaction and tumor cells‐endothelial cells was examined in a model of experimental lung colony formation. Furthermore, we determined TP signaling in promoting colonization through the mobilization of progenitor cells in lung tissue. The present results suggest that TP signaling is required for tumor colonization through the P‐selectin‐mediated interaction of platelets with tumor cells and endothelial cells, and the vascular endothelial growth factor (VEGF) and stromal‐derived factor (SDF)‐1 delivered from activated platelets mediate recruitment of progenitor cells, which might facilitate tumor colonization.
Materials and Methods
Cell lines
B16F1 cells originally isolated from C57Bl/6 mice were cultured at 37°C in RPMI 1640 medium supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2. B16BL‐6 cells were purchased from Riken Brc Cell Bank (RBRV‐RCB2638; Tsukuba, Japan). Retrovirus vector containing GFP and pLEGFP‐N1 (Clontech, Mountain View, CA, USA) was transfected into packaging cells, PT67 (Clontech) using Effectene Transfection Reagent (Qiagen, Tokyo, Japan), then those cells were cultured in Dulbecco's modified medium (SIGMA, Tokyo, Japan) containing 10% of fetal bovine serum (GIBCO, Tokyo, Japan), 1% Streptomycin‐Penicillin (GIBCO) and 0.5 mg/mL of G418 (Roche, Tokyo, Japan), at 37°C with 5% of CO2, for 2 weeks. Retrovirus was recovered from supernatant of the cells, and those RNA and infectious titers were confirmed by RT–PCR and titration with NIH/3T3 cells, respectively.
The resulting 3 × 103 cfu/mL of retrovirus containing GFP, pLEGFP, was infected to 1 × 105 of melanoma cells, B16BL6 were sheeted the previous day, according to the manufacturer's protocol (Clontech), and those cells were cultured in RPMI‐1640 (SIGMA) containing 10% of fetal bovine serum (GIBCO), 1% Streptomycin‐Penicillin (GIBCO) and 0.5 mg/mL of G418 (Roche), at 37°C with 5% of CO2, for 2 weeks. The resulting B16BL6 containing pLEGFP‐N1 was confirmed by fluorescence microscope (Keyence, Osaka, Japan, type BZ‐9000).
Animal and tumor colony formation model
Male C57Bl/6 mice (6–8‐weeks‐old), weighing 25–30 g, were obtained from CLEA Japan (Tokyo, Japan). Male TXA2 receptor knockout mice (TP−/−, 8‐weeks‐old) were developed by us24 and maintained at constant humidity (60 ± 5%) and temperature (20 ± 1°C) on a 12‐h light/dark cycle. All animals were provided food and water ad libitum. All experiments were carried out in accordance with the guidelines for animal experiments of Kitasato University School of Medicine.
B16F1 cells were harvested and washed three times with phosphate‐buffered saline (PBS). The cells were suspended in PBS at a density of 3.0 × 105 cells/mL, and 100 μL of the resulting suspension was injected into the tail vein. On day 21, mice were killed with an excess dose of ether and the lungs were surgically resected. The isolated lungs were fixed with Bouin's solution and the number of surface‐visible metastatic colonies was counted under a light microscope. In some experiments, a P‐selectin antibody (RB40.34, rat IgG1, 30 μg per mouse; BD PharMingen, San Jose, CA, USA) or isotype control antibody (rat IgG) was injected daily into the mice after B16F1 injection.
Peripheral blood analysis
Blood was drawn through the tail vein before and 7, 14 and 21 days after tumor cell injection. Platelet counts were measured by automatic cell counter (MEK‐6450; Nihon Kohden, Tokyo, Japan).
Platelet transfusion
Anesthetized donor wild‐type (WT) mice were bled through a heart puncture into heparinized microtubes. Platelet isolation was carried out according to the method previously reported.25 The platelets were adjusted to 5.0 × 108/mL in PBS. The platelet suspension was injected into the tail vein on the day of tumor cell injection and on days 2 and 5, respectively.
Bleeding time measurements
Bleeding time measurements were carried out according to a previously reported method.26 The bleeding time was defined as the period of time between the incision and the complete cessation of bleeding.
Measurement of plasma levels of VEGF‐A, SDF‐1, proMMP‐9 and stem cell factor
Plasma levels of VEGF‐A, SDF‐1, pro‐MMP‐9 and stem cell factor (SCF) were assessed with specific ELISA kits (R&D Systems, Minneapolis, MN, USA). These experiments were carried out in duplicate.
Flow cytometric analysis
Blood was drawn through the tail vein 1 day after surgery. The white blood cell fraction, including platelets, was obtained by Ficoll separation, and flow cytometric analysis was carried out as described previously.27 Cells were labeled with FITC anti‐mouse CD41 (eBioscience, San Diego, CA, USA) and phycoerythrin (PE)‐labeled anti‐mouse P‐selectin antibodies or FITC labeled anti‐VEGFR1 and PE‐labeled anti‐CXCR4 isotype control antibodies (BD Pharmingen, San Diego, CA, USA) in the presence of the anti‐Fc reception monoclonal antibody 2.4G2 (Becton Dickinson Biosciences, Franklin Lakes, NJ, USA). After washing, cells were analyzed with a FACSC alibur flow cytometer (Becton Dickinson Biosciences) and small cells (with low forward scatter [FSC]) were gated for peripheral blood analysis.
Immunohistochemical studies
For immunofluorescent cytochemistry, lung tissues were immediately fixed with 4% paraformaldehyde in a 0.1‐M phosphate buffer solution (pH 7.4). After fixation, the tissues were dehydrated with a graded ethanol series and then embedded in paraffin. Sections (4 μm) of the paraffin‐embedded tissues were mounted on glass slides, deparaffinized with xyline and placed in cold (4°C) acetone.28, 29
The cryostat sections were blocked with 1% BSA‐PBS and incubated with an anti‐mouse CXCR‐4 antibody (1:200; Lifespan Biosciences, Seattle, WA, USA), an anti‐mouse VEGFR‐1 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), integrin αIIb monoclonal antibody (1:50; Santa Cruz Biotechnology), rabbit anti‐rat VEGF anti‐body (1:500; Santa Cruz Biotechnology) and goat anti‐human CD31 antibody (1:100; Santa Cruz Biotechnology). After washing in PBS, the sections were incubated with Alexa Fluor 488 Goat Anti‐rabbit IgG (1:1000; Molecular Probes, Carlsbad, CA, USA) and Alexa Fluor 568 Goat Anti‐rat IgG (1:1000; Molecular Probes). Negative control staining was carried out by replacing the primary antibodies with 1% BSA‐PBS. Images were captured with a confocal scanning laser microscope (LSM710; Carl Zeiss, Jena, Germany), as described previously.30, 31
Immunohistochemical analysis of platelets deposits around B16BL‐6 cells in early colony formation
On day 7 and 14, mice were killed with an excess dose of ether and the lungs were surgically resected. The primary antibodies used were integrin αIIb monoclonal antibody (1:50; Santa Cruz Biotechnology). Images were captured with a confocal scanning laser microscope (LSM710; Carl Zeiss), as described previously.30, 31
Determination of P‐selectin glycoprotein ligand‐1 mRNA levels in B16F1 cells and Lewis lung carcinoma (LLC) cells using real time PCR
Transcripts encoding P‐selectin glycoprotein ligand‐1 (PSGL‐1) and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) were quantified by real time RT–PCR analysis. After 2‐h treatment with 30 nM U‐46619, B16F1 cells and LLC cells were collected and homogenized with Trizol. The real‐time PCR primers were designed using Primer 3 software (http://primer3.sourceforge.net/) on the basis of data from GenBank. The following primers were used for real‐time PCR: 5′‐GAGAAGATTGCCACCACTGAC‐3′ (sense); and 5′‐GTTGGACGGTCTCTACTGAGG‐3′ (antisense) for PSGL‐1; 5′‐GCCAGAATGTGAATACGTGAG‐3′ (sense); and 5′‐AGCTGCACTGCGAGTTAAAAG‐3′ (antisense) for P‐selectin and 5′‐ACATCAAGAAGGTGGTGAAGC‐3′ (sense); and 5′‐AAGGTGGAAGAGTGGGAGTTG‐3′ (antisense) for GAPDH (Applied Sigma‐Aldrich, Tokyo, Japan).
Determination of PSGL‐1 in B16F1 cells and LLC cells using immunofluorescence analysis
B16F1, LLC and HUVEC were grown from 50% to 60% confluent cultures on Lab‐TekII Chamber slides (Nuno, NY, USA). The monolayers were fixed with 3.7% paraformaldehyde for 10 min, permeabilized in 0.2% Triton X‐100 for 10 min and then incubated with anti PSGL‐1 goat polyclonal antibody (Santa Cruz) overnight. The secondary antibody was Alexa Flour 568‐conjugated donkey anti‐goat IgG purchased from Molecular Probes (Eugene, OR, USA). Sections were then observed using confocal scanning laser microscope.
Determination of PSGL‐1 and P‐selectin mRNA levels in HUVEC using real‐time PCR
Transcripts encoding PSGL‐1 and GAPDH were quantified by real time RT–PCR analysis. After 2‐h treatment with 30 nM U‐46619, HUVEC) were collected and homogenized with Trizol. The real‐time PCR primers were designed using Primer 3 software (http://primer3.sourceforge.net/) on the basis of data from GenBank. The following primers were used for real‐time PCR: 5′‐CAATTTGTCCGTCAACTACCC‐3′ (sense) and 5′‐TGCACACGAAGAAGATAGTGG‐3′ (antisense) for human PSGL‐1; and 5′‐GAAGGTGAAGGACGGACTC‐3′ (sense) and 5′‐GAAGATGGTGATGGGATTTC‐3′ (antisense) for human GAPDH (Applied Sigma‐Aldrich).
Statistical analysis
Data are expressed as means ± standard deviation (SD). Comparisons among multiple groups were carried out by analysis of variance (anova). Comparisons between the two groups were made using the Student's t‐test. Survival experiments were analyzed using the log–rank test and presented as Kaplan–Meier survival curves. A P‐value of <0.05 was considered statistically significant.
Results
Reduced lung B16F1 melanoma metastasis formation in TP −/−
The numbers of lung colonies in TP−/− 21 days after intravenous injection of B16F1 tumor cells were compared with those in WT (Fig. 1a,b). As shown in Fig. 1a, the number of lung colonies in TP−/− (38.7 ± 13.6) was lower than in WT mice (100.0 ± 9.2). Furthermore, TP−/− mice injected with B16F1 cells showed 10% mortality in 6 weeks, whereas WT showed nearly 70% (Fig. 1c; P = 0.036). These results suggested that TP signaling facilitates metastatic formation and is a prognostic factor for the development of metastasis.
Figure 1.
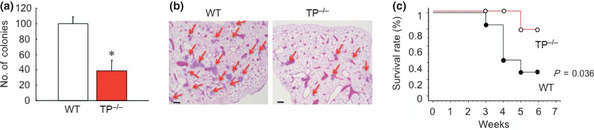
Reduced B16F1 colony formation in thromboxane prostanoid receptor knockout mice (TP −/−). (a) The number of colonies in the lung 21 days after intravenous injection of B16F1 cells. Data are means ± SD for the number of mice (n = 10). *P < 0.05 vs wild‐type mice (WT; Student's t‐test). (b) Typical hematoxylin–eosin staining of the metastatic lung tumors from WT and TP −/− 21 days after intravenous injection of B16F1 cells. Red arrows show the metastatic area. Bar, 250 μm. (c) Mortality rate after intravenous injection of B16F1 cells in WT (n = 20) and TP −/− (n = 20). The number of surviving animals was measured every week until 6 weeks post‐administration. Survival curves were generated using Kaplan–Meier analysis.
Platelet‐enhanced pulmonary metastasis formation
Because platelets play a role in tumor metastasis formation, the changes in the number of platelets during the formation of tumor metastasis after the injection of B16F1 cells were determined (Fig. 2a). The number of platelets did not differ between WT and TP−/− mice at baseline levels. However, the number of platelets in WT at day 14 (15.4 × 106 ± 36.0/μL) and day 21 (17.8 × 106 ± 30.0/μL) were significantly increased in comparison with TP−/− (10.7 × 106 ± 11.5/μL at day 14 and 13.1 × 106 ± 12.3/μL at day 21, respectively). The effect of platelet transfusion on tumor metastasis was further investigated (Fig. 2b). Mice transfused with platelets showed an increased number of colonies at 14 days (115.0 ± 16.9) compared with PBS‐treated mice (85.8 ± 9.7). The bleeding time at 7 days in TP−/− (117 ± 11 s) was prolonged compared with that in WT (101 ± 9 s; Fig. 2c). These results suggested that platelets promoted metastasis formation.
Figure 2.
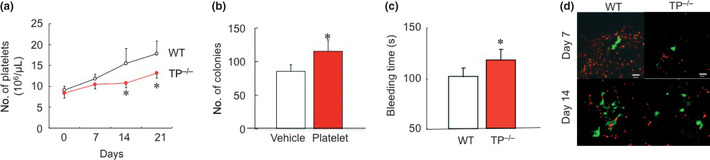
Platelets enhance tumor metastasis. (a) Time course of changes in platelet counts after intravenous injection of B16F1 cells. Data are means ± SD for the number of mice (all n = 10). *P < 0.05 vs wild‐type mice (WT; Student's t‐test). (b) Effect of platelet infusion on the number of colonies formed in the lung 21 days after the injection of B16F1 cells. Data are means ± SD for the number of mice (all n = 10). *P < 0.05 vs PBS‐treated mice (Student's t‐test). (c) Bleeding times were prolonged in thromboxane prostanoid receptor knockout mice (TP −/−). Data are means ± SD for the number of mice (n = 5). *P < 0.05 vs WT (Student's t‐test). (d) The effect of platelets accumulation on colony formation in the lung at 7 and 14 days. Platelets (red) were attached around B16BL‐6 cells (green) in WT, but not in TP −/−.
Platelets accumulate around tumor colony formation
To estimate whether platelets accumulation induces colony formation or not, we injected BL6 melanoma cell that induced more invasion compared with B16F1 melanoma cells. Immunofluorescent study showed WT induced forming platelets accumulation around B16BL‐6 cells at 7 days and almost was completed on day 14 (Fig. 2d). These results suggested that platelets through TP signaling induce colony formation.
P‐selectin activation in platelets through TP signaling during tumor colony formation
P‐selectin is normally stored in the α‐granules of platelets and is rapidly expressed on the surface of activated platelets. To examine whether TP signaling affects platelet activity during tumor metastasis formation, the population of P‐selectin +CD41+ platelets was determined by flow cytometry. The percentage of P‐selectin‐positive platelets in TP−/− mice on day 1 (0.13 ± 0.05%) was significantly reduced compared with that in WT (0.46 ± 0.02%; Fig. 3a). These results showed that TP signaling plays a role in platelet activation during the colonization step of lung metastasis.
Figure 3.
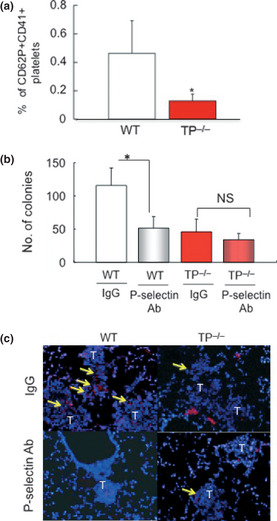
P‐selectin activation through TP signaling enhances pulmonary metastasis. (a) The percentage of P‐selectin+/CD41+ platelets in peripheral blood 7 days after the injection of B16F1 cells. Data are means ± SD for the number of mice (n = 8). *P < 0.05 vs wild‐type mice (WT; Student's t‐test). (b) A neutralizing P‐selectin monoclonal antibody reduced colony formation in WT, but not thromboxane prostanoid receptor knockout mice (TP −/−). Data are means ± SD for the number of mice (n = 5). *P < 0.05 (Student's t‐test). NS, not significant. (c) Immunohistochemical detection of αIIb in metastatic areas. Treatment of WT with a P‐selectin antibody reduced the attachment of platelets to metastatic tumor cells compared with IgG‐treated WT. Platelet attachment to metastatic tumor cells was attenuated in TP −/−. Treatment of TP −/− with a P‐selectin antibody failed to further inhibit the attachment of platelets. Yellow arrows show platelets. Bars, 50 μm.
TP signaling regulated the involvement of P‐selectin in tumor colony formation
To examine the role of P‐selectin in tumor metastasis formation through TP signaling, a P‐selectin neutralizing antibody was injected after the injection of B16F1 cells. P‐selectin neutralizing antibody‐treated WT showed a markedly reduced number of lung colonies (51.7 ± 17.2) compared with IgG‐treated WT (116.2 ± 25.9; Fig. 3b). However, there were no significant differences in the number of lung colonies formed between antibody‐treated TP−/− (34.2 ± 9.5) and IgG‐treated TP−/− mice (45.8 ± 18.9). These results suggested that P‐selectin‐mediated tumor metastasis formation was TP signaling dependent.
P‐selectin‐mediated adhesion of platelets to tumor colonization
To determine whether P‐selectin is involved in the interaction of platelets with metastatic colonies in the lung, platelets were detected by immunohistochemical analysis. Intense staining for the platelet marker, integrin αIIb, was apparent in the metastatic locus in WT, whereas the staining in TP−/− was less pronounced (Fig. 3c). An antibody against P‐selectin reduced the expression of αIIb in the metastatic tumor in WT, but not in TP−/−. This result suggested that TP signaling plays a critical role in P‐selectin‐mediated platelet adhesion to the metastatic tumor locus.
TP signaling induces upregulation of PSGL‐1 in B16F1 and LLC
P‐selectin glycoprotein ligand‐1 is known to be a major ligand for P‐selectin. To confirm whether PSGL‐1 contributes to binding to P‐selectin, the expression of PSGL‐1 was analyzed. LLC was used for positive control. As shown in Fig. 4a, the expression of PSGL‐1 was detected in B16F1 cells and enhanced by U46619, a thromboxane analog (Fig. 4b). These results suggested that the phenomenon of activated platelets, P‐selectin positive platelets, binding to PSGL‐1 on B16F1 cells was probably TP dependent.
Figure 4.
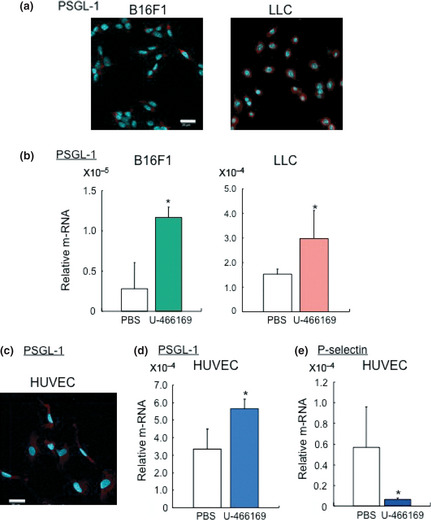
Thromboxane prostanoid receptor (TP) signaling induces P‐selectin glycoprotein ligand‐1 (PSGL‐1) expression on tumor cells and endothelial cells. (a) Immunofluorescence analysis against PSGL‐1 on B16F1 and LLC cells. Bars, 20 μm. (b) The expression of PSGL‐1 on B16F1 and LLC cells treated with U‐46619. (n = 5 per group, *P < 0.05 Student's t‐test). (c) Immunofluorescence analysis against PSGL‐1 on HUVEC. Bars, 50 μm. The effect of (d) PSGL‐1 expression and (e) P‐selectin on HUVEC treated with U‐46619 (n = 4–5 per group, *P < 0.05 Student's t‐test).
TP dependent upregulation of PSGL‐1, but not P‐selectin in endothelial cells
P‐selectin glycoprotein ligand‐1 also expresses on endothelial cells. To prove the activated platelet‐cancer cells interact with endothelial cells, the expression of PSGL‐1 on endothelial cells was carried out. The expression of PSGL‐1 was detected in HUVEC by immunofluorescence analysis (Fig. 4c), and treatment with U‐46619 significantly induced the expression of PSGL‐1 compared with the control (Fig. 4d). In contrast, the expression of P‐selectin in HUVEC was diminished after U‐46619 treatment (Fig. 4e). These results suggested that TP signaling activated expression of PSGL‐1 on endothelial cells causes them to bind to cancer cells through P‐selectin positive platelets.
Reduced plasma levels of hematopoietic cytokines in TP −/−
Because VEGF and SDF‐1 are involved in tumor metastasis and platelets can release these proangiogenic factors on activation,32 the plasma levels of VEGF (Fig. 5a) and SDF‐1 (Fig. 5b) were determined. Plasma levels of VEGF‐A in TP−/− (72.8 ± 21.6 pg/mL) 7 days after the injection of B16F1 cells were found to be lower than in WT (152.0 ± 29.0 pg/mL). In addition, the levels of SDF‐1 in TP−/− (3.3 ± 3.6 ng/mL) were decreased compared with WT (10.7 ± 2.9 ng/mL). Other hematopoietic cytokines including pro‐MMP‐9 (Fig. 5c) and SCF (Fig. 5d) were also measured. Plasma levels of pro‐MMP9 and SCF in TP−/− (0.4 ± 0.2 pg/mL and 0.8 ± 0.2 ng/mL, respectively) 7 days after B16F1 cell injection were lower than in WT (0.6 ± 0.1 ng/mL and 1.0 ± 0.0 pg/ml, respectively). These results suggested that TP signaling is important for tumor metastasis formation through its involvement in the production of hematopoietic cytokines including VEGF‐A, SDF‐1, pro‐MMP9 and SCF.
Figure 5.
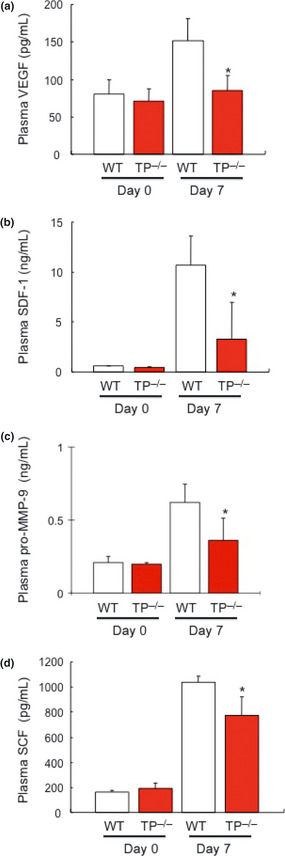
Reduced plasma levels of proangiogenic cytokines in thromboxane prostanoid receptor knockout mice (TP −/−). Plasma levels of (a) vascular endothelial growth factor (VEGF)‐A, (b) stromal‐derived factor (SDF)‐1, (c) pro‐MMP‐9 and (d) stem cell factor (SCF) 7 days after the injection of B16F1 cells. Data are means ± SD for the number of mice (n = 8). *P < 0.05 vs wild‐type mice (WT; Student's t‐test).
TP signaling‐dependent mobilization of CXCR4+ VEGFR1+ cells
Vascular endothelial growth factor is one of the major angiogenic stimulating factors. Immunohistochemical analysis showed that VEGF is localized around the metastasis area in WT (Fig. 6a). An increase in hematopoietic cytokines, especially VEGF and SDF‐1, have been reported to mobilize hematopoietic progenitor cells to promote tumor growth, and VEGFR1+ hematopoietic progenitors are required for the regulation of tumor metastasis.33 Therefore, the involvement of TP signaling in the mobilization of CXCR4+VEGFR1+ cells into the lung tissue was determined (Fig. 6b). Flow cytometry analysis showed that the mobilization of CXCR4+VEGFR1+ cells into the peripheral blood was downregulated in TP−/− (0.3 ± 0.1%) on day 7 in comparison with WT (1.4 ± 0.5%). Double immunostaining for CXCR4+ and VEGFR1+ in lung tissues 7 days after melanoma cell injection showed that CXCR4+VEGFR1+ cells were located around the metastatic tumor (Fig. 6c). The number of CXCR4+VEGFR1+ cells in lung tissues was dramatically reduced in TP−/− as compared with WT (Fig. 6b), although these cells were not detected in naive lung tissue. These results suggested that TP signaling regulates the mobilization as well as the homing of CXCR4+VEGFR1+ cells.
Figure 6.
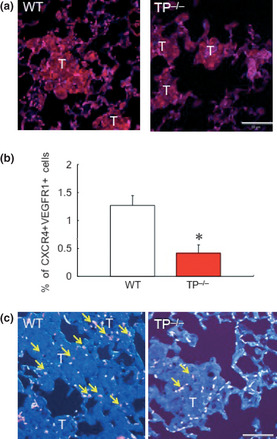
Thromboxane prostanoid receptor (TP) signaling induces the mobilization of VEGFR1+ CXCR4+ cells in the peripheral blood and homing into lung tissues during the colonization of B16F1 cells. (a) Immunohistochemical detection of vascular endothelial growth factor (VEGF) and CD31 in metastatic areas. Wild‐type (WT) mice express VEGF (red) and CD31 (blue) in metastasis area compared with TP knockout mice (TP −/−) mice 7 days after B16F1 cell injection. Bars, 50 μm. (b) The percentage of VEGFR1+ CXCR4+ cells in the peripheral blood 7 days after B16F1 cell injection. Data are means ± SD for the number of mice (n = 8). *P < 0.05 vs WT (Student's t‐test). (c) Reduced homing of CXCR4+ VEGFR1+ cells to the lung tissue in TP −/− 7 days after the injection of B16F1 cells. Yellow arrows show CXCR4 and VEGFR1 double‐positive cells (magenta). T indicates metastatic area. All images are representative of three independent samples. Bars, 50 μm.
Discussion
The objective of the present study was to investigate the role of TP signaling in lung colony formation after the injection of B16F1 melanoma cells and explore the underlying mechanisms of tumor metastasis regulated by the TP signaling pathway. The present results showed that endogenous TXA2 promoted tumor metastasis through P‐selectin‐mediated platelet adhesion to metastatic tumor cells in lung tissues. The results also showed that the TP signaling mediated increasing levels of hematopoietic cytokines and facilitated tumor metastasis through the recruitment of VEGFR1+CXCR4+‐expressing bone marrow‐derived pro‐angiogenic cells into the lung tissues. These results suggest that TP signaling plays a critical role in tumor metastasis through P‐selectin‐mediated interactions of platelets‐tumor cells and tumor cells‐endothelial cells through TP signaling‐dependent production of VEGF and SDF‐1, which are involved in the mobilization of VEGFR1+CXCR4+ cells.
Platelets play a crucial role in tumor metastasis.2, 3, 4, 5, 6 The present study showed that the treatment of mice by platelet transfusion enhanced the development of pulmonary metastasis elicited by the injection of B16F1 melanoma cells (Fig. 2). We also reported that aspirin attenuated pulmonary metastasis in mice.8 Studies have shown that the involvement of platelets in tumor metastasis is mediated by a highly complex process. Tumor cells enter the microvascular system (intravasation), and bind and activate platelets (cohesion). Platelets then assist tumor cell arrest at the endothelium (adhesion) and survival within the microvasculature (immune evasion), which enables exit from the circulation (extravasation), and tumor cell survival and proliferation within the target organs of metastasis.34, 35, 36
Although platelets are known to contain many proangiogenic factors, the mechanisms by which platelets regulate angiogenesis are only partially understood. Among the endogenous factors involved in platelet stimulation, TXA2, an arachidonic acid metabolite, is one of the most significant molecules. Although it has been suggested that the TXA2–TP signaling pathway in tumor cells contributes to tumor metastasis,16, 21 the involvement of endogenous TXA2 in platelets as a part of the tumor metastasis microenvironment remains uncertain. Furthermore, platelets stimulated with TXA2 might supply proangiogenic growth factors to the tumor microenvironment. However, to date, the cellular and molecular mechanisms by which TP signaling enhances tumor colonization through the interaction with platelets have not been fully elucidated. The current study showed that the disruption of the TP gene inhibited the formation of lung metastases in a mouse experimental lung metastasis model, showing that TP signaling is a positive regulator of tumor metastasis. Although melanoma cells injected into TP−/− and WT expressed TP signaling, the inhibition of pulmonary melanoma colonization in TP−/− has been observed. These results suggested that host cells with active TP signaling are a key player in colony formation in this model.
P‐selectin (CD62P) becomes rapidly translocated from intracellular storage vesicles to the platelet surface on activation. P‐selectin, a member of the selectin family of adhesion molecules, has been implicated in tumor metastasis.37, 38 In the present study, the activation status of platelets, as reflected by the expression of P‐selectin on platelets during the development of pulmonary melanoma colonization, was determined. Interestingly, TP−/− showed fewer populations of P‐selectin‐expressing platelets when compared with WT, suggesting that TP signaling is important for P‐selectin expression on platelets. In several in vivo metastasis models, the gene disruption of P‐selectin or the inhibition of P‐selectin function with heparin was found to have a dramatic inhibitory effect on pulmonary metastasis.39, 40 Consistent with these findings, we also showed that treatment with an antibody against P‐selectin reduced pulmonary colony formation in WT (Fig. 3). Of interest, the P‐selectin antibody failed to further inhibit colonization in TP−/−, suggesting that P‐selectin‐mediated pulmonary melanoma colonization is dependent on TP signaling. The results obtained through the blockade of P‐selectin suggest that tumor colony formation involves P‐selectin function and platelets. The specific role of P‐selectin in platelet–tumor complex formation and metastasis was established by the use of P‐selectin‐deficient mice. Both efficient tumor cell–platelet aggregation and the development of experimental lung metastasis were attenuated in P‐selectin‐deficient mice.37, 38 In addition, platelets from P‐selectin‐deficient mice failed to adhere to melanoma cells in vitro.39 The current study also showed that P‐selectin blockade suppressed platelet adhesion to pulmonary colonization of tumors in WT (Fig. 3). We could not detect leukocyte cells around the metastatic area where platelets accumulated.
In addition, TP−/− treated with a P‐selectin antibody failed to show a further reduction in platelet attachment during lung colonization after B16F1 melanoma injection. Taken together, these observations show that TP signaling dependent P‐selectin activation is critical for lung colonization of tumor cells, supporting the concept that P‐selectin‐mediated platelet–tumor cell interactions assist the process of colonization through TP signaling.
In addition to enhanced P‐selectin expression on platelets during colony formation, P‐selectin expression on activated endothelial cells was also shown to contribute to experimental lung metastasis.41 It was already reported that activated platelets swell and adhere to tumor and endothelial cells in the initial colony formation step. We examined whether TP signaling induce platelet activation and contribute to colony formation by adhering to tumor and endothelial cells. P‐selectin is known to bind PSGL‐1 expressed on tumor cells and endothelial cells. We confirmed PSGL‐1 expression by immunofluorescence and PCR analysis. When we added U46619 to B16F1 cell culture, the mRNA level of PSGL‐1 were significantly enhanced compared with the control condition. Furthermore, we also showed that expression of P‐selectin in HUVEC was diminished by U46619 treatment. These results support that TP signaling does not enhance expression of P‐selectin stored in endothelial cells. Activated platelets bind to PSGL‐1 on B16F1 and HUVEC, and that process of activated platelets is TP signaling dependent. As shown in Figs 1a and 2d, TP−/− suppressed colony formation compared with WT. It was shown that deletion of TP signaling on endothelial cells causes inhibition of PSGL‐1 expression that binds to P‐selectin delivered from platelets‐tumor cells.
Proangiogenic factors, including VEGF‐A and SDF‐1, are candidate factors involved in tumor metastasis development.32 In the present study, plasma levels of VEGF‐A and SDF‐1 were increased in WT during pulmonary colony formation, and these factors were significantly suppressed in TP−/− (Fig. 3). Recent evidence, including ours, have shown that VEGF‐A and SDF‐1 released from platelets promote angiogenesis during vascular injury and tumor metastasis.32, 41, 42 Thus, TP signaling might induce the production of VEGF‐A and SDF‐1 in platelets, resulting in tumor colonization in this model. In thrombocytopenic mice, plasma SDF‐1 elevation and the mobilization of CXCR4+VEGFR1+ cells, as well as tissue revascularization, are severely impaired after hind‐limb ischemia.32 In addition, the restoration of platelet production or enforced SDF‐1 expression restores neoangiogenesis, providing evidence for the essential role of platelet‐derived SDF‐1 in regulating neovascularization. Furthermore, we reported that SDF‐1 and VEGF concentrations were markedly reduced by treatment with P‐selectin‐neutralizing antibody in a hind‐limb ischemia model.43 These findings led us to examine SDF‐1 and VEGF delivered from platelets compared with other cells, monocytes and macrophage. We focused on CXCR4+VEGFR1+ cells because VEGF‐A and SDF‐1 induce tumor metastasis by remobilizing bone marrow progenitors cells.40 Subsets of VEGFR1+ hematopoietic progenitors have been reported to promote tumor neoangiogenesis and metastasis. In addition, VEGFR1+ hematopoietic progenitors are distinct from endothelial progenitors, and might contribute to revascularization by releasing angiogenic factors or by positioning themselves perivascularly to stabilize nascent neovessels. SCF induced the release of SDF‐1 from platelets, thereby increasing systemic plasma levels of SDF‐1. The release of SCF through MMP‐9 activation has also been reported to be a critical step in the mobilization of CXCR4+VEGFR1+ cells from the bone marrow.31, 42 The present results showed that increased plasma levels of MMP‐9 and SCF were associated with the mobilization of CXCR4+VEGFR1+ cells. Furthermore, VEGF and SDF‐1, delivered from activated platelets, is also an important factor for the activation of MMP‐9 in the bone marrow and subsequent mobilization of VEGFR1+ myelomonocytic cells.44
The current study also showed that TP signaling facilitates the mobilization of CXCR4+VEGFR1+ cells from the bone marrow to the peripheral blood, as well as the recruitment of hemangiocytes expressing CXCR4+VEGFR1+ to the lung tissue during the evolution of pulmonary metastasis. These results show that the mobilization and recruitment of hemangiocytes might be regulated by TP signaling. The mobilized CXCR4+VEGFR1+ cells release angiopoietin‐2 and have a crucial role in angiogenesis during hind‐limb ischemia,31 as well as tumor metastasis.44 VEGFR1+ hematopoietic progenitor cells are reported to be present at the periphery of the metastatic lesion, likely maintaining tumor survival and growth.42 We showed the expression of angiogenic makers, such as VEGF and CD31, were enhanced in the colony formation area with mobilization of CXCR4+VEGFR1+ cells. In our preliminary experiments, the treatment of WT with antibodies against VEGF or CXCR4 attenuated the numbers of lung colonies elicited by injection of B16F1 melanoma cells, suggesting that the presence of CXCR4+VEGFR1+ cells at premetastatic sites facilitates tumor colonization.
In conclusion, the present study shows that TP signaling plays a critical role in tumor metastasis through P‐selectin‐mediated interactions of activated platelets with tumor cells and endothelial cells, and through the TP signaling‐dependent production of VEGF and SDF‐1, involves the mobilization of hematopoietic cells expressing CXCR4+VEGFR1+ from the bone marrow to facilitate the establishment of tumor colonization (Fig. 7). Therefore, highly selective TP antagonists might become a useful therapeutic tool for cancer treatment.
Figure 7.
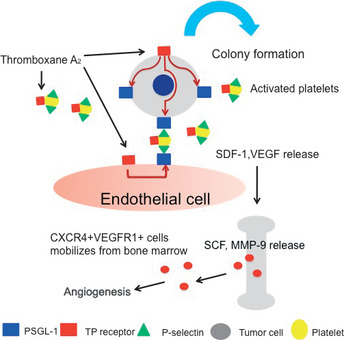
Role of thromboxane prostanoid receptor (TP) signaling in the enhancement of tumor colony formation. TP signaling regulates the P‐selectin‐mediated adhesion of activated platelets to P‐selectin glycoprotein ligand‐1 (PSGL‐1) on metastatic tumor cells and endothelial cells, and induce colony formation. Vascular endothelial growth factor (VEGF) and stromal‐derived factor (SDF)‐1 delivered from accumulated platelets induce the mobilization and recruitment of hemangiocytes expressing CXCR4 and VEGFR1 in the lung tissues.
Disclosure Statement
There are no financial conflicts of interest.
References
- 1. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9: 239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta P. Potential role of platelets in the pathogenesis of tumor metastasis. Blood 1984; 63: 55–63. [PubMed] [Google Scholar]
- 3. Honn KV, Tang DG, Crissman JD. Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Rev 1992; 11: 325–51. [DOI] [PubMed] [Google Scholar]
- 4. Pinedo HM, Verheul HM, D'Amato RJ et al Involvement of platelets in tumour angiogenesis? Lancet 1998; 352: 1775–7. [DOI] [PubMed] [Google Scholar]
- 5. Erpenbeck L, Schön MP. Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 2010; 115: 3427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gay LJ, Felding‐Habermann B. Contribution of platelets to tumour metastasis. Nat Review Cancer 2011; 11: 123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sierko E, Wojtukiewicz MZ. Inhibition of platelet function: does it offer a chance of better cancer progression control? Semin Thromb Hemost 2007; 33: 712–21. [DOI] [PubMed] [Google Scholar]
- 8. Amano H, Ito Y, Suzuki T et al Roles of a prostaglandin E type receptor, EP3, in up‐regulation of matrix metalloproteinase‐9 and vascular endothelial growth factor during enhancement of tumor metastasis. Cancer Sci 2009; 100: 2318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nieswandt B, Hafner M, Echtenacher B et al Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 1999; 59: 1295–300. [PubMed] [Google Scholar]
- 10. Varki NM, Varki A. Heparin inhibition of selectin‐mediated interactions during the hematogenous phase of carcinoma metastasis: rationale for clinical studies in humans. Semin Thromb Hemost 2002; 28: 53–66. [DOI] [PubMed] [Google Scholar]
- 11. Browder T, Folkman J, Pirie‐Shepherd S. The hemostatic system as a regulator of angiogenesis. J Biol Chem 2000; 275: 1521–4. [DOI] [PubMed] [Google Scholar]
- 12. Pradono P, Tazawa R, Maemondo M et al Gene transfer of thromboxane A(2) synthase and prostaglandin I(2) synthase antithetically altered tumor angiogenesis and tumor growth. Cancer Res 2002; 62: 63–6. [PubMed] [Google Scholar]
- 13. Moussa O, Yordy JS, Abol‐Enein H et al Prognostic and functional significance of thromboxane synthase gene overexpression in invasive bladder cancer. Cancer Res 2005; 65: 11581–7. [DOI] [PubMed] [Google Scholar]
- 14. Sakai H, Suzuki T, Takahashi Y et al Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett 2006; 580: 3368–74. [DOI] [PubMed] [Google Scholar]
- 15. Honn KV. Inhibition of tumor cell metastasis by modulation of the vascular prostacyclin/thromboxane A2 system. Clin Exp Metastasis 1983; 1: 103–14. [DOI] [PubMed] [Google Scholar]
- 16. Nie D, Lamberti M, Zacharek A et al Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun 2000; 267: 245–51. [DOI] [PubMed] [Google Scholar]
- 17. Cathcart MC, Reynolds JV, Kenneth J et al The role of prostacyclin synthase and thromboxane synthase signaling in the development and progression of cancer. Biochimica Biophysica Acta 2010; 1805: 153–66. [DOI] [PubMed] [Google Scholar]
- 18. Yoshimoto A, Kasahara K, Kawashima A et al Characterization of the prostaglandin biosynthetic pathway in non‐small cell lung cancer: a comparison with small cell lung cancer and correlation with angiogenesis, angiogenic factors and metastases. Oncol Rep 2005; 13: 1049–57. [PubMed] [Google Scholar]
- 19. Yokoyama I, Hayashi S, Kobayashi T et al Prevention of experimental hepatic metastasis with thromboxane synthase inhibitor. Res Exp Med 1995; 195: 209–15. [DOI] [PubMed] [Google Scholar]
- 20. Watkins G, Dagulas‐Jones A, Mansel RE et al Expression of thromboxane synthase, TBXAS1 and the thromboxane A2 receptor, TBXA2R, in human breast cancer. Int Semin Surg Oncol 2005; 2: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cathcart MC, Gately K, Cummins R et al Examination of thromboxane synthase as a prognostic factor and therapeutic target in non‐small cell lung cancer. Mol Cancer 2011; 10: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moussa O, Ashton AW, Fraig M et al Novel role of thromboxane receptors beta isoform in bladder cancer pathogenesis. Cancer Res 2008; 68: 4097–104. [DOI] [PubMed] [Google Scholar]
- 23. Nie D, Guo Y, Yang D et al Thromboxane A2 receptors in prostate carcinoma: expression and its role in regulating cell motility via small GTPase Rho. Cancer Res 2008; 68: 115–21. [DOI] [PubMed] [Google Scholar]
- 24. Takayama K, Yuhki K, Ono K et al Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat Med 2005; 11: 562–6. [DOI] [PubMed] [Google Scholar]
- 25. Amano H, Hackett NR, Rafii S et al Thrombopoietin gene transfer‐mediated enhancement of angiogenic responses to acute ischemia. Circ Res 2005; 97: 337–45. [DOI] [PubMed] [Google Scholar]
- 26. Ma H, Hara A, Xiao CY et al Increased bleeding tendency and decreased susceptibility to thromboembolism in mice lacking the prostaglandin receptor subtype EP3. Circulation 2001; 104: 1176–80. [DOI] [PubMed] [Google Scholar]
- 27. Eshima K, Suzuki H, Shinohara N. Cross positive selection of thymocytes expressing a single TCR by multiple major histocompatibility complex molecules of both classes: implication for CD4+ versus CD8+ lineage commitment. J Immunol 2006; 176: 1628–36. [DOI] [PubMed] [Google Scholar]
- 28. Amano H, Ando K, Minamida S et al Adenylate cyclase/protein kinase A signaling pathway enhances angiogenesis through induction of vascular endothelial growth factor in vivo. Jpn J Pharmacol 2001; 87: 181–8. [DOI] [PubMed] [Google Scholar]
- 29. Amano H, Hayashi I, Endo H et al Host prostaglandin E(2)‐EP3 signaling regulates tumor‐associated angiogenesis and tumor growth. J Exp Med 2003; 197: 221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katoh H, Hosono K, Ito Y et al COX‐2 and Prostaglandin EP3/EP4 Signaling Regulate the Tumor Stromal Proangiogenic Microenvironment via CXCL12‐CXCR4 Chemokine Systems. Am J Pathol 2010; 176: 1469–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hosono K, Suzuki T, Tamaki H et al Roles of prostaglandin E2‐EP3/EP4 receptor signaling in the enhancement of lymphangiogenesis during fibroblast growth factor‐2‐induced granulation formation. Arterioscler Thromb Vasc Biol 2011; 31: 1049–58. [DOI] [PubMed] [Google Scholar]
- 32. Jin DK, Shido K, Kopp HG et al Cytokine –mediated development of SDF‐1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 2006; 12: 557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaplan RN, Riba RD, Zacharoulis S et al VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 2005; 438: 820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 2006; 127: 679–95. [DOI] [PubMed] [Google Scholar]
- 35. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12: 895–904. [DOI] [PubMed] [Google Scholar]
- 36. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer 2009; 9: 285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim YJ, Borsig L, Han HL et al Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am J Pathol 1999; 155: 461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borsig L, Wong R, Feramisco J et al Heparin and cancer revisited: mechanistic connections involving platelets, P‐selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA 2001; 98: 3352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luwig RJ, Boehme B, Padda M et al Endothelial P‐selectin as a target of heparin action in experimental melanoma. Cancer Res 2004; 64: 2743–50. [DOI] [PubMed] [Google Scholar]
- 40. Kohker S, Ullrich S, Richter U et al E‐/P‐selectin and colon carcinoma metastasis: first in vivo evidence for their crucial role in clinically relevant model of spontaneous metastasis formation in lung. Br J Cancer 2010; 102: 602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanagi R. Carbohydrate antigen sialyl Lewis a – its pathophysiological significance and induction mechanism in cancer progression. Chang Gung Med J 2007; 30: 189–209. [PubMed] [Google Scholar]
- 42. Mohle R, Green D, Moore MAS et al Constitutive production and thrombin‐induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci USA 1997; 94: 663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kato S, Amano H, Ito Y et al Effect of erythropoietin on angiogenesis with the increased adhesion of platelets to the microvessels in the hind‐limb ischemia model in mice. J Pharmacol Sci 2010; 112: 167–75. [DOI] [PubMed] [Google Scholar]
- 44. Wiesner T, Bugl S, Mayer F et al Differential changes in platelet VEGF, Tsp, CXCL12, and CXCL4 in patients with metastatic cancer. Clin Exp Metastasis 2010; 27: 141–9. [DOI] [PubMed] [Google Scholar]