

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors, such as gefitinib and erlotinib, are effective for non‐small cell lung cancer with activating EGFR mutations. However, even in patients with an initial dramatic response to such a drug, acquired resistance develops after 6–12 months. A secondary mutation of T790M in EGFR and amplification of the MET gene account for this resistance; however, the mechanism(s) of approximately 30% of acquired resistance cases remain unknown. We established an erlotinib‐resistant lung cancer cell line named PC‐9/ER3 that harbors an EGFR mutation after continuously exposing PC‐9 cells to erlotinib. PC‐9/ER3 cells were 136‐fold more resistant to erlotinib than the parental cells. Although the PC‐9/ER3 cells did not carry the T790M mutation or MET amplification and had similar levels of phosphorylated (p) STAT3, pJAK2 increased in the resistant cells. It was found in the present study that 3–12 h of exposure to erlotinib in both cell lines did not affect pJAK2 expression, but did result in increased pSTAT3 expression. pAkt in PC‐9/ER3 cells was less suppressed than in PC‐9 cells, although pEGFR and pMAPK were markedly suppressed in both cell lines. The combined treatment of erlotinib plus a JAK2 inhibitor (JSI‐124) suppressed pAkt in PC‐9/ER3 cells. Similarly, the combination of erlotinib plus JSI‐124 or siRNA against JAK2 restored sensitivity to erlotinib in PC‐9/ER3 cells. The combination of erlotinib plus JSI‐124 was also effective for reducing PC‐9/ER3 tumors in a murine xenograft model. Our results suggest that the activation of JAK2 partially accounts for acquired erlotinib resistance.(Cancer Sci, doi: 10.1111/j.1349‐7006.2012.02363.x, 2012)
Lung cancer, the leading cause of cancer‐related death in the USA, accounted for 29% of all male cancer deaths and 26% of all female cancer deaths in 2011.1 The overall 5‐year survival rate of patients with metastatic disease remains <15%.2 However, somatic mutations have been discovered to exist in the epidermal growth factor receptor (EGFR) tyrosine kinase in a subset of patients with non‐small cell lung cancer (NSCLC).3, 4, 5 Remarkably, these mutations strongly sensitize the cancer cells to the growth suppressive effects of the EGFR‐tyrosine kinase inhibitors (TKI), gefitinib and erlotinib, leading to clinical responses.3, 4, 6, 7 However, the majority of NSCLC initially sensitive to gefitinib or erlotinib become resistant to these agents within 1 year.8 Once EGFR mutant lung cancer develops into progressive disease during treatment with EGFR‐TKI, no optimal therapy has yet been established. Several possible mechanisms for the acquired resistance have been identified, the most common being the development of an EGFR T790M gatekeeper mutation in approximately 50% of cases.9 Other mechanisms of acquired resistance include bypass signaling, such as MET amplification,10 PTEN loss11 and hepatocyte growth factor overexpression.12 Approximately 30% of cases remain for which the mechanism of acquired resistance is presently unknown.9 Clinical trials testing the tolerance for changing treatment regimens to include irreversible TKI, such as BIBW2992, to prevent acquired resistance via T790M or combining EGFR‐TKI with a MET inhibitor to prevent acquired resistance via amplification of MET, have been performed.13, 14
Phosphoinositide 3 kinase (PI3K)/Akt, Ras/mitogen‐activated protein kinase (MAPK), and signal transducer and activator of transcription 3 (STAT3) are three major downstream pathways activated by EGFR phosphorylation.15 STAT3 is reported to be a critical mediator of the oncogenic effects of EGFR mutations.16 Non‐receptor tyrosine kinases, such as Src and JAK2, also phosphorylate STAT3.17 In this study, we isolated one cell line (PC‐9/ER3) from five erlotinib‐resistant clones generated in vitro from parental PC‐9 cells that harbored the activating EGFR mutation by chronically exposing the cell line to erlotinib. As PC‐9/ER3 cells harbored neither the T790M mutation nor MET gene amplification, we investigated EGFR signaling abnormalities in these cells, including those involving JAK2, STAT3, Akt and MAPK.
Materials and Methods
Establishment of an erlotinib‐resistant cell line
The human NSCLC cell line PC‐9 was derived from an untreated Japanese patient with pulmonary adenocarcinoma that carried an in‐frame deletion in EGFR exon 19 (del E746‐A750) and was highly sensitive to EGFR‐TKI.18 PC‐9 cells were purchased from Immuno‐Biological Laboratories (Gunma, Japan) and were cultured at 37°C with 5% CO2 in RPMI‐1640 medium supplemented with 10% heat‐inactivated FBS. To establish an erlotinib‐resistant subline, the cells were treated with gradually increasing concentrations of erlotinib, starting at 0.02 μM/L, which was near the 50% inhibitory concentration (IC50) of the drug in PC‐9 cells (Fig. 1A). After 12 weeks, the cells were able to grow in 5 μM/L erlotinib. Then, we performed single‐cell cloning by limiting dilution and obtained five erlotinib‐resistant cell lines.
Figure 1.
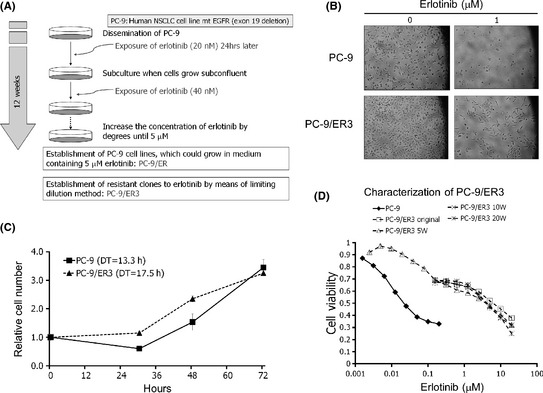
Establishment of an erlotinib‐resistant lung cancer cell line. (A) Overview of the strategy to establish erlotinib‐resistant cells from PC‐9 cells. (B) A light microscopic (×100) image of PC‐9 and PC‐9/ER3 cells. Cells were exposed to erlotinib (1 μM) for 72 h. (C) Relative cell numbers of PC‐9 and PC‐9/ER3 are shown in the culture medium for 72 h. PC‐9 and PC‐9/ER3 cells were seeded on 6‐cm dishes (4 × 105 per dish) in the absence of erlotinib. Cells were trypsinized and counted in triplicate. DT, doubling time. Data are presented as the mean ± SD. (D) Cells (3 × 103 per well) were seeded onto 96‐well plates in quadruplicate and grown in the absence or presence of the indicated concentration of erlotinib. After 96 h, the cells were subjected to MTT assays. PC‐9/ER3 cells were maintained in culture medium without erlotinib for 5 (5W), 10 (10W) or 20 (20W) weeks. EGFR, epidermal growth factor receptor; NSCLC, non‐small cell lung cancer.
Sensitivity test
Growth inhibition was measured by an MTT assay.19 Briefly, the cells were plated onto 96‐well plates at a density of approximately 3 × 103 cells per well and exposed to erlotinib for 96 h. Each assay was done in quadruplicate and the mean ± SD of the IC50 was calculated. Among the five erlotinib‐resistant clones, the PC‐9/ER3 cell line did not harbor the T790M mutation, and its resistance was stable for at least 20 weeks. Thus, we used PC‐9/ER3 cells to investigate acquired resistance to erlotinib.
Reagents and antibodies
Erlotinib was purchased from Chemie Tek (Indianapolis, IN, USA). JSI‐124 (cucurbitacin I),17 a selective JAK2 inhibitor, and Stattic,20 an inhibitor of STAT3 activation, were acquired from Calbiochem (San Diego, CA, USA). LY294002,21 a potent inhibitor of PI3K, was obtained from Cell Signaling Technology (Danvers, MA, USA). Rabbit antisera against EGFR, pEGFR (pY1068), pSTAT3 (pY705), ERK1/2, pERK (pT202/pY204), pAkt (pSer473), total Akt and GAPDH were purchased from Cell Signaling Technology. Polyclonal anti‐pJAK2 (pY1007/1008) antibody was obtained from Millipore (Billerica, MA, USA). Other polyclonal antibodies against JAK2, STAT3, survivin and c‐MYC were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Sequencing of the EGFR gene
To determine the EGFR sequence, DNA was extracted from each cell line using a QIAamp DNA Mini Kit (Qiagen, Tokyo, Japan), and the exons encoding the intracellular domain (exons 18–22) were amplified by PCR. Primer sequences and amplification conditions were as described previously.5 PCR products were processed with a BigDye Terminator Cycle sequencing kit (Applied Biosystems, Tokyo, Japan) and analyzed in both the sense and antisense directions for the presence of mutations on an ABI 3100 sequencer (Applied Biosystems).
Quantitative PCR
Quantitative PCR was performed on a GeneAmp 5700 (Applied Biosystems). The copy number ratio of MET to GAPDH, a housekeeping gene, was calculated using a genomic DNA sample. The sequences of the Taqman probe and primers for MET and GAPDH were as follows: human MET, 5′‐FAM‐TGCCTGCGAAGTGAAGGGTCTCCG‐TAMRA‐3′ (Taqman probe), 5′‐CCAATTTCTGACCGAGGGAATC‐3′ (forward primer) and 5′‐GTCCTACCAT‐ACATGAAACATGGA‐3′ (reverse primer); and human GAPDH, 5′‐FAM‐TCAAGGTGGGGAGGGAGGTAGAGGGG‐TAMRA‐3′ (Taqman probe), 5′‐GGCTCCCACCTTTCTCATCC‐3′ (forward primer) and 5′‐GATGTGGGGAGTACGCTGC‐3′ (reverse primer).
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay buffer (1% Triton X‐100, 0.1% SDS, 50 mM Tris–HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM β‐glycerol‐phosphate, 10 mM NaF and 1 mM Na‐orthovanadate) containing the respective protease inhibitor tablet (Roche, Tokyo, Japan). Whole cell lysates were separated with SDS‐PAGE and then transferred to a membrane and detected by antibodies using ECL Plus Western Blotting detection reagents (GE Healthcare Biosciences, Tokyo, Japan). Each protein was incubated with an appropriate primary antibody and detected by HRP‐mediated chemiluminescence (ECL Plus).
Xenograft model
Female BALB/c nu/nu mice at 7 weeks of age were purchased from Japan Charles River Co. (Yokohama, Japan). All mice were provided with sterilized food and water and housed in a barrier facility under a 12‐h L:D cycle. Cells (2 × 106) were injected subcutaneousy into the backs of the mice. At 1 week after injection, the mice were randomly assigned to one of four groups (five to six mice per group) that received vehicle, 25 mg/kg/day erlotinib, 1 mg/kg/day JSI‐124, or 25 mg/kg/day erlotinib plus 1 mg/kg/day JSI‐124. Vehicle and erlotinib were administered once a day, five times a week by gavage. JSI‐124 (1 mg/kg) was administered once a day, five times a week intraperitoneally. Tumor volume (width2 × length/2) was determined periodically.
All experiments involving animals were performed under the auspices of the Institutional Animal Care and Research Advisory Committee of the Department of Animal Resources, Okayama University Advanced Science Research.
siRNA gene knockdown
MISSION predesigned small interfering (si)RNA (Sigma, St. Louis, MO, USA) targeting STAT3 and JAK2 sequences were 5′‐GGAUAACGUCAUUAGCAGA[dT][dT]‐3′ and 5′‐UCUGCUAAUGACGUUAUCC[dT][dT]‐3′ and 5′‐GAUAGGUGCCCUAGGGUUU[dT][dT]‐3′ and 5′‐AAACCCUAGGGCACCUAUC[dT][dT]‐3′, respectively. MISSION siRNA Universal Negative Control (Sigma) was used as a non‐targeting control for siRNA experiments. PC‐9 and PC‐9/ER3 cells were transiently transfected with the combination of two siRNA duplexes (5 nM STAT3‐specific siRNA and 30 nM JAK2‐specific siRNA) using Lipofectamine 2000 (Invitrogen, Tokyo, Japan) according to the manufacturer's protocol. Assays for silencing were performed on confluent monolayers 24 or 36 h after transfection. Protein expression after siRNA knockdown was evaluated by western blot analysis. Proliferation of PC‐9/ER3 and PC‐9 cells, in which STAT3 or JAK2 was knocked down by siRNA, was measured using the MTT assay as described above.
Statistical analysis
The differences between the groups were compared using Student's t‐test. P < 0.05 was considered statistically significant. All data were analyzed using Microsoft Office Excel 2007 (Microsoft Japan Corporation, Tokyo, Japan).
Results
PC‐9/ER3 cells were established after continuous exposure of PC‐9 cells to erlotinib
By microscopic observation, PC‐9/ER3 cells showed the same morphology as the parental PC‐9 cells (Fig. 1B). The rate of proliferation of PC‐9/ER3 cells was similar to that of PC‐9 cells, with doubling times of 13.3 and 17.5 h (P = 0.43), respectively (Fig. 1C). The parental PC‐9 cells could not proliferate in the presence of 1 μM/L erlotinib, whereas PC‐9/ER3 cells continued to grow under the same condition (Fig. 1B). The IC50 values of erlotinib in the PC‐9 and the PC‐9/ER3 cells were 0.0089 ± 0.0001 and 1.21 ± 0.11 μM/L, respectively. PC‐9/ER3 cells were 136‐fold more resistant to erlotinib than the parental PC‐9 cells. The resistance was stable for at least 20 weeks without exposure to erlotinib (Fig. 1D). The IC50 values for gefitinib were 0.011 ± 0.001 μM/L in PC‐9 cells and 2.78 ± 0.42 μM/L in PC‐9/ER3 cells, indicating that PC‐9/ER3 cells showed cross‐resistance to gefitinib that was 252‐fold higher than in PC‐9 cells. Both PC‐9/ER4 and PC‐9/ER5 cells were also resistant to erlotinib and gefitinib (Fig. S1).
Mechanism of erlotinib resistance is not due to the T790M point mutation or MET gene amplification
To examine genetic alterations, including the well‐known T790M mutation, we conducted direct sequencing of EGFR at exons 18–22. The T790M mutation at exon 20 was not observed in the PC‐9/ER3 cells (Fig. 2A). No other genetic differences in EGFR DNA sequences between PC‐9 and PC‐9/ER3 cells were detected.22 Next, we used a more sensitive assay for the EGFR T790M mutation: the peptide nucleic acid‐locked nucleic acid (PNA‐LNA) PCR clamp‐based detection test (Mitsubishi Chemical Medience, Tokyo, Japan), which can detect mutations present in 0.1–1% of samples.23, 24 This method also failed to detect the T790M mutation in PC‐9/ER3 cells, confirming the results of direct sequencing. In addition, the PCR‐Invader method (Bio Medical Laboratories, Tokyo, Japan), which was more sensitive than the direct sequencing and was as sensitive as the PNA‐LNA PCR clamp‐based detection test,25 also failed to detect T790M in the other resistant cell lines (PC‐9/ER1, 2, 3, 4 and 5).
Figure 2.
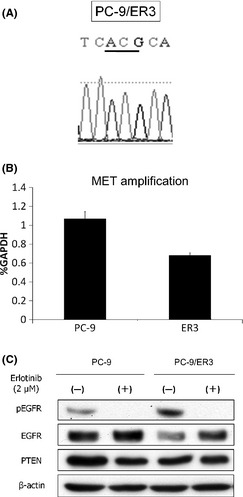
Characterization of PC‐9/ER3 cells. (A) The T790M mutation was not found in PC‐9/ER3 cells by direct sequencing. (B) MET gene copy number was examined by quantitative PCR using genomic DNA extracted from PC‐9 and PC‐9/ER3 cells. MET gene copy number relative to GAPDH was measured in three independent experiments. Bars, SD. (C) Cells were incubated with or without 2 μM erlotinib for 6 h and subjected to western blotting. pEGFR was suppressed, and PTEN expression was similar in both cell lines.
The second most common cause of acquired resistance of NSCLC to EGFR‐TKI in vitro and in vivo involves the amplification of MET.10, 26, 27 We examined differences in MET amplification between PC‐9 and PC‐9/ER1, 2, 3, 4 and 5 cells using a quantitative PCR method. No MET gene amplification was detected in PC‐9/ER3 cells (Fig. 2B) or the other resistant cells (PC‐9/ER1, 2, 4 and 5) (Fig. S2). In addition, PTEN expression was similar in both PC‐9 and PC‐9/ER3 cell lines (Fig. 2C).
pJAK2 increased in PC‐9/ER3 cells
PC‐9 and PC‐9/ER3 cells were treated with erlotinib (0.05 μM) for various lengths of time (0–12 h). pEGFR and pMAPK in both cell lines were markedly suppressed by erlotinib. However, pAkt, which is an effector molecule downstream of EGFR, was not inhibited by erlotinib, and pJAK2 increased more in PC‐9/ER3 than in the parental PC‐9 cells (Fig. 3). JAK2 in both PC‐9/ER4 and PC‐9/ER5 cells was also activated (Fig. S3). Phosphorylation of STAT3, which is a downstream effector of JAK2, increased time‐dependently in both PC‐9 and PC‐9/ER3 cells after treatment with erlotinib. Although expression of survivin, the anti‐apoptotic gene that is downstream of STAT3, did not change during erlotinib treatment, PC‐9/ER3 cells had more expressions than PC‐9 cells as a base line (Fig. 3).
Figure 3.
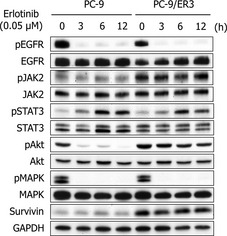
Protein expression in PC‐9 and PC‐9/ER3 cells treated with erlotinib for various lengths of time. pEGFR and pMAPK in both cell lines were markedly suppressed. pAkt was not inhibited by erlotinib, and pJAK2 increased in PC‐9/ER3 cells more than in the parental PC‐9 cells. pSTAT3 increased in both PC‐9 and PC‐9/ER3 cells in a time‐dependent manner. Although expression of survivin did not change during erlotinib treatment, PC‐9/ER3 cells had more expressions than PC‐9 cells as a base line.
Inhibition of JAK2 rather than STAT3 restores the sensitivity of PC‐9/ER3 cells to erlotinib
We hypothesized that the inhibition of JAK2/STAT3 or PI3K/Akt might restore sensitivity to erlotinib in PC‐9/ER3 cells. The differences in sensitivity to treatment with erlotinib plus or minus JSI‐124, LY294002 or Stattic were evaluated using the MTT assay (Fig. 4A–F). JSI‐124, LY294002 and Stattic suppressed pJAK2, pAkt and pSTAT3, respectively, in both PC‐9 and PC‐9/ER3 cells (Figs 5,S4). Cells were treated with the indicated concentration of erlotinib in combination with the approximate IC50 concentration of each drug (0.05 μM JSI‐124, 15 μM LY294002 or 5 μM Stattic) for 96 h. The respective sensitivities of PC‐9 and PC‐9/ER3 cells to JSI‐124 alone were quite similar (Fig. 4A). The sensitivity of the PC‐9/ER3 cells to erlotinib was restored by the combined treatment of erlotinib plus JSI‐124 to a level comparable to that of PC‐9, while the sensitivity of PC‐9 cells to erlotinib did not increase with the same treatment (Fig. 4B). The sensitivity of PC‐9/ER3 cells to LY294002 was slightly higher than that of PC‐9 cells (Fig. 4C). The erlotinib sensitivity of PC‐9/ER3 cells was moderately restored upon treatment with erlotinib plus LY294002, but that of PC‐9 cells was not affected (Fig. 4D). The sensitivity of both cell lines to Stattic was similar (Fig. 4E). However, the sensitivity of PC‐9/ER3 cells to erlotinib was nearly unaffected by the presence of static, while the sensitivity of the PC‐9 cells to erlotinib in the presence of static decreased (Fig. 4F).
Figure 4.
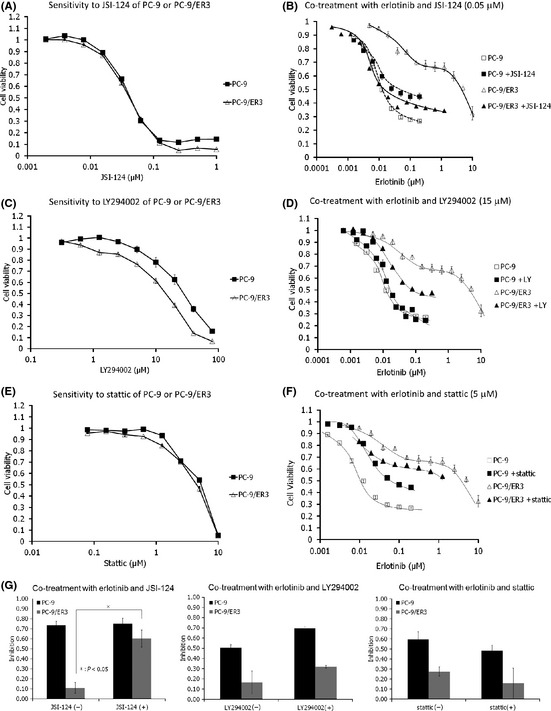
Comparison of cell growth after treatment with drugs alone or in combination. (A) Cells (3 × 103 per well) were seeded in 96‐well plates in quadruplicate and grown in the absence or presence of the indicated concentration of JSI‐124. After 96 h, the cells were subjected to MTT assays. (B) Cells (3 × 103 per well) were seeded in 96‐well plates in quadruplicate and grown in the absence or presence of the indicated concentration of erlotinib with or without 0.05 μM JSI‐124. After 96 h, the cells were subjected to MTT assays. (C, D) Same as in (A) and (B), respectively, but with LY294002. The concentration of LY290442 in (D) was 15 μM. (E, F) Same as in (A) and (B), respectively, but with Stattic. The concentration of Stattic in (f) was 5 μM. (G) Cells were treated with 10 nM erlotinib plus or minus 30 nM JSI‐124 (left panel), 5 μM LY294002 (center panel) or 2 μM Stattic (right panel) for 96 h. Growth inhibition relative to erlotinib‐untreated cells with or without JSI‐124, LY294002 or Stattic are shown. Data are representative of two independent experiments. Bars, standard error.
Figure 5.
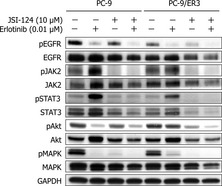
Protein expression in PC‐9 and PC‐9/ER3 cells upon treatment with erlotinib plus or minus JSI‐124. Extracts from PC‐9 and PC‐9/ER3 cell lines, treated with erlotinib (0.01 μM) or JSI‐124 (10 μM) or both for 6 h, were subjected to western blotting. Treatment with erlotinib alone inhibited pMAPK in both cell lines and pAkt in PC‐9, but not PC‐9/ER3, cells. Treatment with JSI‐124 alone inhibited pJAK2, pSTAT3 and pMAPK in both cell lines and pAkt in PC‐9/ER3, but not in PC‐9, cells. pAkt in PC‐9 cells treated with both erlotinib and JSI‐124 was similar to that with erlotinib alone. However, in PC‐9/ER3 cells, JSI‐124 moderately suppressed pAkt. The combination of both drugs suppressed pAkt as much as that in PC‐9 cells treated with erlotinib alone.
To confirm the interaction of erlotinib (0.01 μM) with JSI‐124 (0.03 μM), LY294002 (5 μM) or Stattic (2 μM), the number of viable cells after drug exposure for 96 h was counted. As shown in Figure 4(G), the growth inhibition ratio after treatment with erlotinib plus JSI‐124 of PC‐9/ER3 cells increased significantly (P < 0.05). That was also proved in PC‐9/ER5 cells (Fig. S5). However, the combination of erlotinib with LY294002 or stattic was not effective in the resistant cells.
These results suggest that the combination of erlotinib with a JAK2 inhibitor was more effective than the combination of erlotinib with a STAT3 inhibitor in PC‐9/ER3 cells.
Combination of erlotinib with JSI‐124 suppresses pAkt in PC‐9/ER3 cells
To determine whether the combined effect of erlotinib with JSI‐124 correlated with changes in MAPK or Akt signaling pathways, cells were treated with erlotinib alone, JSI‐124 alone, or the combination of both drugs, and the lysates were processed for western blotting with antibodies specific for pJAK2, pSTAT3, pAkt and pMAPK. Figure 5 shows that treatment with erlotinib alone inhibited pMAPK in both cell lines as well as pAkt in PC‐9, but not in PC‐9/ER3 cells. We used 10 μM of JSI‐124, which seemed high compared to the results from MTT assays. However, treatment duration of the drug was 6 h in western blot analysis and 96 h in MTT assays. The concentration of JSI‐124 was referred from the report of Blaskovich et al.17 Treatment with JSI‐124 alone inhibited pJAK2, pSTAT and pMAPK in both cell lines as well as pAkt in PC‐9/ER3 cells, but not in PC‐9 cells. As expected, the phosphorylation of Akt in PC‐9 cells treated with both erlotinib and JSI‐124 was similar to that with erlotinib alone. However, in PC‐9/ER3 cells, JSI‐124 moderately suppressed pAkt. In addition, the combination of both drugs suppressed pAkt as much as that detected in PC‐9 cells treated with erlotinib alone. The combination of si‐JAK2 with erlotinib suppressed pAkt in PC‐9/ER3 cells (Fig. S6).
siRNA knockdown of JAK2 also recovers sensitivity to erlotinib in PC‐9/ER3 cells
We examined whether siRNA knockdown of STAT3 or JAK2 would restore the sensitivity to erlotinib in PC‐9/ER3 cells (Fig. 6). Accordingly, gene‐specific siRNA were transfected into PC‐9 and PC‐9/ER3 cells. The mock (control) transfected cells received transfection reagents without siRNA. siRNA specific for STAT3 and JAK2 lowered STAT3 and JAK2 expression, respectively (Fig. 6A,C). Subsequently, we evaluated the growth inhibition of each cell line by erlotinib using MTT assays. The knockdown of STAT3 did not increase the sensitivity to erlotinib of either cell line (Fig. 6B). The knockdown of JAK2 did not influence the sensitivity of PC‐9 cells to erlotinib, but it did restore the sensitivity of PC‐9/ER3 cells to erlotinib (Fig. 6D). These results suggest that the resistance of PC‐9/ER3 cells to erlotinib was not induced by the activation of the JAK2/STAT3 pathway, but rather a JAK2‐related pathway, such as the PI3K/Akt pathway.
Figure 6.

siRNA knockdown of STAT3 and JAK2. (A) STAT3 was knocked down in PC‐9 and PC‐9/ER3 cells by STAT3‐specific siRNA. (B) Cells (3 × 103 per well) were seeded in 96‐well plates in quadruplicate and grown with the indicated concentration of erlotinib. After 96 h, the cells were subjected to MTT assays. The knockdown of STAT3 did not affect the sensitivity to erlotinib of either cell line. (C) JAK2‐specific siRNA lowered JAK2 expression. (D) The knockdown of JAK2 did not influence the sensitivity of PC‐9 cells to erlotinib, but restored the sensitivity of PC‐9/ER3 cells to erlotinib.
Combination of erlotinib with JSI‐124 to treat PC‐9/ER3 tumors in a xenograft model is effective
The antitumor effect of the combination of erlotinib with JSI‐124 was examined in vivo. PC‐9/ER3 xenograft model mice were treated with JSI‐124 alone, erlotinib alone, erlotinib in combination with JSI‐124, or vehicle alone (Fig. 7). JSI‐124 alone did not inhibit tumor growth. Erlotinib alone did not significantly affect tumor volume. The two‐drug combination induced significant tumor shrinkage compared with either drug alone (P < 0.05). These results showed that the combination of erlotinib and JSI‐124 restored the sensitivity of PC‐9/ER3 cells to erlotinib in vivo.
Figure 7.
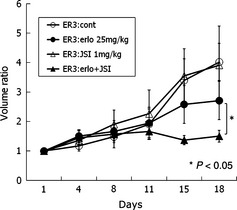
Growth curves of PC‐9/ER3 xenograft tumors. PC‐9/ER3 xenograft model mice were treated with JSI‐124 (1 mg/kg/day) alone, erlotinib (25 mg/kg/day) alone, erlotinib in combination with JSI‐124, or vehicle alone. JSI‐124 alone did not inhibit tumor growth. Erlotinib alone did not significantly affect the tumor volume. The two‐drug combination resulted in significant tumor shrinkage compared to either drug alone (P < 0.05). Differences in tumor volume were compared using Student's t‐test. Bars, standard error.
Discussion
Several EGFR‐related proteins were evaluated to identify the signal abnormalities of erlotinib‐resistant PC‐9/ER3 cells, which had JAK2 activation without STAT3 activation. We found that JAK2 phosphorylation followed by the activation of Akt partially accounted for acquired erlotinib resistance and that resistance could be overcome by treatment with a combination of erlotinib and a JAK2 inhibitor.
The STAT family of transcription factors consists of seven proteins in humans (STAT1–STAT4, STAT5A, STAT5B and STAT6) that are encoded by separate genes. STAT3 and STAT5 are the STAT most often implicated in human cancer progression.28 Activated STAT3 and STAT5 were expressed in approximately 55 and 33% of NSCLC tumors, respectively.29, 30 STAT3 has been the subject of more investigations than STAT5.28 Our previous study showed that pSTAT3 was less suppressed compared to EGFR, despite the administration of gefitinib in our mutant EGFR‐transgenic mice. This suggests that signals from upstream might activate STAT3 even in EGFR‐driven lung cancer.31 Although in the present study we focused on STAT3, STAT5 will be further examined in our future experiments. Aberrant STAT3 activation was shown to be required for the survival of human cancer cells by promoting the overexpression of genes that encode anti‐apoptotic proteins, cell‐cycle regulators and angiogenic factors.32, 33, 34 STAT activation by cytokines is mediated through JAK, which include four family members, JAK1, JAK2, JAK3 and Tyk2.35 In PC‐9/ER3 cells, STAT3 did not seem to play a critical role in erlotinib resistance. Activation of Akt was inhibited by blocking activation of JAK2 in PC‐9/ER3 cells (Fig. 5). Although a direct relationship between the JAK2 and Akt pathways remains unclear, our data indicated a connection. Vogt and Hart supposed that two branches of oncogenic signal initiated by PI3K (Akt‐mTOR and BMX‐STAT3 pathways) were networked.36 Although it has never been proved, Akt from JAK2 axis in EGFR resistance might emerge in the network.
Recent studies in breast cancer, lung cancer and diffuse large B cell lymphoma cell lines have demonstrated a central role for JAK family kinases in mediating IL‐6 signaling in these cells.29, 37, 38 Our study provides a molecular reason for JAK2 activation and highlights JAK2 as one of several mechanisms of acquired drug resistance and a potential target for recovery from resistance. No difference was observed in IL‐6 protein levels evaluated by enzyme‐linked immunosorbent assay between PC‐9 and PC‐9/ER3 cells (data not shown). Lee et al.39 report that JAK1 and JAK2 activation participates in IL‐5‐induced upregulation of c‐MYC in a human hematopoietic progenitor cell line. In Bcr‐Abl+ chronic myelogenous leukemia cells, the activation of JAK2 did not lead to STAT5 activation, which was activated by Bcr‐Abl.40, 41 One major effect of the activation of JAK2 by the Bcr‐Abl oncoprotein is increased c‐MYC expression, which is required for leukemia induction.40, 41, 42, 43 Inhibition of JAK2 resulted in decreased pAkt and c‐MYC in imatinib‐resistant chronic myelogenous leukemia cells, and JAK2 was identified as a potentially important therapeutic target for imatinib‐resistant chronic myelogenous leukemia.43 MYC is a classical oncogene in lung cancer, and its amplification in adenocarcinoma of the lung occurs in both late and early stages of lung cancer progression and serves as a prognostic molecular marker.44 Thus, we expected that inhibition of JAK2 would result in decreased pAkt and c‐MYC, which would lead to PC‐9/ER3 cell death independently of STAT3 suppression. In reality, JAK2‐specific siRNA inhibited c‐MYC expression, and the combination of erlotinib and JSI‐124 efficiently suppressed c‐MYC expression in PC‐9/ER3 cells (Fig. S7). The relationships among JAK2, Akt and MYC as downstream signals of EGFR in erlotinib‐resistant lung cancer cells should be pursued further.
Our study had some limitations. Only one cell line (PC‐9/ER3) was extensively investigated. The activation of JAK2 was also considered to be one of the various EGFR‐TKI‐resistant mechanisms in the other resistant line (PC‐9/ER5) derived from PC‐9. However, this mechanism may be applied only to PC‐9. We should establish further EGFR‐TKI‐resistant cell lines derived from other sensitive cell lines, such as HCC827 and H3255, and need to verify whether activated JAK2 was really related with the resistant mechanism. In addition, the resistant cells were 136‐fold more resistant to erlotinib than the parental cells, which is not easily translatable to a clinical situation. However, such a big difference in sensitivity to erlotinib between the cells might have helped us identify a new mechanism of resistance. Patients with NSCLC who undergo surgery and have high pJAK2 expression have a significantly worse overall survival rate compared with those with low pJAK2 expression.45 Future studies should examine pJAK2 expression in EGFR‐TKI‐resistant clinical samples.
Unexpectedly, PC‐9/ER3 cells did not carry the T790M mutation, although PC‐9 cells easily developed the T790M mutation after continuous exposure to gefitinib46 or vandetanib.47 It was found that 5 (12.8%) in 39 erlotinib‐resistant tumors in mutated EGFR‐driven transgenic mice expressed the T790M mutation.48 The reason why PC‐9/ER3 cells selectively bypassed JAK2 signaling but did not develop the T790M mutation or MET overexpression is unknown.
In conclusion, we were able to show participation of the JAK2 pathway as one of the mechanisms of acquired erlotinib resistance and that acquired erlotinib resistance could be overcome by suppression of the JAK2 pathway both in vitro and in vivo.
Disclosure Statement
Drs Takigawa and Kiura were paid an honorarium for lecturing from Chugai Company, Japan. The other authors report no conflict of interest.
Supporting information
Fig. S1. IC 50 values of erlotinib and gefitinib in PC‐9 and PC‐9/ER3, 4 and 5 cells determined by MTT assay. Fig. S2. MET gene copy number relative to GAPDH in PC‐9 and PC‐9/ER1, 2, 3, 4 and 5 cells. Fig. S3. Protein expression in PC‐9 and PC‐9/ER3, 4 and 5 cells treated with or without erlotinib for 6 h. Fig. S4. Target protein expression in PC‐9 and PC‐9/ER3 cells treated with each indicated concentrations of LY294002 or Stattic. Fig. S5. Growth inhibition ratio after treatment with erlotinib plus JSI‐124 of PC‐9/ER5 cells. Fig. S6. The combination of erlotinib with inhibition of JAK2 using siRNA in PC‐9/ER3 cells suppressed pAkt in PC‐9/ER3 cells. Fig. S7. Inhibition of JAK2 suppresses c‐MYC expression.
Acknowledgments
This work was supported, in part, by a Grant‐in‐Aid from The Ministry of Education, Culture, Sports, Science, and Technology of Japan (No. 21591182 and No. 23390221).
(Cancer Sci 2012; 103: 1795– 1802)
References
- 1. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011; 61: 212–36. [DOI] [PubMed] [Google Scholar]
- 2. Goldstraw P, Crowley J, Chansky K et al The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol 2007; 2: 706–14. [DOI] [PubMed] [Google Scholar]
- 3. Paez JG, Janne PA, Lee JC et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 4. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 5. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sordella R. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 7. Tsao MS, Sakurada A, Cutz JC et al Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 8. Inoue A, Suzuki T, Fukuhara T et al Prospective phase II study of gefitinib for chemotherapy‐naive patients with advanced non‐small‐cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006; 24: 3340–6. [DOI] [PubMed] [Google Scholar]
- 9. Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res 2011; 17: 5530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bean J, Brennan C, Shih JY et al MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamamoto C, Basaki Y, Kawahara A et al Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib‐resistant lung cancer cells harboring epidermal growth factor receptor‐activating mutations. Cancer Res 2010; 70: 8715–25. [DOI] [PubMed] [Google Scholar]
- 12. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 13. Sequist LV, von Pawel J, Garmey EG et al Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non‐small‐cell lung cancer. J Clin Oncol 2011; 29: 3307–15. [DOI] [PubMed] [Google Scholar]
- 14. Takezawa K, Okamoto I, Tanizaki J et al Enhanced anticancer effect of the combination of BIBW2992 and thymidylate synthase‐targeted agents in non‐small cell lung cancer with the T790M mutation of epidermal growth factor receptor. Mol Cancer Ther 2010; 9: 1647–56. [DOI] [PubMed] [Google Scholar]
- 15. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007; 98: 1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non‐small‐cell lung cancer‐associated mutations of the epidermal growth factor receptor. Cancer Res 2006; 66: 3162–8. [DOI] [PubMed] [Google Scholar]
- 17. Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI‐124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res 2003; 63: 1270–9. [PubMed] [Google Scholar]
- 18. Ono M, Hirata A, Kometani T et al Sensitivity to gefitinib (Iressa, ZD1839) in non‐small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal‐regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther 2004; 3: 465–72. [PubMed] [Google Scholar]
- 19. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55–63. [DOI] [PubMed] [Google Scholar]
- 20. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small‐molecule inhibitor of STAT3 activation and dimerization. Chem Biol 2006; 13: 1235–42. [DOI] [PubMed] [Google Scholar]
- 21. Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 1994; 269: 5241–8. [PubMed] [Google Scholar]
- 22. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–81. [DOI] [PubMed] [Google Scholar]
- 23. Miyazawa H, Tanaka T, Nagai Y et al Peptide nucleic acid‐locked nucleic acid polymerase chain reaction clamp‐based detection test for gefitinib‐refractory T790M epidermal growth factor receptor mutation. Cancer Sci 2008; 99: 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagai Y, Miyazawa H, Huqun et al Genetic heterogeneity of the epidermal growth factor receptor in non‐small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid‐locked nucleic acid PCR clamp. Cancer Res 2005; 65: 7276–82. [DOI] [PubMed] [Google Scholar]
- 25. Naoki K, Soejima K, Okamoto H et al The PCR‐invader method (structure‐specific 5′ nuclease‐based method), a sensitive method for detecting EGFR gene mutations in lung cancer specimens; comparison with direct sequencing. Int J Clin Oncol 2011; 16: 335–44. [DOI] [PubMed] [Google Scholar]
- 26. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 27. Lutterbach B, Zeng Q, Davis LJ et al Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res 2007; 67: 2081–8. [DOI] [PubMed] [Google Scholar]
- 28. Lai SY, Johnson FM. Defining the role of the JAK‐STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist Updat 2010; 13: 67–78. [DOI] [PubMed] [Google Scholar]
- 29. Gao SP, Mark KG, Leslie K et al Mutations in the EGFR kinase domain mediate STAT3 activation via IL‐6 production in human lung adenocarcinomas. J Clin Invest 2007; 117: 3846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanchez‐Ceja SG, Reyes‐Maldonado E, Vazquez‐Manriquez ME, Lopez‐Luna JJ, Belmont A, Gutierrez‐Castellanos S. Differential expression of STAT5 and Bcl‐xL, and high expression of Neu and STAT3 in non‐small‐cell lung carcinoma. Lung Cancer 2006; 54: 163–8. [DOI] [PubMed] [Google Scholar]
- 31. Takata S, Takigawa N, Segawa Y et al STAT3 expression in activating EGFR‐driven adenocarcinoma of the lung. Lung Cancer 2012; 75: 24–9. [DOI] [PubMed] [Google Scholar]
- 32. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000; 19: 2474–88. [DOI] [PubMed] [Google Scholar]
- 33. Grandis JR, Drenning SD, Zeng Q et al Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A 2000; 97: 4227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Niu G, Wright KL, Huang M et al Constitutive Stat3 activity up‐regulates VEGF expression and tumor angiogenesis. Oncogene 2002; 21: 2000–8. [DOI] [PubMed] [Google Scholar]
- 35. Schindler C, Darnell JE Jr. Transcriptional responses to polypeptide ligands: the JAK‐STAT pathway. Annu Rev Biochem 1995; 64: 621–51. [DOI] [PubMed] [Google Scholar]
- 36. Vogt PK, Hart JR. PI3K and STAT3: a new alliance. Cancer Discov 2011; 1: 481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berishaj M, Gao SP, Ahmed S et al Stat3 is tyrosine‐phosphorylated through the interleukin‐6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res 2007; 9: R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lam LT, Wright G, Davis RE et al Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor‐{kappa}B pathways in subtypes of diffuse large B‐cell lymphoma. Blood 2008; 111: 3701–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee WH, Liu FH, Lin JY et al JAK pathway induction of c‐Myc critical to IL‐5 stimulation of cell proliferation and inhibition of apoptosis. J Cell Biochem 2009; 106: 929–36. [DOI] [PubMed] [Google Scholar]
- 40. Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c‐Myc induction by Bcr‐Abl. Oncogene 2002; 21: 7137–46. [DOI] [PubMed] [Google Scholar]
- 41. Nieborowska‐Skorska M, Wasik MA, Slupianek A et al Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med 1999; 189: 1229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992; 70: 901–10. [DOI] [PubMed] [Google Scholar]
- 43. Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res 2006; 66: 6468–72. [DOI] [PubMed] [Google Scholar]
- 44. Iwakawa R, Kohno T, Kato M et al MYC amplification as a prognostic marker of early‐stage lung adenocarcinoma identified by whole genome copy number analysis. Clin Cancer Res 2011; 17: 1481–9. [DOI] [PubMed] [Google Scholar]
- 45. Zhao M, Gao FH, Wang JY et al JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non‐small‐cell lung cancer. Lung Cancer 2011; 73: 366–74. [DOI] [PubMed] [Google Scholar]
- 46. Ogino A, Kitao H, Hirano S et al Emergence of epidermal growth factor receptor t790m mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res 2007; 67: 7807–14. [DOI] [PubMed] [Google Scholar]
- 47. Ichihara E, Ohashi K, Takigawa N et al Effects of vandetanib on lung adenocarcinoma cells harboring epidermal growth factor receptor T790M mutation in vivo. Cancer Res 2009; 69: 5091–8. [DOI] [PubMed] [Google Scholar]
- 48. Politi K, Fan PD, Shen R, Zakowski M, Varmus H. Erlotinib resistance in mouse models of epidermal growth factor receptor‐induced lung adenocarcinoma. Dis Model Mech 2010; 3: 111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. IC 50 values of erlotinib and gefitinib in PC‐9 and PC‐9/ER3, 4 and 5 cells determined by MTT assay. Fig. S2. MET gene copy number relative to GAPDH in PC‐9 and PC‐9/ER1, 2, 3, 4 and 5 cells. Fig. S3. Protein expression in PC‐9 and PC‐9/ER3, 4 and 5 cells treated with or without erlotinib for 6 h. Fig. S4. Target protein expression in PC‐9 and PC‐9/ER3 cells treated with each indicated concentrations of LY294002 or Stattic. Fig. S5. Growth inhibition ratio after treatment with erlotinib plus JSI‐124 of PC‐9/ER5 cells. Fig. S6. The combination of erlotinib with inhibition of JAK2 using siRNA in PC‐9/ER3 cells suppressed pAkt in PC‐9/ER3 cells. Fig. S7. Inhibition of JAK2 suppresses c‐MYC expression.