

Abstract
Priming of CD8+ T cells requires two signals, one produced by T‐cell receptor recognition of antigen, and a second that is often provided by the innate immune response. In this context, antigens non‐covalently or covalently associated with heat shock proteins (HSP) are internalized and processed in antigen‐presenting cells (APC) to be presented by MHC I molecules to CD8+ T cells, thus, signal 1 has been well characterized in this pathway of cross‐presentation. Signal 2 is not fully understood, although there are reports that Toll‐like receptors (TLRs) interact with HSP and activate APC. The ability of HSP to activate APC through TLRs is, however, controversial because of the possibility of endotoxin contamination. Using a variety of TLR KO mice, we present evidence that TLRs (TLR2, 3, 4, 7, and 9) and their adaptor molecules MyD88 and IRAK4 are dispensable in cross‐priming by a mycobacterial HSP70–antigen (ovalbumin as a model antigen) fusion protein; in contrast, MyD88/IRAK4, but not TLRs, are required for tumor rejection induced by the same reagent. Our results indicate that HSP‐mediated cross‐priming uses a second signal produced by mechanisms other than TLR cascades. We hypothesize that efficient cross‐priming by HSP70 alone is insufficient for tumor rejection and that MyD88/IRAK4‐dependent inflammatory stimulation, which might contribute to maintenance of the initially primed effector cells, is required to eradicate tumor burden. (Cancer Sci 2012; 103: 851–859)
Heat shock protein (HSP) acts as a vehicle to deliver its associated antigen (Ag) to antigen‐presenting cells (APC), such as dendritic cells, and this then facilitates Ag cross‐presentation to CD8+ T cells.1, 2, 3 Various aspects of this pathway have been well characterized. For example, certain scavenger receptors such as CD91,4 LOX‐1,5 and SREC‐16 on APC are used for internalization of extracellular HSP for the cross‐presentation pathway.7 This receptor‐mediated endocytosis mechanism to internalize HSP‐Ag complexes contributes to extremely efficient antigen cross‐presentation, compared to Ag alone.
In addition to its role in the adaptive immune response, HSP has been implicated as a natural ligand for a variety of receptors involved in the activation of innate immunity. There are reports showing that Toll‐like receptor (TLR)2 and TLR4 are receptors for Gp96 and HSP70 for activation of the NF‐κB signaling pathway.8, 9 CD4010 and CCR511 also serve as receptors for HSP70. The interaction between HSP and these receptors is important in terms of providing a second signal required for full activation of T cells, together with the first signal provided by Ag recognition through the TCR. Despite a significant number of reports describing the importance of TLRs and the MyD88 adaptor molecule in HSP‐mediated cross‐priming,12 there are other conflicting reports. Very highly purified HSP did not activate APC, although it was able to stimulate cross‐presentation,13 thus contamination with endotoxin‐like molecules in HSP preparations used in many experiments is the most critical point to be carefully evaluated.14 Moreover, most results implicating TLRs have been obtained by in vitro experiments. Therefore, the potential involvement of TLRs in HSP‐induced cross‐priming needs to be revisited.
Given this controversy regarding TLR involvement, we decided to use a variety of TLR KO mice to define whether TLRs and their adaptor molecules MyD88 and IRAK4 are required for in vivo cross‐priming and tumor rejection elicited by a mycobacterial HSP70–ovalbumin (OVA) fusion protein. We found that in vivo cross‐priming, as evaluated by proliferation, γ‐interferon (IFN‐γ) production, and specific cytolytic activity of adoptively transferred OT‐I CD8+ T cells, did not require TLR2, 3, 4, 7, or 9, or MyD88/IRAK4. Moreover, generation of OVA‐specific CTLs from naïve mice immunized with mycobacterial HSP70‐OVA again revealed MyD88 dispensability. In striking contrast, however, we found that MyD88 and IRAK4 are indispensable in mycobacterial HSP70‐OVA‐dependent rejection of a tumor expressing endogenous OVA. These results reveal an unexpected complexity in the requirement for the TLR‐mediated signaling cascade: it is not essential for cross‐priming by HSP70 but is required for tumor rejection.
Materials and Methods
Cells
Bone marrow was harvested from C57BL/6 mice to generate bone marrow‐derived dendritic cells (BMDC). The BMDC were cultured in 24‐well plates at a density of 1 × 106 cells/mL with RPMI‐1640 (Sigma, St. Louis, MO, USA) containing 10% FCS, 2 mM l‐glutamine, 1 mM sodium pyruvate, 0.1 mM non‐essential amino acid, penicillin–streptomycin, 2‐mercaptoethanol, and 20 ng/mL granulocyte‐macrophage colony‐stimulating factor (GM‐CSF; R&D Systems, Minneapolis, MN, USA). E.G7 is an OVA cDNA transfected derivative of the EL4 (methylchoranthlene‐induced thymoma of C57BL/6 [H‐2b] origin) cell line.15
Mice
C57BL/6 mice were purchased from CLEA Japan (Tokyo, Japan) and kept in the animal facilities of the Research Center for Allergy and Immunology, RIKEN (Yokohama, Japan). The CD11c‐diphtheria toxin receptor (DTR) mice have been described previously.16 Knock‐out and transgenic mice described below were kindly provided by Dr Carbone (OT‐I; University of Melbourne, Melbourne, Australia),17 Dr Akira (TLRs KO, MyD88 KO, IL‐18KO, IL18R KO; Osaka University, Osaka, Japan),18, 19, 20 Dr Yeh (IRAK4 KO; University of Toronto, Toronto, Canada),21 Dr Kikutani (CD40 KO; Osaka University),22 and Dr Tonegawa (TAP1 KO; MIT, Cambridge, MA, USA).23
Reagents
Synthetic OVA257–264 (SIINFEKL) peptide was purchased from Qiagen (Tokyo, Japan). Expression and purification of recombinant His‐tagged full‐length OVA fused with Mycobacterium tuberculosis HSP70 (OVA‐HSP70) and control His‐tagged full‐length OVA (OVA) were carried out as described previously.24
Proliferation of transferred OT‐I CD8+ T cells
The OT‐I spleen cells were purified by sequentially negative (CD11b) and positive (CD8) BD IMag purification systems (BD Biosciences, Franklin Lakes, NJ, USA) selection. Purified OT‐I CD8+ T cells were labeled with 10 μM carboxyfluorescein succinimidyl ester (CFSE) (Dojindo, Kumamoto, Japan) and then adoptively transferred into recipient mice by tail vein injection. The recipient mice were simultaneously injected with 10 μg or the indicated dose of OVA‐HSP70 or OVA into the tail vein. Three days later, spleen was determined by CFSE dilution as assessed by flow cytometry. With CD11c‐DTR mice, the CD11c+ dendritic cells (DC) were depleted by i.p. injection of diphtheria toxin (DT), 10 μg/kg (day −1) and 5 μg/kg (day 1). CD4+ and natural killer (NK) cells were depleted with GK1.5 and PK136 antibodies on day −1 (500 μg/mouse via tail vein).
Intracellular cytokine staining of transferred OT‐I CD8+ T cells
Transfer of CFSE‐labeled OT‐I CD8+ T cells and OVA‐HSP70 were carried out as described above. Three days later, the recipients were killed and the spleen cells were stimulated with 1 μM OVA257–264 in the presence of 5 μg/ml brefeldin A at 37°C for 4 h. Intracellular IFN‐γ staining was carried out per the manufacturer's instructions (BD Biosciences). Production of IFN‐γ and proliferation of OT‐I CD8+ T cells were determined by flow cytometry.
In vitro cross‐presentation assay
Bone marrow dendritic cells (1 × 106) were suspended in 500 μL complete RPMI containing indicated doses of OVA‐HSP70. The samples were incubated for 3 h at 37°C then fixed with 0.5% paraformaldehyde for 10 min at room temperature followed by 30 min quenching with 0.1 M glycine. The OT‐I CD8+ T cells were purified with the BD IMag purification system. Bone marrow‐derived dendritic cells (1 × 104/well) or the indicated number of fixed BMDC and 1 × 105/well OT‐I CD8+ T cells were co‐cultured in 96‐well round bottom plates with complete RPMI. Supernatant was collected 24–72 h later and the amount of IFN‐γ was measured by an ELISA assay.
Vaccination and induction of CTL
Mice were vaccinated i.v. three times within a 1‐week interval with 10 μg of OVA‐HSP70. Seven days after the final immunization, the spleen cells were stimulated in vitro with 10 μM OVA257–264 for 1 h, then cultured for 5 days at 37°C. Induction of antigen‐specific CTL was determined by tetramer antibody (MBL, Nagoya, Japan) staining and a standard 51Cr‐release assay.
Cross‐presentation assay and 51 Cr release assay with splenic DC
After collagenase treatment and red blood cell removal, C57BL/6 spleen cells were stained with FITC‐CD8 and PE‐CD11c. Then, CD8+ CD11c+ DC and CD8− CD11c+ DC were purified with a FACSAria (BD Biosciences). The cross‐presentation assay was carried out as described above in “In vitro cross‐presentation assay”. Briefly, DC were pulsed with 150 μg/mL OVA‐HSP70 for 3 h. The indicated number of fixed BMDC and 1 × 105/well OT‐I CD8+ T cells were co‐cultured in a 96‐well round plate. After the cross‐presentation assay, a 51Cr release assay was carried out with co‐cultured OT‐I CD8+ T cells (originally 1 × 105/well) as effectors. Specific cell lysis activity toward E.G7 (5 × 103/well) was measured with a standard in 51 Cr release assay.
In vivo CTL assay
On day 0, recipient mice were injected i.v. with 1 × 106 OT‐I CD8+ T cells mixed with 10 μg OVA‐HSP70. On day 3, mice were injected i.v. with differentially CSFE‐labeled spleen cells as target cells. The red blood cells had been removed from these spleen cells by osmotic lysis, then the cells were split into two populations. One was labeled with a high concentration of CFSE (10 μM), and pulsed with 1 μM OVA257–264 at 37°C for 1 h. The other population (control) was labeled with a low concentration of CFSE (1 μM). An equal number of the two target cells was mixed and a total of 2 × 107 cells was injected i.v. Twenty hours later, the two types of target cells in the spleen were analyzed by flow cytometry.
Immunotherapy against an established tumor
On day 0, recipient mice were inoculated intradermally with 1 × 106 E.G7 cells on the right side of the back. On day 6, mice were injected with 1 × 106 OT‐I CD8+ T cells into the tail vein. On days 6, 8, 10, and 12, mice were vaccinated i.v. with 10 μg OVA‐HSP70. Tumor sizes were measured with calipers, and the average tumor diameters were calculated using the formula:
Average tumor diameter = [longest tumor diameter + shortest tumor diameter]/2
Results
Antigen fused with HSP70 is more efficient in cross‐priming of T cells than antigen alone
As it has been noted that a histidine tag by itself is immunogenic,25 we prepared histidine‐tagged recombinant OVA as a control protein. Thus, 6× His‐tagged recombinant OVA and OVA‐HSP70 fusion proteins (Fig. 1a, left panel) were expressed in Escherichia coli and purified with Ni2+‐NTA agarose. The purified proteins were further applied on Detoxi‐Gel AffinityPak Prepacked Column (Thermo Scientific, Waltham, MA, USA) to remove endotoxin. We confirmed that the level of endotoxin in the final preparation was <0.5 EU/mL of solution using a test from Mitsubishi Chemical Medience (Tokyo, Japan) (data not shown). Both OVA, purchased from Sigma, and the recombinant proteins were separated by SDS‐PAGE and stained with Coomassie Brilliant Blue to assess purity (Fig. 1a, right panel). Graded doses of the recombinant proteins were injected i.v. into C57BL/6 (B6) mice, thereafter, CFSE‐labeled OT‐I CD8+ T cells were adoptively transferred i.v. to those mice. Three days later, the transferred T cells were recovered from the spleen and analyzed for in vivo proliferation by flow cytometry. We observed that 10 pmol OVA‐HSP70 induced significant division of the transferred T cells, whereas 100 pmol OVA protein was unable to stimulate division; 1000 pmol was required (Fig. 1b). These results indicate that OVA‐HSP70 is at least 10‐fold more efficient in priming naïve OT‐I CD8+ T cells than OVA alone.
Figure 1.
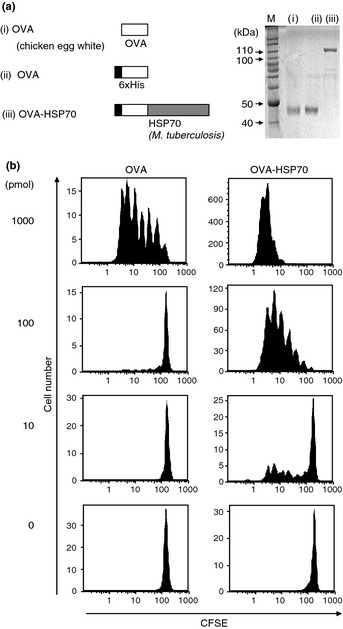
Antigen fused with heat shock protein (HSP)70 is more efficient in cross‐priming T cells than antigen alone. Recombinant proteins used in this study and their purity as assessed by SDS‐PAGE and Coomassie Brilliant Blue staining (a). Carboxyfluorescein succinimidyl ester (CFSE)‐labeled OT‐I CD8+ T cells were adoptively transferred into C57BL/6 mice and simultaneously the indicated dose of ovalbumin (OVA) or OVA‐HSP70 was injected. Three days later, division of OT‐I CD8+ T cells in recipient mice spleens was determined by assessing CFSE dilution by flow cytometry (b).
Cross‐priming induced by HSP70 is dependent on DC and the transporter associated with antigen processing (TAP) complex
To examine cells responsible for cross‐priming by OVA‐HSP70, we first used CD11c‐DTR mice in which DT eliminates CD11c+ cells, thus DC, in vivo.16 We found that DT‐induced depletion of CD11c+ cells led to abrogation of division of transferred OT‐I CD8+ T cells (Fig. 2a, upper column). The same experiments with WT mice indicated the toxicity of DT had no influence on the division of transferred OT‐I CD8+ T cells. (Fig. 2a, lower column). Proliferation of OT‐I CD8+ T cells was also completely abolished in TAP1 KO mice, whereas in vivo elimination of CD4+ T cells or NK cells by injection of depleting antibodies had no effect on the proliferation (Fig. 2b,c). To further identify cells to which OVA‐HSP70 is targeted, Alexa Fluor 633‐labeled fusion protein was injected into mice. Three hours post‐injection, the labeled protein was found mostly accumulated on CD11c+ B220− cells (close to 25% of the total DC population; Fig. 3c) but not on other cells (Fig. 3a,b), and the frequency of positive cells and the level of accumulation declined by 24 h (Fig. 3c), suggesting that in vivo OVA‐HSP70 is internalized mainly by DC. Note that OVA alone did not accumulate on CD11c+ B220− cells (data not shown). Taken together, these results indicate that in vivo HSP70 uses the conventional cross‐presentation pathway, which requires TAP and DC.
Figure 2.
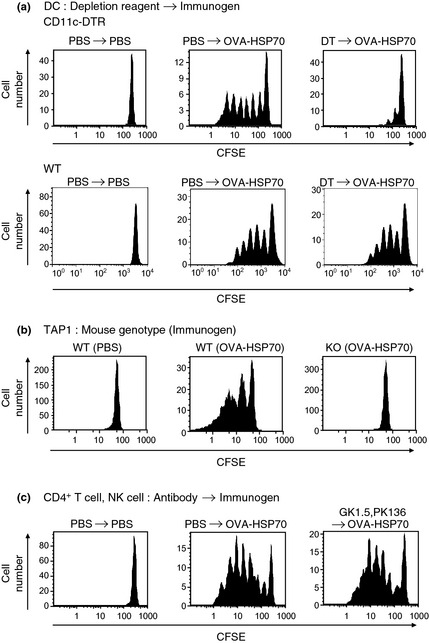
Heat shock protein (HSP)‐mediated cross‐presentation in vivo is dependent on CD11c+ dendritic cells (DC) and transporter associated with antigen processing (TAP). The HSP‐mediated proliferation of transferred carboxyfluorescein succinimidyl ester (CFSE)‐labeled OT‐I CD8+ T cells was measured in DC depleted (a), TAP1 KO (b), and CD4+ T cell and natural killer (NK) cell depleted (c) recipient mice. Injection of CFSE‐labeled OT‐I CD8+ T cells and ovalbumin (OVA)‐HSP70 (10 μg = 100 pmol) and flow cytometry were carried out. In CD11c–diphtheria toxin receptor (DTR) mice, but not WT mice, CD11c+ cells were depleted by injection of DT on days −1 and 1 (a). CD4+ and NK cells were depleted with injection of GK1.5 (anti‐CD4) and PK136 (anti‐NK1.1) antibodies on day −1 (c).
Figure 3.
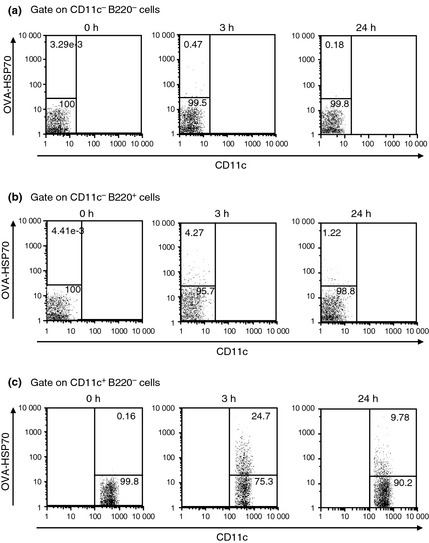
CD8+ dendritic cells are responsible for heat shock protein (HSP)70‐mediated cross‐priming in vivo. C57BL/6 mice were injected i.v. with 100 μg Alexa Fluor 633‐labeled ovalbumin (OVA)‐HSP70. Three hours or 24 h later, spleen cells of recipient mice were recovered and stained with FITC‐CD11c and PE‐B220, then OVA‐HSP70 uptake was measured by flow cytometry. The numbers indicate the frequency of cells within a particular gate.
CD8+ dendritic cells responsible for Hsp70‐mediated cross‐priming in vivo
As CD8+ and not CD8− DC are primarily involved in cross‐priming in vivo,26 we next investigated whether this was also the case for HSP70‐induced cross‐priming. Based on the results of the studies in Figure 3(c), we further separated CD11c+ cells into CD8+ and CD8− populations and examined them for incorporation of Alexa Fluor 633‐labeled OVA‐HSP70. We found that ~28% CD8+ DC bound the fusion proteins, whereas only ~18% of the CD8− DC did so. Co‐culture assays using OT‐I CD8+ T cells with sorted CD8+ or CD8− DC allowed us to show that CD8+ DC are more potent in stimulating OT‐I CD8+ T cells to secrete IFN‐γ (Fig. 4b, left panel) and to differentiate into CTL capable of killing EL4 cells pulsed with OVA257–264 (Fig. 4b, right panel). Based on these results, we concluded that HSP70‐mediated cross‐priming is mainly dependent on CD8+ DC and, to a lesser extent, on CD8− DC.
Figure 4.
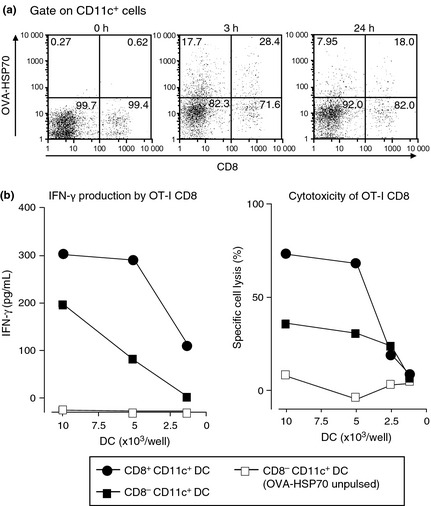
CD8+ dendritic cells (DC) showed more efficient ovalbumin–heat shock protein 70 (OVA‐HSP70) uptake and HSP‐mediated cross‐priming than CD8− DC. Uptake of Alexa Fluor 633‐labeled OVA‐HSP70 by CD8+ and CD8− DC was detected. Both FITC‐CD8 and PE‐CD11c were used for staining (a). The FACS‐purified CD8+ and CD8− DCs were pulsed with 150 μg/mL OVA‐HSP70 for 3 h and fixed. The fixed DC and OT‐I CD8+ T cells were co‐cultured in 96‐well round‐bottom plates. Supernatants were collected and the amount of γ‐interferon (IFN‐γ) was measured by ELISA assay (b, left panel). After supernatants were recovered, the co‐cultured OT‐I CD8+ T cells were used as effectors in a 51Cr release assay against E.G7 target cells (b, right panel).
Toll‐like receptors, MyD88/IRAK4 and CD40, are dispensable for cross‐priming with OVA‐HSP70 in vivo
There is a report suggesting that mycobacterial (bacillus Calmette‐Guerin) HSP65‐mediated cross‐priming requires MyD88,12 leading us to investigate whether this is the case for HSP70‐mediated cross‐priming. To this end, we used a panel of KO mice as hosts and examined the proliferation and production of IFN‐γ of adoptively transferred, CFSE‐labeled OT‐I CD8+ T cells. As shown in Figure 5, none of the tested TLRs (TLR2, 3, 4, 7, and 9), nor the adaptor molecule MyD88, or IRAK4 signaling molecule, or CD40 were essential in the proliferation or IFN‐γ production of OT‐I CD8+ T cells stimulated by vaccination with OVA‐HSP70. Next, mice lacking the indicated TLRs, MyD88/IRAK4, and CD40 were adoptively transferred with OT‐I CD8+ T cells and spleen cells pulsed with/without OVA257–264 as target cells (Fig. 6a). We found that none of these molecules was required for inducing cytolytic activity, as indicated by an in vivo CTL assay. Moreover, we generated GM‐CSF‐stimulated BMDC from WT or MyD88 KO mice and pulsed them with graded doses of OVA or OVA‐HSP70. After fixation, the DC were cultured with naïve OT‐I CD8+ T cells. As expected, based on the in vivo studies depicted in Figure 1, we observed that OVA‐HSP70 was much more efficient than OVA in activating T cells to secrete IFN‐γ (Fig. 6b). Consistent with the results of the in vivo CTL assay, this T‐cell response was independent of MyD88.
Figure 5.
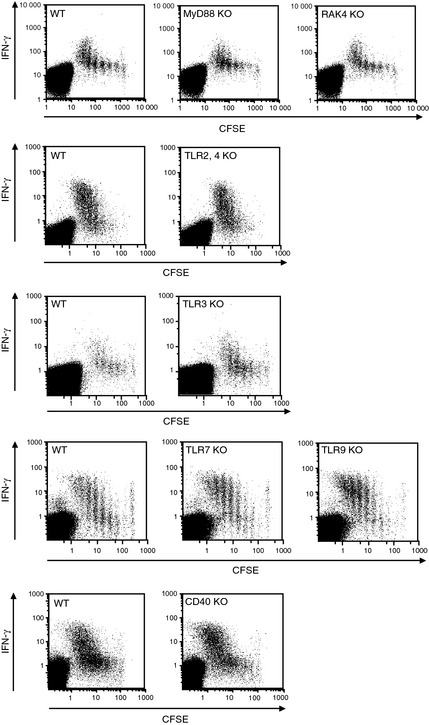
Heat shock protein (HSP)‐mediated cross‐priming in vivo is independent of Toll‐like receptors (TLR)/MyD88 and CD40. Carboxyfluorescein succinimidyl ester (CFSE)‐labeled OT‐I CD8+ T cells were adoptively transferred into C57BL/6 or the indicated KO mice. Simultaneously the mice were injected with 10 μg ovalbumin (OVA)‐HSP70. Three days later, recipient spleen cells were stimulated with OVA 257–264 in the presence of brefeldin A at 37°C for 4 h. After intracellular γ‐interferon (IFN‐γ) staining, IFN‐γ production and proliferation of transferred OT‐I CD8+ T cells were determined by flow cytometry.
Figure 6.

Ovalbumin–heat shock protein 70 (OVA‐HSP70) can induce specific CTLs without Toll‐like receptors (TLR)/MyD88 and CD40 signaling. On day 0, recipient mice were transferred with OT‐I CD8+ T cells and simultaneously vaccinated with 10 μg OVA‐HSP70. On day 3, mice were injected i.v. with two kinds of target cells. One was labeled with a high concentration of carboxyfluorescein succinimidyl ester (CFSE) and pulsed with 1 μM OVA 257–264, the other was labeled with a low concentration of CFSE and not pulsed with peptide. Twenty hours later, recipient mice were killed and the frequency of the two types of target cells remaining in the spleen was analyzed by flow cytometry (a). Bone marrow dendritic cells from C57BL/6 or MyD88 KO mice were incubated with OVA‐HSP70 for 3 h then fixed. The bone marrow dendritic cells and OT‐I CD8+ T cells were then co‐cultured in 96‐well round‐bottom plates. Supernatants were collected and the amount of γ‐interferon (IFN‐γ) was measured by ELISA assay (b). C57BL/6 and MyD88 KO mice were vaccinated three times in a 1‐week interval with 10 μg OVA‐HSP70. Seven days after the final immunization, recipient mice were killed and the spleen cells were stimulated with OVA 257–264 in vitro. Induction of antigen‐specific CD8+ T cells was detected with H‐2K b/OVA 257–264‐specific tetramers by flow cytometry (c), and CTL activity of the splenocytes was measured by 51 Cr release assay (d). E.G7, OVA cDNA transfected derivative of the EL4 cell line; EL4, methylchoranthlene‐induced thymoma of C57BL/6 (H‐2b) origin; E/T, effector/target.
It remained a possibility that experiments using OT‐I CD8+ T cells are biased to bypass the requirement for TLRs in cross‐priming. Therefore, we immunized WT and MyD88 KO mice with OVA‐HSP70, and the spleen cells were then stimulated in vitro with OVA257–264 peptide for 5 days. The resulting effector cells were stained with anti‐CD8 antibody and a tetramer consisting of Kb‐OVA257–264. We found that antigen‐specific CTLs were generated in both WT and MyD88 KO mice (Fig. 6c), and that both effector cells showed comparable cytolytic activity against EL4 cells pulsed with OVA peptide and E.G7 cells, which express OVA endogenously (Fig. 6d). Taken together, these results lead us to conclude that TLR pathway molecules including MyD88/IRAK4 and CD40 are not essential for HSP70‐mediated cross‐priming.
MyD88 and IRAK4 are indispensable in HSP70‐mediated tumor rejection
Based on the above results, we set up a final series of experiments that we expected to confirm that TLRs, MyD88/IRAK4, and CD40 were also not required for another well‐established HSP70‐mediated cross‐priming activity, the antitumor effect. For this purpose, we challenged our panel of KO mice with live E.G7 tumor cells and on day 6, OT‐I CD8+ T cells were adoptively transferred and the mice were vaccinated four times with OVA‐HSP70 (Fig. 7). We expected that, like WT mice, all the KO mice would reject the growing tumors after receiving OT‐I CD8+ T cells and OVA‐HSP70. Accordingly, TLR2/4‐, TLR3‐, and TLR7/9‐deficient mice completely rejected the tumors; however, to our surprise, mice lacking MyD88 or IRAK4 were unable to reject the tumors. CD40‐deficient mice rejected the tumors.
Figure 7.
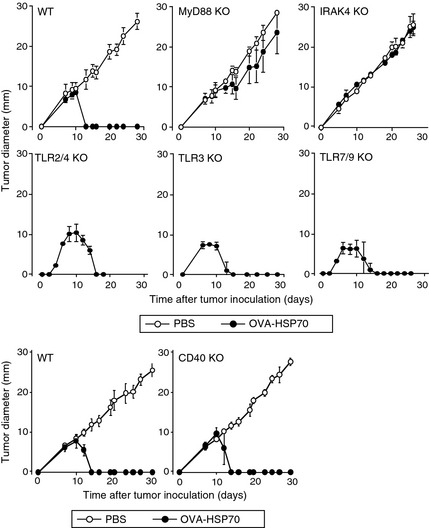
MyD88 and IRAK4 are indispensable in heat shock protein (HSP)70‐mediated tumor rejection. On day 0, recipient mice were inoculated intradermally with E.G7 cells. On day 6, mice were transferred with OT‐I CD8+ T cells into the tail vein. On days 6, 8, 10, and 12, mice were vaccinated i.v. with 10 μg ovalbumin (OVA)‐HSP70. Tumor sizes were measured and the average tumor diameters were calculated with the formula: Average tumor diameter = [longest tumor diameter + shortest tumor diameter]/2. TLR, Toll‐like receptor.
Discussion
The possible contribution of endotoxin‐like molecules to HSP‐induced antitumor immunity has long been an enigma. The results presented here, at least in part, should help to resolve this issue. Mycobacterial HSP70‐induced cross‐priming as well as tumor rejection was not impaired in TLR2/4 double‐KO mice, clearly indicating that these immune responses are not caused by a contaminating endotoxin, such as E. coli LPS. Moreover, TLR7/9 double‐KO mice and TLR3 KO mice also showed no reduction in cross‐priming or tumor rejection (Figs 5, 7). Strikingly, MyD88, which is a key adaptor molecule for most of the TLRs, was dispensable in proliferation and IFN‐γ production by transferred OT‐ICD8+ T cells and CTL generation in naïve mice (Figs 5, 6). These results strongly argue that the putative second signal provided by HSP‐mediated immunity is not linked with the TLR/MyD88 signaling cascade.
Also implicated as a receptor for HSP70, CD40 facilitates endocytosis as well as activating APC.27 However, we observed no reduction of cross‐priming ability or tumor rejection in CD40 KO mice (Figs 5, 6, 7). Involvement of other receptors, IL‐18R and IL‐1R, included upstream of MyD88, has not yet been tested, which is an important question to be answered in future experiments.
Recently, Pawaria and Binder have shown that CD91 serves as a signaling receptor for Gp96, HSP70, and calreticulin, promoting maturation of APC, secretion of cytokines, and priming of T‐helper cells.28 Interestingly, interaction of Gp96 or calreticulin with CD91 triggers phosphorylation of membrane‐proximal tyrosine 4508, whereas HSP70 binding results in phosphorylation of both tyrosine 4508 and a membrane‐distal tyrosine 4474 of the CD91β chain to transduce signals.28 Although these results were obtained using RAW264.7 cells and peritoneal macrophages, our cross‐priming results are consistent, because CD91 by itself, but not the TLRs, acts as a signaling receptor for HSP70.
Nonetheless, tumor rejection was absolutely dependent on MyD88/IRAK4, although TLR2, 3, 4, 7, and 9 were dispensable (Fig. 7). In this regard, it is possible that cross‐priming by HSP70 is the minimum essential event but, by itself, is insufficient for tumor rejection. Keeping HSP70‐primed T cells upfront as effector cells efficient enough to continuously kill cancer cells might require additional factors such as inflammatory signals that are likely provided by MyD88/IRAK4‐dependent signaling. In this context, there is a report suggesting that ISCOMATRIX vaccine led to normal cross‐presentation of DC isolated from draining lymph node in MyD88 KO mice, as determined by ex vivo proliferation of OT‐ICD8+ T cells in the co‐culture system.29 However, enodogenous T cell responses were significantly reduced in MyD88 KO, but not in TLR4 KO, mice,29 suggesting requirements of inflammatory signals provided by the MyD88‐dependent cascade. In any case, the precise mechanisms underlying cross‐priming and the resulting tumor rejection remain to be elucidated.
In summary, we have shown that TLRs, MyD88/IRAK4, and CD40 are not essential for HSP70‐mediated cross‐priming. We have also shown that MyD88/IRAK4, but not TLRs or CD40, are required for tumor rejection induced by the same reagents.
Disclosure Statement
The authors have no conflicts of interest.
Acknowledgments
We thank Mr. Matsuura (Laboratory for Immunochaperones, Research Center for Allergy and Immunology, RIKEN Yokohama Institute, Yokohama, Japan) for purification of recombinant proteins. This work was supported by a Grant‐in‐Aid for Scientific Research Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan.
References
- 1. Udono H, Srivastava PK. Heat shock protein 70‐associated peptides elicit specific cancer immunity. J Exp Med 1993; 178: 1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein‐chaperoned peptides. Science 1995; 269: 1585–8. [DOI] [PubMed] [Google Scholar]
- 3. Ishii T, Udono H, Yamano T et al Isolation of MHC class I‐restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 1999; 162: 1303–9. [PubMed] [Google Scholar]
- 4. Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol 2000; 1: 151–5. [DOI] [PubMed] [Google Scholar]
- 5. Delneste Y, Magistrelli G, Gauchat J et al Involvement of LOX‐1 in dendritic cell‐mediated antigen cross‐presentation. Immunity 2002; 17: 353–62. [DOI] [PubMed] [Google Scholar]
- 6. Zhou Z, Hartwieg E, Horvitz HR. CED‐1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 2001; 104: 43–56. [DOI] [PubMed] [Google Scholar]
- 7. Calderwood SK, Theriault J, Gray PJ, Gong J. Cell surface receptors for molecular chaperones. Methods 2007; 43: 199–206. [DOI] [PubMed] [Google Scholar]
- 8. Vabulas RM, Braedel S, Hilf N et al The endoplasmic reticulum‐resident heat shock protein Gp96 activates dendritic cells via the Toll‐like receptor 2/4 pathway. J Biol Chem 2002; 277: 20847–53. [DOI] [PubMed] [Google Scholar]
- 9. Asea A, Rehli M, Kabingu E et al Novel signal transduction pathway utilized by extracellular HSP70: role of toll‐like receptor (TLR) 2 and TLR4. J Biol Chem 2002; 277: 15028–34. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y, Kelly CG, Karttunen JT et al CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC‐chemokines. Immunity 2001; 15: 971–83. [DOI] [PubMed] [Google Scholar]
- 11. Floto RA, MacAry PA, Boname JM et alDendritic cell stimulation by mycobacterial Hsp70 is mediated through CCR5. Science 2006; 20: 314. [DOI] [PubMed] [Google Scholar]
- 12. Palliser D, Ploegh H, Boes M. Myeloid differentiation factor 88 is required for cross‐priming in vivo. J Immunol 2004; 172: 3415–21. [DOI] [PubMed] [Google Scholar]
- 13. Bausinger H, Lipsker D, Ziylan U et al Endotoxin‐free heat‐shock protein 70 fails to induce APC activation. Eur J Immunol 2002; 32: 3708–13. [DOI] [PubMed] [Google Scholar]
- 14. Marincek BC, Kühnle MC, Srokowski C et al Heat shock protein‐antigen fusions lose their enhanced immunostimulatory capacity after endotoxin depletion. Mol Immunol 2008; 46: 181–91. [DOI] [PubMed] [Google Scholar]
- 15. Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988; 54: 777–85. [DOI] [PubMed] [Google Scholar]
- 16. Jung S, Unutmaz D, Wong P et al In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell‐associated antigens. Immunity 2002; 17: 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 18. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010; 11: 373–84. [DOI] [PubMed] [Google Scholar]
- 19. Takeda K, Tsutsui H, Yoshimoto T et al Defective NK cell activity and Th1 response in IL‐18‐deficient mice. Immunity 1998; 8: 383–90. [DOI] [PubMed] [Google Scholar]
- 20. Hoshino K, Tsutsui H, Kawai T et al Generation of IL‐18 receptor‐deficient mice: evidence for IL‐1 receptor‐related protein as an essential IL‐18 binding receptor. J Immunol 1999; 162: 5041–4. [PubMed] [Google Scholar]
- 21. Suzuki N, Suzuki S, Duncan GS et al Severe impairment of interleukin‐1 and Toll‐like receptor signalling in mice lacking IRAK‐4. Nature 2002; 416: 750–6. [DOI] [PubMed] [Google Scholar]
- 22. Kawabe T, Naka T, Yoshida K et al The immune responses in CD40‐deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994; 1: 167–78. [DOI] [PubMed] [Google Scholar]
- 23. Van Kaer L, Ashton‐Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4‐8+ T cells. Cell 1992; 71: 1205–14. [DOI] [PubMed] [Google Scholar]
- 24. Mizukami S, Kajiwara C, Ishikawa H, Katayama I, Yui K, Udono H. Both CD4+ and CD8+ T cell epitopes fused to heat shock cognate protein 70 (hsc70) can function to eradicate tumors. Cancer Sci 2008; 99: 1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takakura Y, Takemoto S, Nishikawa M. Hsp‐based tumor vaccines: state‐of‐the‐art and future directions. Curr Opin Mol Ther 2007; 9: 385–91. [PubMed] [Google Scholar]
- 26. den Haan JM, Lehar SM, Bevan MJ. CD8+ but not CD8− dendritic cells cross‐prime cytotoxic T cells in vivo. J Exp Med 2000; 192: 1685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becker T, Hartl FU, Wieland F. CD40, an extracellular receptor for binding and uptake of Hsp70‐peptide complexes. J Cell Biol 2002; 158: 1277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pawaria S, Binder RJ. CD91‐dependent programming of T‐helper cell responses following heat shock protein immunization. Nat Commun. 2011; 2: 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilson NS, Yang B, Morelli AB et al ISCOMATRIX vaccines mediate CD8+T‐cell cross‐priming by a MyD88‐dependent signaling pathway. Immunol Cell Biol 2011; doi: 10.1038/icb.2011.71 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]