

Abstract
Chronic inflammation is a strong risk factor for the development of cancer. Many previous studies have demonstrated that a transcriptional factor, nuclear factor (NF)‐κB, plays an important role in the association between inflammation and cancer development, particularly tumor promotion and tumor progression. Although it is well recognized that cancer develops via stepwise accumulation of genetic aberrations, the mechanisms underlying the generation of these genetic alterations in normal epithelial cells under inflammatory conditions are not known. We recently demonstrated that pathogenic bacterial or viral factors and the subsequent inflammatory reactions lead to the aberrant expression of a DNA mutator enzyme, activation‐induced cytidine deaminase (AID), in various epithelial cells via NF‐κB activation, which causes the accumulation of genetic alterations in tumor‐related genes. AID activation is widely observed in gastrointestinal tissues with cancer‐associated inflammation, such as chronic viral hepatitis, Helicobacter pylori‐related gastritis, Barrett's esophagus and inflammatory bowel disease. Furthermore, a deficiency of endogenous AID expression reduces both accumulation of somatic mutations in tumor‐related genes and tumor incidence in a mouse model of inflammation‐associated cancer development. These findings strongly suggest that AID plays an integral role in inflammation‐associated carcinogenesis and is therefore a potential target molecule for the prevention and treatment of cancers. (Cancer Sci 2012; 103: 1201–1206)
Cancer cells have various genetic alterations, including somatic mutations, chromosomal rearrangements, copy number alterations and epigenetic changes.1 Recent genome technology innovations, such as ultra‐deep sequencing and comprehensive genome hybridization analyses, have allowed investigators to better understand the landscape of genetic alterations in cancer tissues.2 Using these technologies, comprehensive genome analyses of various cancer tissues have clarified that cancer cells have numerous nucleotide alterations, including “passenger mutations, which might not be involved in cancer development, and ‘driver mutations”, which directly contribute to carcinogenesis.3 In contrast, chronic inflammation plays important roles in the development of various human cancers, and tumor cells are considered to be generated from stepwise accumulation of genetic alterations in various genes during the process of inflammation‐associated carcinogenesis. In some diseases, including hereditary non‐polyposis colorectal cancer, genetic abnormalities in the DNA repair system result in accumulation of genetic alterations in various genes, leading to colorectal carcinogenesis.4, 5 However, in most sporadic cancers the molecular mechanisms for acquiring genetic alterations under inflammatory conditions are unknown.
Nuclear Factor (NF)‐κB is a Key Molecule in Inflammation‐Associated Carcinogenesis
Many epidemiological studies have clearly demonstrated that chronic inflammation predisposes to tumor formation in various organs, including hepatocellular carcinoma (HCC) caused by hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, gastric cancer caused by Helicobacter pylori infection, colorectal cancer caused by inflammatory bowel diseases (IBD), bile duct cancer caused by primary sclerosing cholangitis and esophageal cancer caused by Barrett's esophagus.6, 7, 8, 9, 10, 11 Accordingly, over 25% of human cancer cases are thought to be associated with chronic inflammation.12
Chronic inflammation is characterized by a continued active inflammatory response and tissue destruction, followed by irreversible tissue remodeling. Various cytokines, chemokines and transcription factors contribute to pathogenesis of chronic inflammation, and are also involved in the carcinogenesis process.13 One of the most well known molecules is the transcription factor NF‐κB, which is heavily involved in the processes of both tumor promotion and tumor progression and is considered a key regulator of cancer‐associated inflammation. NF‐κB is activated by various proinflammatory cytokines and microbial products, and regulates various cytokines and chemokines related to the determination of cell fate by binding their gene promoter.14 Although NF‐κB is physiologically associated with immune and inflammatory cell function, activated NF‐κB proteins are detected in epithelial cells under many chronic inflammatory conditions related to carcinogenesis, such as H. pylori‐related chronic gastritis, HCV‐associated chronic liver disease and IBD.15, 16, 17, 18 NF‐κB activation in inflamed epithelial cells, premalignant cells and malignant cells is involved not only in regulating cell survival, proliferation and growth, but also in the epithelial‐to‐mesenchymal transition.19 In addition to these crucial processes that could contribute to tumorigenesis, a novel molecule, activation‐induced cytidine deaminase (AID), a nucleotide‐editing enzyme that is directly involved in DNA instability, has been recently identified to be regulated by NF‐κB20 (Fig. 1).
Figure 1.
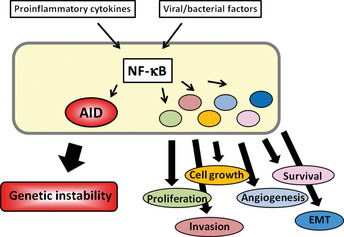
Link between inflammation and carcinogenesis. Nuclear factor (NF)‐κB is activated by various proinflammatory cytokines and microbial products and regulates various cytokines and chemokines related to the determination of cell fate. NF‐κB activation in epithelial cells and malignant cells is involved in generating genetic instability via aberrant activation‐induced cytidine deaminase (AID) activation, as well as cell growth, survival, proliferation, angiogenesis and epithelial‐to‐mesenchymal transition (EMT).
AID Triggers Both Somatic Mutations and DNA Double‐Strand Breaks
Several molecules that possess the capacity to induce mutations in target DNA or RNA have been identified. Most of these molecules are members of the apolipoprotein B mRNA‐editing enzyme, catalytic polypeptide‐like (APOBEC) family.21 Among the APOBEC family members, AID is the only molecule that can induce genetic alterations in human DNA sequences.22 AID is a key molecule for generating immune diversity and is essential for inducing both somatic hypermutation, which occurs in variable regions of the immunoglobulin (Ig) genes, and class‐switch recombination, which occurs in switch regions of the Ig genes (Fig. 2A).23, 24 It is considered that AID acts on single‐strand DNA during the transcriptional stage, resulting in the conversion of cytosine (C) to uracil (U). The generated U:G mismatches can usually be repaired to C:G by the high‐fidelity repair system. However, if the U:G mismatch is not repaired before the onset of DNA replication and is replicated over, it give rise to C:G to T:A transitions.25 In contrast, nicks in the near sites of both strand sequences of switch regions are generated by the repair process of AID‐induced U:G mismatches, resulting in DNA double‐strand breaks that are required for class‐switch recombination26 (Fig. 3).
Figure 2.
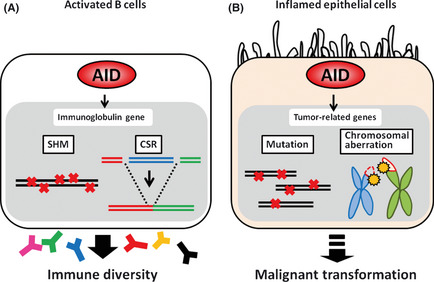
Activation‐induced cytidine deaminase (AID) function under physiological and inflammatory conditions. (A) Under physiological conditions, AID is expressed only in activated B cells and is a key molecule for generating immune diversity by inducing somatic hypermutation (SHM) and class‐switch recombination (CSR) in immunoglobulin genes. (B) Under inflammatory conditions, AID is aberrantly expressed in epithelial cells. AID can induce somatic mutations and chromosomal aberrations in tumor‐related genes, contributing to malignant transformation.
Figure 3.
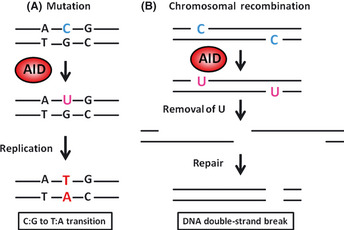
Molecular mechanisms of the induction of mutation and chromosomal recombination by activation‐induced cytidine deaminase (AID). (A) AID acts on single‐strand DNA during the transcriptional stage. AID binds to the target DNA and deaminates deoxycytidine (dC) to deoxyuracil (U), creating a U–G mismatch. The general DNA replication without repair pathways can convert U to T and G to A, generating C:G to T:A mutations. (B) DNA double‐strand breaks are generated through the repair process of AID‐induced U:G mismatches through the production of abasic sites. Class‐switch recombination (CSR) occurs by recombination of double‐strand breaks.
Constitutive AID Expression Contributes to Tumorigenesis
Under physiological conditions, AID has a strong preference for targeting the Ig genes in activated B cells, but it also mutates a number of non‐Ig genes, including Bcl6, Pax5, mirR142, Pim1 and c‐myc.27, 28 Strikingly, approximately 25% of all genes expressed in germinal center B cells are mutated by AID activity.29 Additionally, a number of studies demonstrated that high AID expression is frequently observed in various types of human B‐cell lymphomas and leukemias, and AID expression is associated with various gene mutations and chromosomal translocations.30, 31, 32, 33, 34 These findings call attention to the concept that while the nucleotide‐editing function of AID is physiologically essential for immune diversity in activated B cells, AID could be involved in the malignant transformation of lymphocytes via the accumulation of genetic alterations in tumor‐related genes. This hypothesis was confirmed by generating a transgenic (Tg) mouse model with constitutive and ubiquitous AID expression.35 All AID‐Tg mice developed T cell lymphomas via the accumulation of mutations in non‐Ig genes, such as T cell receptor and c‐myc. Notably, the AID‐Tg mice not only developed malignant lymphomas but also various epithelial tumors, including liver, lung and gastric cancers.36 Thus, the cancer phenotypes of AID‐Tg mice further suggest that epithelial cells with constitutive AID expression are transformed into malignant cells via the accumulation of genetic alterations in tumor‐related genes (Fig. 2B).
Role of AID During Inflammation‐Associated Human Carcinogenesis
Under physiological conditions, expression of the AID gene Aicda is strictly regulated by many factors, because AID has a dangerous ability to induce genomic instability.37 Four regions with transcription factor‐binding motifs have been identified in and around the Aicda locus.38 Among these regions, the transcription enhancer activity of region 4, the area approximately 8 kb upstream of the transcription start site, has a primary role in the induction of AID by environmental stimuli.39 The major transcription factors that mediate the signals initiated by CD40L, interleukin (IL)‐4 and transforming growth factor‐β stimuli are identified as NF‐κB, STAT6 and Smad3/4, respectively. Furthermore, NF‐κB‐binding sites are also found immediately upstream of the transcription start site in region 1.40, 41 The NF‐κB site in region 4 is considered to have a main role against CD40 stimulation in activated B cells, while the site in region 1 is responsible for the cell's response to viral infection and tumor necrosis factor (TNF)‐α (Fig. 4).39 These findings indicate that NF‐κB plays a key role in the regulation of AID expression and suggest that activation of NF‐κB in epithelial cells under many inflammatory conditions might induce AID, leading to genetic instability.
Figure 4.
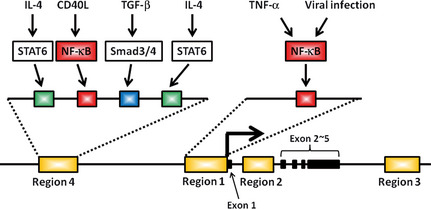
Transcriptional regulation of activation‐induced cytidine deaminase (AID) expression induced by environmental stimuli. The transcriptional regulatory elements for AID are localized to four regions. Nuclear factor (NF)‐κB‐binding sites are present in region 4 and region 1. The site in region 4 has a main role against CD40 stimulation in activated B cells, while the site in region 1 might be responsible for the cell's response to viral infection and tumor necrosis factor (TNF)‐α.
Liver carcinogenesis related to chronic HCV infection
Epidemiological studies have demonstrated that most HCC arise in the setting of chronic liver disease with features of chronic hepatitis or liver cirrhosis. HCV infection is one of the leading causes of chronic liver damage and induces the development of HCC at an annual rate of 0.5–1.0% in chronic hepatitis and 5–8% in liver cirrhosis.6 Recent studies demonstrated that a number of genetic alterations, including the tumor‐suppressor TP53 gene, are present not only in human HCC but also in the surrounding hepatitis or liver cirrhosis.42 Strikingly, aberrant AID expression is observed in both HCC cells and the surrounding non‐cancerous hepatocytes, while no AID expression is observed in normal liver tissues.42 Previous studies have revealed that high TNF‐α expression levels are found in the liver of patients with chronic viral hepatitis, which activates the NF‐κB classical pathway, and the HCV core protein has the potential to induce NF‐κB activation in human hepatocytes.43, 44 Consistent with the regulation of AID by NF‐κB in activated B cells, AID expression is induced in response to TNF‐α stimulation in cultured human hepatocytes through the NF‐κB pathway.45 Likewise, AID is expressed in cultured human hepatocytes expressing HCV core protein via the NF‐κB pathway. Furthermore, aberrant expression of AID in hepatocytes results in the accumulation of genetic alterations of various genes, including the TP53 gene.45 These findings provide the concept that both the response to chronic inflammation and HCV infection itself induce the aberrant expression of AID in hepatocytes, contributing to the genomic instability required for hepatocarcinogenesis.
Gastric carcinogenesis related to H. pylori infection
Helicobacter pylori causes chronic gastric inflammation and is defined as a class one carcinogen for human gastric cancer.46 Patients with a previous history of H. pylori‐related gastric cancer are at a higher risk of subsequent development of gastric cancers.47 Although eradication of H. pylori reduces the development of metachronous gastric cancer, H. pylori‐infected mucosa might already have a number of genetic alterations, leading to multicentric cancer development.48, 49 Interestingly, AID protein is aberrantly expressed in a substantial proportion of H. pylori‐associated human gastric epithelium and gastric cancer tissues, although no AID expression is observed in normal gastric mucosa or in gastric tissues after H. pylori eradication.50
Helicobacter pylori can be subclassified into “cag” pathogenicity island (cagPAI)‐positive and cagPAI‐negative strains based on the presence or absence of cagPAI, a 40‐kb genome fragment containing 31 genes.51 The cagPAI‐positive isolates are more virulent strains that produce severe pathological infection in humans and are deeply associated with an increased risk for gastric cancer.52 Previous studies showed that infection with cagPAI‐positive H. pylori is associated with increased expression of NF‐κB in gastric epithelial cells both in vitro and in vivo.53, 54 Intriguingly, infection with cagPAI‐positive H. pylori ectopically induces a high expression of AID in human gastric epithelial cell lines, while cagPAI‐negative H. pylori has no effect on AID expression. Similar to cagPAI‐positive H. pylori infection, TNF‐α stimulation increases the expression of endogenous AID protein in gastric epithelial cells through the NF‐κB pathway. Furthermore, aberrant AID expression in gastric epithelial cells causes a number of somatic mutations in tumor‐related genes, including the TP53 gene, and knockdown of endogenous AID significantly reduces the number of TP53 mutations observed in H. pylori‐infected cells.50
In contrast, it is reasonable to assume that AID is capable of inducing chromosomal aberrations in epithelial cells because of its ability to trigger DNA double‐strand breaks. Consistent with this hypothesis, AID expression in gastric epithelial cells causes chromosomal aberrations, mainly submicroscopic deletions, at various chromosomal loci.55 Among these deleted loci, the recurrently deleted chromosomal regions harbored the tumor‐suppressor genes cyclin‐dependent kinase inhibitor (CDKN)2A/CDKN2B. Oral infection of wild‐type mice with H. pylori reduces the copy number of the Cdkn2b‐Cdkn2a locus, whereas no such changes are observed in the gastric mucosa of H. pylori‐infected AID‐deficient mice.55 Taken together, these findings suggest that both H. pylori infection and the resulting inflammatory responses induce aberrant AID expression in gastric epithelial cells via NF‐κB activation, leading to accumulation of genetic alterations, including somatic mutations and submicroscopic deletions in tumor‐related genes during H. pylori‐related gastric carcinogenesis.
Colitis‐associated carcinogenesis
Inflammatory bowel diseases is an important etiological risk for the development of colorectal cancer.56 It is well known that the risk of colitis‐associated colorectal cancer increases according to the number of years after disease onset.57 The cumulative risk of developing colorectal cancer in patients with ulcerative colitis (UC) is estimated to be 1.6% at 10 years, 8.3% at 20 years and 18.4% at 30 years from disease onset.9 Recent studies have shown that patients with Crohn's disease are also at high risk of developing colon cancer.58 Importantly, colitis‐associated colorectal cancer has several distinct characteristics compared with sporadic colorectal cancers. In most sporadic colorectal cancers, adenomatous polyps first develop through inactivation of the APC gene, and the subsequent acquisition of other gene abnormalities, such as KRAS, TP53 and DCC genes, contribute to cancer development.59 In contrast, mutations in the TP53 gene are already present in dysplastic lesions with a background of UC, suggesting that colonic inflammation induces the accumulation of mutations in the TP53 gene.60, 61 Interestingly, AID protein is expressed in colonic epithelial cells of patients with IBD, while AID is not detected in normal colonic mucosa.62
Colonic mucosal inflammation is usually mediated by either an excessive T helper cell (Th) 1 T‐cell response associated with increased interferon‐γ and IL‐12 secretion, or an excessive Th2 T‐cell response related to increased IL‐4, IL‐5 and IL‐13 secretion.63 Although the concentration of the Th2 cell‐driven cytokine IL‐4 varies in UC colon tissue, UC is considered to have a Th2 profile.64 In addition to the finding that the proinflammatory cytokine TNF‐α induces AID expression through the NF‐κB pathway, Th2 cytokines IL‐4 and IL‐13 enhance the aberrant expression of AID via STAT6 activation in cultured colonic epithelial cells.62 Moreover, constitutive AID expression in colonic epithelial cells generates a number of mutations of the TP53 gene.
Recent studies have revealed that IL‐10 has a pivotal role in mediating the signals that control inflammation in the human gut.65 Moreover, a mouse model with target disruption of the IL‐10 gene spontaneously develops intestinal inflammation followed by colon cancer, and thus this mouse model has been used to represent human IBD.66, 67 We found that AID expression is observed in the inflamed cecal mucosa of IL‐10‐deficient mice, and mice deficient in both IL‐10 and AID do not develop colon cancer in the cecum, whereas IL‐10‐deficient mice develop spontaneous colon cancers.68 Furthermore, somatic mutation rates of Trp53 in the cecal mucosa of mice deficient in both IL‐10 and AID are significantly lower than those in the mucosa of mice deficient in IL‐10 only, regardless of the same inflammation conditions. Therefore, it is conceivable that aberrant AID expression in the inflamed colonic mucosa plays an integral role in the development of colitis‐associated cancers via accumulation of genetic abberations.
Barrett's esophageal carcinogenesis related to bile acid exposure
Barrett's esophagus is a well known high‐risk factor for the development of esophageal adenocarcinoma.11 Barrett's esophagus is a metaplastic change from the normal stratified squamous epithelium of the lower esophagus to a columnar‐lined epithelium with intestinal‐type differentiation due to chronic duodenogastro‐esophageal reflux and resultant inflammation.69 Several studies have demonstrated that NF‐κB is activated by bile acid components, resulting in the upregulation of various genes involved in the development of metaplasia of Barrett's esophagus and cancer formation.70 Interestingly, AID is aberrantly expressed in the columnar cell‐lined Barrett's esophagus and Barrett's adenocarcinoma, whereas weak or no expression of AID is observed in normal squamous epithelial cells of the esophagus.71 In vitro experiments using human non‐neoplastic esophageal squamous‐derived cells showed that exposure to deoxycholic acid induces endogenous AID upregulation via NF‐κB activation. Moreover, constitutive AID expression in esophageal squamous cells causes the accumulation of mutations in TP53.71 Taken together, these findings suggest that bile acid reflux‐mediated aberrant AID expression in Barrett's columnar‐lined epithelium enhances the susceptibility to genetic alterations, contributing to the development of Barrett's adenocarcinoma.
Conclusion
Recent epidemiological studies have revealed that removing the causes of chronic inflammation, such as eradication of HCV or H. pylori, reduced cancer development, suggesting that regulation of inflammation contributes to suppressing cancer development.48 To date, we have demonstrated that aberrant AID expression in various epithelial cells induces the accumulation of genetic alterations, and a deficiency of endogenous AID reduces the accumulation of somatic mutations in various genes in inflamed tissues, resulting in reduced incidence of inflammation‐associated cancer development.68 These findings might provide a novel strategy for cancer prevention by targeting AID‐related pathways. Recently, several studies have shown that deamination activity of AID promote active DNA demethylation via the base excision repair pathway, suggesting that AID is involved in the epigenetic regulation of various genes.72, 73 Thus, it is hoped that further elucidation of the link between AID and the DNA methylation system will lead to a better understanding of the mechanisms of inflammation‐associated carcinogenesis.
Disclosure Statement
The authors declare no potential conflict of interest.
References
- 1. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–99. [DOI] [PubMed] [Google Scholar]
- 2. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second‐generation sequencing. Nat Rev Genet 2010; 11: 685–96. [DOI] [PubMed] [Google Scholar]
- 3. Bentley DR, Balasubramanian S, Swerdlow HP et al Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008; 456: 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363: 558–61. [DOI] [PubMed] [Google Scholar]
- 5. Fishel R, Lescoe MK, Rao MR et al The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75: 1027–38. [DOI] [PubMed] [Google Scholar]
- 6. Kiyosawa K, Umemura T, Ichijo T et al Hepatocellular carcinoma: recent trends in Japan. Gastroenterology 2004; 127: S17–26. [DOI] [PubMed] [Google Scholar]
- 7. Ikeda K, Marusawa H, Osaki Y et al Antibody to hepatitis B core antigen and risk for hepatitis C‐related hepatocellular carcinoma: a prospective study. Ann Intern Med 2007; 146: 649–56. [DOI] [PubMed] [Google Scholar]
- 8. Chiba T, Seno H, Marusawa H, Wakatsuki Y, Okazaki K. Host factors are important in determining clinical outcomes of Helicobacter pylori infection. J Gastroenterol 2006; 41: 1–9. [DOI] [PubMed] [Google Scholar]
- 9. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta‐analysis. Gut 2001; 48: 526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fevery J, Verslype C. An update on cholangiocarcinoma associated with primary sclerosing cholangitis. Curr Opin Gastroenterol 2010; 26: 236–45. [DOI] [PubMed] [Google Scholar]
- 11. Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett's oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer 2010; 10: 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer 2007; 121: 2373–80. [DOI] [PubMed] [Google Scholar]
- 13. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140: 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature 2006; 441: 431–6. [DOI] [PubMed] [Google Scholar]
- 15. Viala J, Chaput C, Boneca IG et al Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5: 1166–74. [DOI] [PubMed] [Google Scholar]
- 16. Elsharkawy AM, Mann DA. Nuclear factor‐kappaB and the hepatic inflammation‐fibrosis‐cancer axis. Hepatology 2007; 46: 590–7. [DOI] [PubMed] [Google Scholar]
- 17. Greten FR, Eckmann L, Greten TF et al IKKbeta links inflammation and tumorigenesis in a mouse model of colitis‐associated cancer. Cell 2004; 118: 285–96. [DOI] [PubMed] [Google Scholar]
- 18. Pikarsky E, Porat RM, Stein I et al NF‐kappaB functions as a tumour promoter in inflammation‐associated cancer. Nature 2004; 431: 461–6. [DOI] [PubMed] [Google Scholar]
- 19. Ben‐Neriah Y, Karin M. Inflammation meets cancer, with NF‐kappaB as the matchmaker. Nat Immunol 2011; 12: 715–23. [DOI] [PubMed] [Google Scholar]
- 20. Marusawa H, Takai A, Chiba T. Role of activation‐induced cytidine deaminase in inflammation‐associated cancer development. Adv Immunol 2011; 111: 109–41. [DOI] [PubMed] [Google Scholar]
- 21. Cascalho M. Advantages and disadvantages of cytidine deamination. J Immunol 2004; 172: 6513–8. [DOI] [PubMed] [Google Scholar]
- 22. Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol 2002; 20: 165–96. [DOI] [PubMed] [Google Scholar]
- 23. Muramatsu M, Sankaranand VS, Anant S et al Specific expression of activation‐induced cytidine deaminase (AID), a novel member of the RNA‐editing deaminase family in germinal center B cells. J Biol Chem 1999; 274: 18470–6. [DOI] [PubMed] [Google Scholar]
- 24. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000; 102: 553–63. [DOI] [PubMed] [Google Scholar]
- 25. Liu M, Schatz DG. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol 2009; 30: 173–81. [DOI] [PubMed] [Google Scholar]
- 26. Revy P, Muto T, Levy Y et al Activation‐induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper‐IgM syndrome (HIGM2). Cell 2000; 102: 565–75. [DOI] [PubMed] [Google Scholar]
- 27. Kotani A, Okazaki IM, Muramatsu M et al A target selection of somatic hypermutations is regulated similarly between T and B cells upon activation‐induced cytidine deaminase expression. Proc Natl Acad Sci USA 2005; 102: 4506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL‐6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 1998; 280: 1750–2. [DOI] [PubMed] [Google Scholar]
- 29. Liu M, Duke JL, Richter DJ et al Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008; 451: 841–5. [DOI] [PubMed] [Google Scholar]
- 30. Pasqualucci L, Neumeister P, Goossens T et al Hypermutation of multiple proto‐oncogenes in B‐cell diffuse large‐cell lymphomas. Nature 2001; 412: 341–6. [DOI] [PubMed] [Google Scholar]
- 31. Greeve J, Philipsen A, Krause K et al Expression of activation‐induced cytidine deaminase in human B‐cell non‐Hodgkin lymphomas. Blood 2003; 101: 3574–80. [DOI] [PubMed] [Google Scholar]
- 32. Pasqualucci L, Guglielmino R, Houldsworth J et al Expression of the AID protein in normal and neoplastic B cells. Blood 2004; 104: 3318–25. [DOI] [PubMed] [Google Scholar]
- 33. Chiarle R, Zhang Y, Frock RL et al Genome‐wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011; 147: 107–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klein IA, Resch W, Jankovic M et al Translocation‐capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 2011; 147: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okazaki IM, Hiai H, Kakazu N et al Constitutive expression of AID leads to tumorigenesis. J Exp Med 2003; 197: 1173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morisawa T, Marusawa H, Ueda Y et al Organ‐specific profiles of genetic changes in cancers caused by activation‐induced cytidine deaminase expression. Int J Cancer 2008; 123: 2735–40. [DOI] [PubMed] [Google Scholar]
- 37. Nagaoka H, Tran TH, Kobayashi M, Aida M, Honjo T. Preventing AID, a physiological mutator, from deleterious activation: regulation of the genomic instability that is associated with antibody diversity. Int Immunol 2010; 22: 227–35. [DOI] [PubMed] [Google Scholar]
- 38. Yadav A, Olaru A, Saltis M, Setren A, Cerny J, Livak F. Identification of a ubiquitously active promoter of the murine activation‐induced cytidine deaminase (AICDA) gene. Mol Immunol 2006; 43: 529–41. [DOI] [PubMed] [Google Scholar]
- 39. Tran TH, Nakata M, Suzuki K et al B cell‐specific and stimulation‐responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol 2010; 11: 148–54. [DOI] [PubMed] [Google Scholar]
- 40. Gourzi P, Leonova T, Papavasiliou FN. Viral induction of AID is independent of the interferon and the Toll‐like receptor signaling pathways but requires NF‐kappaB. J Exp Med 2007; 204: 259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen‐Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med 2009; 206: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kou T, Marusawa H, Kinoshita K et al Expression of activation‐induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer 2007; 120: 469–76. [DOI] [PubMed] [Google Scholar]
- 43. Tai DI, Tsai SL, Chen YM et al Activation of nuclear factor kappaB in hepatitis C virus infection: implications for pathogenesis and hepatocarcinogenesis. Hepatology 2000; 31: 656–64. [DOI] [PubMed] [Google Scholar]
- 44. Marusawa H, Hijikata M, Chiba T, Shimotohno K. Hepatitis C virus core protein inhibits Fas‐ and tumor necrosis factor alpha‐mediated apoptosis via NF‐kappaB activation. J Virol 1999; 73: 4713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Endo Y, Marusawa H, Kinoshita K et al Expression of activation‐induced cytidine deaminase in human hepatocytes via NF‐kappaB signaling. Oncogene 2007; 26: 5587–95. [DOI] [PubMed] [Google Scholar]
- 46. Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2002; 2: 28–37. [DOI] [PubMed] [Google Scholar]
- 47. Aoi T, Marusawa H, Sato T, Chiba T, Maruyama M. Risk of subsequent development of gastric cancer in patients with previous gastric epithelial neoplasia. Gut 2006; 55: 588–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fukase K, Kato M, Kikuchi S et al Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open‐label, randomised controlled trial. Lancet 2008; 372: 392–7. [DOI] [PubMed] [Google Scholar]
- 49. Maehata Y, Nakamura S, Fujisawa K et al Long‐term effect of Helicobacter pylori eradication on the development of metachronous gastric cancer after endoscopic resection of early gastric cancer. Gastrointest Endosc 2012; 75: 39–46. [DOI] [PubMed] [Google Scholar]
- 50. Matsumoto Y, Marusawa H, Kinoshita K et al Helicobacter pylori infection triggers aberrant expression of activation‐induced cytidine deaminase in gastric epithelium. Nat Med 2007; 13: 470–6. [DOI] [PubMed] [Google Scholar]
- 51. Tomb JF, White O, Kerlavage AR et al The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature 1997; 388: 539–47. [DOI] [PubMed] [Google Scholar]
- 52. Blaser MJ. Helicobacter pylori and gastric diseases. BMJ 1998; 316: 1507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peek RM Jr. IV. Helicobacter pylori strain‐specific activation of signal transduction cascades related to gastric inflammation. Am J Physiol Gastrointest Liver Physiol 2001; 280: G525–30. [DOI] [PubMed] [Google Scholar]
- 54. Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF‐kappa B in gastric epithelial cells. Gastroenterology 1997; 113: 1099–109. [DOI] [PubMed] [Google Scholar]
- 55. Matsumoto Y, Marusawa H, Kinoshita K, Niwa Y, Sakai Y, Chiba T. Up‐regulation of activation‐induced cytidine deaminase causes genetic aberrations at the CDKN2b‐CDKN2a in gastric cancer. Gastroenterology 2010; 139: 1984–94. [DOI] [PubMed] [Google Scholar]
- 56. Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002; 347: 417–29. [DOI] [PubMed] [Google Scholar]
- 57. Mellemkjaer L, Olsen JH, Frisch M, Johansen C, Gridley G, McLaughlin JK. Cancer in patients with ulcerative colitis. Int J Cancer 1995; 60: 330–3. [DOI] [PubMed] [Google Scholar]
- 58. Jess T, Gamborg M, Matzen P, Munkholm P, Sorensen TI. Increased risk of intestinal cancer in Crohn's disease: a meta‐analysis of population‐based cohort studies. Am J Gastroenterol 2005; 100: 2724–9. [DOI] [PubMed] [Google Scholar]
- 59. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–67. [DOI] [PubMed] [Google Scholar]
- 60. Yin J, Harpaz N, Tong Y et al p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 1993; 104: 1633–9. [DOI] [PubMed] [Google Scholar]
- 61. Kern SE, Redston M, Seymour AB et al Molecular genetic profiles of colitis‐associated neoplasms. Gastroenterology 1994; 107: 420–8. [DOI] [PubMed] [Google Scholar]
- 62. Endo Y, Marusawa H, Kou T et al Activation‐induced cytidine deaminase links between inflammation and the development of colitis‐associated colorectal cancers. Gastroenterology 2008; 135: 889–98, 98 e1‐3. [DOI] [PubMed] [Google Scholar]
- 63. Fuss IJ, Neurath M, Boirivant M et al Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN‐gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL‐5. J Immunol 1996; 157: 1261–70. [PubMed] [Google Scholar]
- 64. Inoue S, Matsumoto T, Iida M et al Characterization of cytokine expression in the rectal mucosa of ulcerative colitis: correlation with disease activity. Am J Gastroenterol 1999; 94: 2441–6. [DOI] [PubMed] [Google Scholar]
- 65. Glocker EO, Kotlarz D, Boztug K et al Inflammatory bowel disease and mutations affecting the interleukin‐10 receptor. N Engl J Med 2009; 361: 2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 1993; 75: 263–74. [DOI] [PubMed] [Google Scholar]
- 67. Berg DJ, Davidson N, Kuhn R et al Enterocolitis and colon cancer in interleukin‐10‐deficient mice are associated with aberrant cytokine production and CD4(+) TH1‐like responses. J Clin Invest 1996; 98: 1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Takai A, Marusawa H, Minaki Y et al Targeting activation‐induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 2011; 31: 1733–42. [DOI] [PubMed] [Google Scholar]
- 69. Kazumori H, Ishihara S, Kinoshita Y. Roles of caudal‐related homeobox gene Cdx1 in oesophageal epithelial cells in Barrett's epithelium development. Gut 2009; 58: 620–8. [DOI] [PubMed] [Google Scholar]
- 70. O'Riordan JM, Abdel‐latif MM, Ravi N et al Proinflammatory cytokine and nuclear factor kappa‐B expression along the inflammation‐metaplasia‐dysplasia‐adenocarcinoma sequence in the esophagus. Am J Gastroenterol 2005; 100: 1257–64. [DOI] [PubMed] [Google Scholar]
- 71. Morita S, Matsumoto Y, Okuyama S et al Bile acid‐induced expression of activation‐induced cytidine deaminase during the development of Barrett's oesophageal adenocarcinoma. Carcinogenesis 2011; 32: 1706–12. [DOI] [PubMed] [Google Scholar]
- 72. Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5‐methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011; 145: 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cortellino S, Xu J, Sannai M et al Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination‐base excision repair. Cell 2011; 146: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]