

Abstract
Ras‐GTPase‐activating protein SH3 domain‐binding proteins (G3BP) are overexpressed in various human tumors and participate in several signaling pathways involved in growth, differentiation and apoptosis. G3BP interact with RasGAP (Ras‐GTPase activating protein) only in growing cells and depend on Ras activation, and participate in the Ras signal pathway. Therefore, the blockage and downregulation of G3BP may be a new strategy for cancer therapy. In this report, we demonstrate that a novel peptide GAP161 blocked the functions of G3BP and markedly suppressed HCT116 cell growth through the induction of apoptosis. The peptide bound with G3BP, which interfered with the interaction of G3BP1 with RasGAP and further suppressed Ras signaling pathways. GAP161 downregulated G3BP1 and G3BP2 proteins. Similarly, the knockdown of G3BP substantially decreased the proliferation of HCT116 cells and inhibited Ras signal pathways. Furthermore, the downregulation of G3BP could enhance cisplatin‐induced apoptosis and growth inhibition of HCT116 cells. We also found that GAP161 suppressed the growth of BALB/c mice bearing colon CT26 tumors and nude mice bearing HCT116 xenografts. These results suggest that downregulation of G3BP might be useful in cancer therapy and that GAP161 is a promising new therapeutic agent for cancers. (Cancer Sci, doi: 10.1111/j.1349‐7006.2012.02361.x, 2012)
The G3BP protein family contains G3BP1 and G3BP2, with similar molecular structures. Both proteins are dramatically overexpressed in human cancers, especially in breast, head and neck, colon and lung.1, 2, 3 G3BP are involved in a variety of growth‐related signaling pathways that are involved in carcinogenesis and metastasis,4, 5 including NF‐κB, Ras signaling and the ubiquitin proteasome system.1, 6, 7, 8 The expression level of G3BP1 changes reciprocally to the level of phosphatase and tensin homolog deleted on chromosome ten (PTEN). The negative effect of PTEN expression on the protein level of G3BP1 is recapitulated by direct inhibition of PI3K.9 G3BP1 and G3BP2 also interact with p53 and MDM2 to modulate their expression and localization.10 In addition, G3BP1 is an important marker of stress granules (SG). The NTF2‐like domain of G3BP1 mediates its recruitment to SG influenced by Ras.11 Stresses like hypoxia, heat shock and chemotherapy could induce SG formation, and SG could inhibit apoptosis by suppressing stress‐responsive MAPK pathways.12 Taken together, several lines of evidence suggest that the overexpressed G3BP in a wide range of cancers could be a target for anticancer therapy.
It is believed that RasGAP, a regulator of Ras and Rho GTP‐binding proteins,13, 14 have dual functions: the negative effect of GTPase pathways and downstream signaling transduction using effectors such as Raf‐1, PI3K and MAPK.15 The SH3 domain of RasGAP is contributed to binding of G3BP and is essential for Ras downstream signaling. Two independent groups found that p120‐RasGAP was cleaved by caspases, which induced both anti‐apoptotic and pro‐apoptotic signals, depending on the extent of its cleavage by caspases.16, 17 Cui et al.18 report that two peptides derived from RasGAP SH3 domain were cytotoxicitic to tumor cells without significant effects on normal cells. The peptides also showed an obvious sensitizing effect to cisplatin (CDDP). CDDP could activate multiple intracellular pathways, and CDDP‐induced NF‐κB activation contributed to the resistance of cancer cells to cisplatin.19, 20, 21, 22 During NF‐κB activation, phosphorylation of p65 subunit on Ser 536 is required for NF‐κB to be fully activated.23, 24
In this study, we report a new peptide called GAP161, which blocked the cellular functions of G3BP, and triggered HCT116 cells to undergo apoptosis and potentiated the cytotoxicity of CDDP. Several G3BP‐related molecules of transduction signaling pathways were detected that include Ras, ERK, NF‐κB and Akt pathways. Meanwhile, we found that downregulation of G3BP inhibited HCT116 cell growth by inducing apoptosis similar to the effects of GAP161. We conclude that GAP161 can selectively target G3BP and promote apoptosis. Similar effects of GAP161 on HCT116 cells were also observed in mice bearing xenograft tumors.
Materials and Methods
Peptide synthesis
GAP161, a cell‐permeable peptide GAP161 was synthesized at Wuhan KatyGen Pharmaceuticals (Hubei, China). The molecular weight of GAP161 was 3811.6 Da. The peptides were dissolved in deionized water at a final concentration of 10 mM and stored at −20°C until further use.
Cell culture and reagents
Human colon carcinoma HCT116 cells were purchased from ATCC (Manassas, VA, USA). Cells were grown in DMEM (Gibco, Grand Island, NE, USA) supplemented with 10% FBS, 2 mM l‐glutamine and 1% penicillin–streptomycin at 37°C in the presence of 5% CO2. Cisplatin (Sigma‐Aldrich, St Louis, MO, USA) was diluted in PBS at a final concentration of 20 mM and stored at −20°C. U0126 and LY294002 were from Cell Signaling Technology (Beverly, MA, USA) and diluted in DMSO.
Confocal microscopy
Cells were grown on glass coverslips and incubated at 37°C for 24 h. Cells were fixed and permeabilized by ice‐cold ethanol and then blocked with 1% BSA at room temperature. Coverslips were stained with the anti‐G3BP1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by anti‐mouse rhodamine–conjugated secondary antibody, and examined by Zeiss LSM710 confocal microscopy (Zeiss, Oberkochen, Germany).
Cell proliferation and cell cytotoxicity assay
Cell proliferation was determined by trypan blue dye exclusion assay. Cytotoxicity was determined by sulforhodamine B assay, as described previously.25 For colony formation, cells were harvested after indicated treatment and seeded into six‐well plates at a density of 1000 cells per well. Cells were fed with new culture medium every 4 days and colonies were counted and photographed after 9 days.
siRNA transfection
The sequences used for siRNA were synthesized by Ribobio Technology (Guangzhou, China). The siRNA sequence targeting G3BP1 was 5′‐GCA ACA GUA UUU CGG UAU AdT dT‐3′, and the target sequence for G3BP2 was 5′‐GUA AGA AAG UAG CCA UAA AdT dT‐3′. The control mock siRNA sequence was 5′‐UUC UCC GAA CGU GUC ACG UdT dT‐3′. Cells at 40–60% confluence in six‐well culture dishes were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions.
Annexin V‐FITC/propidium iodide staining and terminal deoxyribonucleotide transferase‐mediated dUTP nick end‐labeling assays
To quantify apoptosis, cells were stained with Annexin V and PI using an Annexin V‐FITC/PI Apoptosis Kit (Roche, Mannheim, Germany), following the protocol provided by the manufacturer. For TUNEL assay, the tissues were treated as indicated and TUNEL staining was performed in situ using the DeadEnd fluorometric TUNEL system kit (Promega, Madison, WI, USA), according to the manufacturer's protocol. The images were captured by an image analysis system (Eclipse TE2000‐U, Nikon, Japan).
Quantification of mRNAs using RT‐qPCR
Total RNA was isolated from the cells and cDNA was generated by RT‐PCR using a cDNA synthesis kit (TaKaRa, Dalian, China). The quantitative real‐time PCR reactions were performed using 2× SYBR Premix Ex Taq II (TaKaRa). All samples were processed and measured using the CFX96TM Real‐Time PCR Detection System (Bio‐rad, Hercules, CA, USA). The following forward (F) and reverse (R) primers were used to amplify G3BP1, G3BP2 and GAPDH cDNAs: F‐G3BP1: 5′‐GAGAAGCCTAGTCCCCTGCT‐3′, R‐G3BP1: 5′‐CCATTTGAATCCAATCCCCCA‐3′; F‐G3BP2: 5′‐TTCAGTGACCAGTAAAAACCTGC‐3′, R‐G3BP2: 5′‐GTGCTTTAACATGGGGTGGAA‐3′ and F‐GAPDH 5′‐CATGAGAAGTATGACAACAGCCT‐3′, R‐GAPDH: 5′‐AGTCCTTCCACGATACCAAAGT‐3′. Relative expression levels of G3BP mRNA were determined using the comparative ΔΔC t method, with GAPDH as a control and relative to the not‐transfected or not‐treated cells.
Caspase‐3/7 activity assay
After 48 h treatment, cells were subjected to caspase‐3/7 activity measurement using the Caspase‐Glo assay kit (Promega), according to the manufacturer's protocol. Caspase activities were expressed as increased fold compared to untreated cells.
Ras activation assay
The active form of Ras in cells was analyzed using a Ras activation assay kit according to the manufacturer's protocol (Upstate Biotechnology, Lake Placid, NY, USA). In brief, the treated cells were disrupted with Mg2+ lysis/wash buffer (MLB), and then the lysate was incubated at 4°C for 60 min, with beads coated with the GST fusion protein, Raf‐1 Ras‐binding domain, which specifically binds to Ras‐GTP. The beads were then washed with MLB thrice, and the washed beads were suspended in 2× Laemmli sample buffer. SDS‐PAGE and an immunoblot assay using anti‐Ras were used to detect the relative amount of Ras‐GTP.
Western blot and coimmunoprecipitation analysis
Whole cell lysates were used for immunoblot, as previously described.26 For co‐immunoprecipitation assay, after desired treatment, cells were collected and lysed in RIPA lysis buffer. Cell lysates were incubated at 4°C overnight with anti‐G3BP1 antibodies, anti‐G3BP2 antibodies or anti‐RasGAP antibodies, followed by the incubation of protein A/G plus Agarose (Santa Cruz Biotechnology) for 6 h to overnight at 4°C. The beads were then washed with cold lysis buffer followed by centrifugation at 3000 g at 4°C for 5 min. The antibody binding proteins were resolved by SDS‐PAGE and analyzed by western blot.
Primary antibodies were as follows: anti‐caspase‐3, anti‐caspase‐9, anti‐cleaved caspase‐7, anti‐PARP, anti‐phospho‐MEK, anti‐phospho‐ERK1/2, anti‐ERK1/2, anti‐phospho‐Akt, anti‐Akt, anti‐NF‐κB p65, anti‐phospho‐NF‐κB p65(Ser536) (Cell Signaling Technology), anti‐G3BP1, anti‐Bcl‐2 and anti‐RasGAP (Santa Cruz Biotechnology), anti‐G3BP2 (Abcam) and anti‐β‐actin (Sigma).
GST pull‐down assay
For binding assays, GST ‐G3BP proteins were preincubated with 30 μM GAP161 before HCT116 cell lysates were added. Client proteins associated with GST‐G3BP were captured by glutathione Sepharose beads (Pierce, Rockford, IL, USA), while unbound proteins were removed by wash buffer. The fraction that was bound to the beads was analyzed by SDS‐PAGE followed by immunoblotting with antibodies specific to G3BP1, G3BP2 and RasGAP.
In vivo tumor mouse model
For the mouse tumor model, female BALB/c mice were injected with 1.5 million mouse colon carcinoma CT26 cells subcutaneously on the right flank. The following day and thereafter daily for GAP161 and every other day for CDDP, the mice were injected intraperitoneally with PBS, 25, 50 and 100 mg/kg of GAP161, 1 mg/kg CDDP, or a combination of GAP161 and CDDP. After 11 days, all mice were weighed and killed, and the tumors were excised. Tumors were weighed, and the mean tumor weight was calculated.
For the HCT116 xenograft tumor model, female BALB/c nude mice (18–22 g) were implanted by subcutaneous injection of 5 × 106 cells on the right flank. After 3 weeks, tumors were aseptically dissected and pieces of tumor tissue (2 mm3 in size) were transplanted subcutaneously by trocars into mice. When tumor size was over 100 mm3, mice were divided into groups (n = 6) and injected intraperitoneally with PBS and GAP161 each day, or 0.5 mg/kg CDDP every other day, or a combination of these. Tumor size was measured and tumor volume was determined by length × width2/2. At day 24, tumors were taken from mice and weighed. The tumor growth inhibition (TGI) for each group was calculated as 1 – tumor weight (treatment)/tumor weight (control) × 100%.
Immunohistochemistry
For histologic examination, the final tumors were cut into two. Half was for western blot analysis; the other half was formalin‐fixed and embedded in paraffin blocks and then cut into thin sections. The tissue sections were either immunostained with antibody against G3BP and NF‐κB. Stained sections were observed using a Leica microscope (×400; Leica, Wetzlar, Germany).
Results
GAP161 suppresses HCT116 cell growth and induces apoptosis
Based on the structural model of G3BP‐RasGAP complex built by Cui et al.18 we designed a peptide by simulation of its interaction with G3BP. To increase cell permeability, the designed peptide was fused with a short peptide derived from the human immunodeficiency virus TAT protein (HIV48–57). The amino acid sequence of the final designed peptide is shown in (Fig. 1a) and is named GAP161. To determine the potential effect on cell growth, HCT116 cells were treated with increasing concentrations of GAP161 for various periods of time. As shown in the left panel of Figure 1(b), GAP161 inhibited the proliferation of HCT116 cells in a dose‐dependent and a time‐dependent manner. The long‐term cell viability was measured by colony formation assay (Fig. 1b, right panel), which also showed the cytotoxicity in a dose‐dependent manner. FACS analysis showed that GAP161 markedly increased accumulation of cells in the sub‐G1 phase (Fig. 1c), indicating that GAP161 suppressed cell growth by promoting apoptosis. To test this hypothesis, Annexin V/PI staining was performed and the results showed that GAP161 significantly induced apoptosis in HCT116 cells (Fig. 1c, lower panel). As shown in Figure 1(d), GAP161 markedly induced the cleavage of caspase‐3 and 7 and downregulated the expression of Bcl‐2 protein. Together, these results showed that GAP161 could inhibit the growth of cultured HCT116 cells by activating the apoptotic signal pathway.
Figure 1.
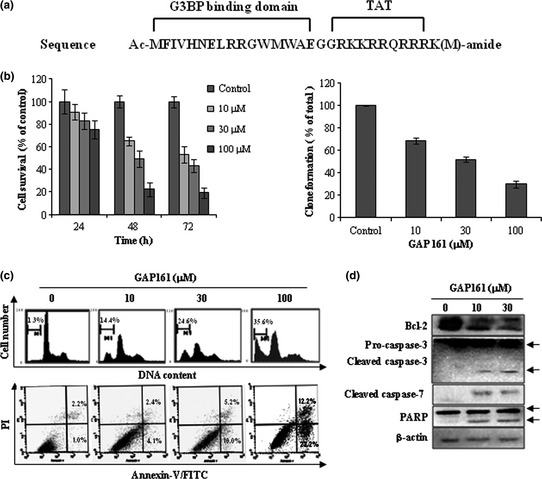
GAP161 suppresses HCT116 cell growth in vitro. (a) Schematic structure of GAP161. (b) Cells were treated with the indicated concentrations of GAP161 for the indicated times, and then cell survival was determined by trypan blue dye (left), and long‐term cell viability was assessed by colony formation assay (right). (c) After the indicated treatments for 48 h, cells were stained by PI or Annexin V‐FITC/PI, and then analyzed by flow cytometry. (d) The changes of apoptotic molecules after GAP161 treatment for 48 h.
GAP161 binds and downregulates G3BP in HCT116 cells
To assess the cellular uptake and distribution of GAP161, the peptide was labeled with FITC and incubated with HCT116 cells for various periods of time. As shown in Figure 2(a), GAP161 began to localize on the cell membrane after only 15 min incubation. After 30 min incubation, GAP161 entered cells and mostly located in the cytoplasm. When cells were incubated for 24 h, some of the peptides were distributed into the nucleus. To see whether the distribution of GAP161 was correlated with G3BP, cells were treated with the FITC‐labeled GAP161 for 60 min and then fixed. The fixed cells were incubated with anti‐G3BP antibody followed by an incubation with rhodamine (red)‐conjugated secondary antibody for monitoring G3BP. The stained cells were examined under a confocal microscopy. We found that GAP161 and G3BP were co‐localized mainly in the cytoplasm (Fig. 2b). To investigate whether GAP161 directly interacted with G3BP, immunoprecipitation (anti‐G3BP antibody), western blot and Typhoon scanning were used to analyze FITC‐GAP161‐treated HCT116 cells (Fig. 2c). Consistent with their co‐localization inside the cells, the immunoprecipitation assay showed that GAP161 directly interacted with G3BP.
Figure 2.
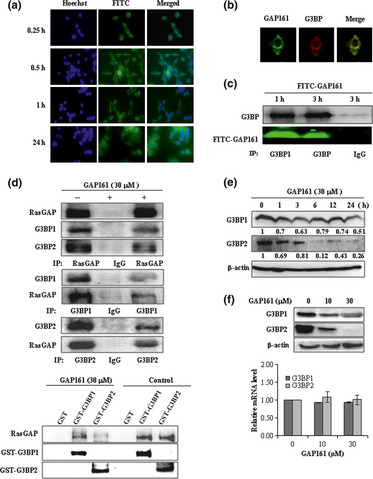
GAP161 penetrates cell membrane and downregulate G3BP in HCT116 cells. (a) Cells were treated with 5 μM FITC‐labeled GAP161 (green) for 15 min, 30 min and 1 h and 24 h, and then nucleus were stained with Hoechst (blue). (b) Confocal images of co‐localization of GAP161 (green) and G3BP1 (red) in 1 h treated HCT116 cells. (c) In vivo interaction between GAP161 and G3BP tested by co‐immunoprecipitation. Cells were treated with 5 μM FITC‐labeled GAP161 for 1 h or 3 h, and total lysates were immunoprecipitated by specific anti‐G3BP1 antibody and then G3BP1 was detected by western blot, and FITC‐labeled GAP161 was detected by typhoon scanning. (d) GAP161 decreased the binding of RasGAP to G3BP. Up panel, HCT116 cells exposed with or without GAP161 for 48 h, and then were used for immunoprecipitation (IP). Bottom, GST, GST‐G3BP were mixed with HCT116 cell lysate in the presence of GAP161 or not. The G3BP complexes were isolated by GST‐pull‐down. The presence of RasGAP was revealed by western blot analysis. (e) Time course of G3BP downregulation after GAP161 treatment. The blots were quantified and protein band intensities normalized relative β‐actin. The numbers under the blots represent fold change relative to control. (f) GAP161 downregulated G3BP protein (up panel) but not mRNA level (bottom).
After confirming the interaction between GAP161 and G3BP, we investigated whether GAP161 affected the interaction of G3BP with RasGAP. As shown in Figure 2(d), RasGAP interacted with G3BP in HCT116 cells and a reciprocal co‐IP experiment indicated that GAP161 decreased the binding of RasGAP to G3BP. GAP161 did not affect the level of RasGAP protein, but downregulated the level of G3BP1 and G3BP2 (Fig. 2d). Moreover, a GST fusion G3BP affinity chromatography assay was used to examine the ability of GAP161 to disrupt the G3BP association with RasGAP. The addition of GAP161 to the cell lysates led to the release of RasGAP. GAP161 appeared to decrease the binding of RasGAP to G3BP (Fig. 2d). Western blot assay showed that GAP161 significantly decreased G3BP level in a dose‐dependent and a time‐dependent manner (Fig. 2e). However, the level of G3BP mRNA did not change after the treatment, indicating that the decrease of G3BP protein was not due to transcription (Fig. 2f).
Knockdown of G3BP inhibits HCT116 cell growth through induction of apoptosis
The results described above suggest that GAP161‐induced apoptosis was caused by the downregulation of G3BP proteins. To test this possibility, we designed specific siRNA against G3BP1 and G3BP2. RT‐qPCR and western blotting analyses showed that transcription and expression of G3BP in HCT116 cells were dramatically downregulated by siRNA (Fig. 3a). Notably, the knockdown of G3BP significantly inhibited HCT116 cell growth and colony formation (Fig. 3b). Furthermore, Annexin V staining revealed that G3BP siRNA induced HCT116 cells to undergo apoptosis (Fig. 3c). Meanwhile, apoptosis‐related Bcl‐2 protein was downregulated and the active cleavages of caspasse‐3, 9 and PARP were upregulated (Fig. 3d).
Figure 3.
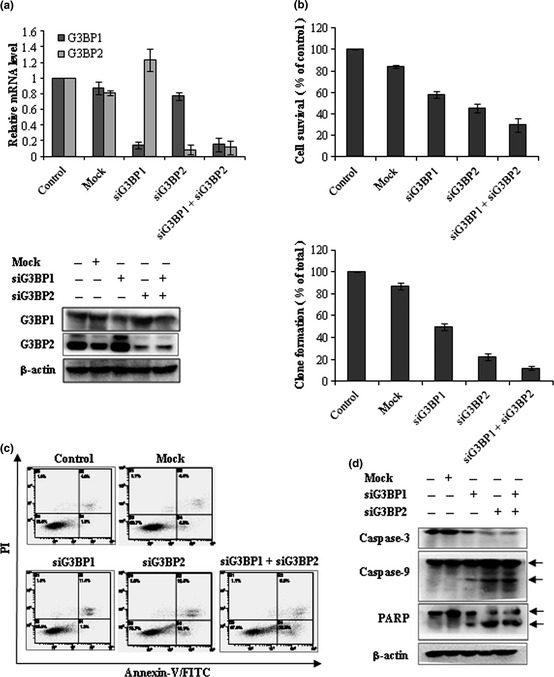
Knockdown of G3BP inhibits HCT116 cells growth through induction of apoptosis. (a) HCT116 cells were treated with 100 nM G3BP‐siRNA for 48 h and harvested for the detection of G3BP by RT‐qPCR (up panel) and western blot (down panel). (b) Knockdown of G3BP suppressed cell proliferation measured by sulforhodamine B (SRB) (up panel) and clone formation described in Materials and Methods (bottom). (c) Detection of apoptotic cells after treatment of G3BP siRNA. Apoptosis was measured by Annexin V‐FITC/PI labeling. (d) Western blotting analysis of apoptosis related protein expression in G3BP silenced HCT116 cells.
Downregulation of G3BP impaires Ras signaling pathway
Ras is a small GTPase responsible for transducing molecular signals. The function of G3BP is important in the oncogenic Ras signaling pathway.3 We found that GAP161 could decrease the abundance of RasGTP, but did not affect the cellular level of Ras protein (Fig. 4a). The key signal pathway from activated Ras is the ERK/MAPK cascade and PI3K pathway. Western blot analyses showed that GAP161 clearly inhibited MEK, ERK activity and Akt phosphorylation, but had no significant effect on total ERK and Akt protein levels (Fig. 4b). It has been reported that NF‐κB activity could be modulated by ERK and Akt.27 Interestingly, we also found that GAP161 significantly downregulated the phosphorylation and expression of NF‐κB.
Figure 4.
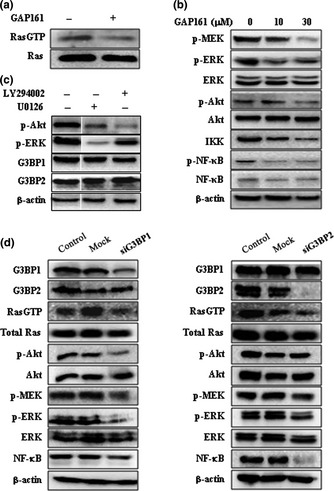
Downregulate G3BP impairs Ras activity. (a) The effect of GAP161 on Ras activity was evaluated in HCT116 cells. The cell lysates were harvested and analyzed as described in the Materials and Methods using a Ras activation kit. Equal protein loading for reactions to assess Ras activity was confirmed by Western blot of total cell lysates. (b) GAP161 inhibited activity of Ras downstream signaling proteins. (c) G3BP work upstream of MEK and Akt. MEK inhibitor U0126 and PI3K inhibitor LY294002 did not affect G3BP protein. (d) Knockdown of G3BP decreased Ras activity and inhibited Ras signaling pathway.
To confirm these results, we monitored Ras activity in G3BP‐silenced HCT116 cells. We found that knockdown of G3BP deceased the level of RasGTP and impaired Ras activity (Fig. 4d). Consequently, the activities of downstream signaling proteins, such as MEK/ERK, Akt and NF‐κB, were downregulated. In addition, the MEK and PI3K inhibitors U0126 and LY294002 did not affect the cellular level of G3BP proteins, indicating that G3BP were upstream of MEK and Akt (Fig. 4c).
Downregulation of G3BP sensitizes HCT116 cells to CDDP
It has been reported that concurrent blockade of the NF‐κB and Akt pathways potently sensitizes cancer cells to chemotherapeutics.19, 28 We found that a combination of GAP161 and CDDP significantly inhibited the growth and colony formation of HCT116 cells as compared with the treatment of CDDP or GAP161 alone (Fig. 5a). Thus, GAP161 synergistically enhanced CDDP‐induced cytotoxicity in HCT116 cells. The knockdown of G3BP by siRNA also significantly sensitized HCT116 cells to CDDP (Fig. 5b).
Figure 5.
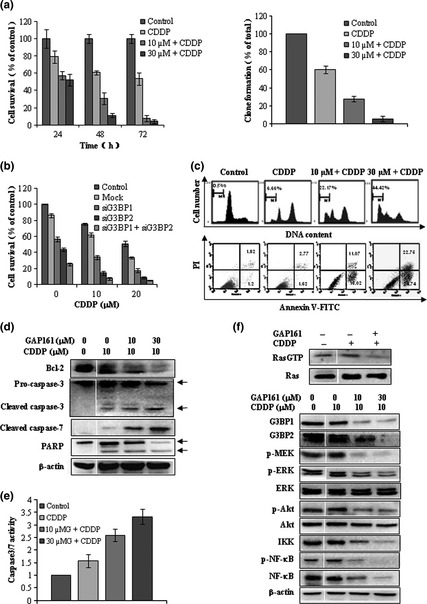
Downregulate G3BP induces and potentiates CDDP‐mediated cytotoxcity and apoptosis. (a) Cells were treated with the indicated concentrations of GAP161 with 10 μM CDDP, and cell viability was determined by trypan blue dye (left) and colony formation assay (right). (b) Knockdown of G3BP potentiates CDDP‐induced cytotoxicity. (c) Cells were treated with GAP161 and CDDP, the amounts of sub‐G1 cells and apoptotic cells were determined, respectively. (d) Detection of key molecules in apoptosis activation 48 h after GAP161 combined with CDDP treatment. (e) Caspase3/7 enzyme activity in GAP161 and CDDP combination exposed HCT116 Cells. (f) GAP161 inhibited Ras signal to sensitize CDDP treated HCT116 cells.
Consistent with the cell viability results, GAP161 markedly potentiated CDDP‐induced sub‐G1 phase accumulation and Annexin V–positive cells (Fig. 5c). Western blotting analysis showed that CDDP modestly induced activations of caspase‐3 and caspase‐7 (Fig. 5d). However, GAP161 markedly enhanced CDDP‐induced cleavages of caspase‐3 and caspase‐7 (Fig. 5d). These results were confirmed by caspase‐3 and caspase‐7 activity assays (Fig. 5e). Bcl‐2 was also downregulated in the combination‐treated cells compared with GAP161 treatment (Fig. 5d). These results further confirmed that the synergetic effect of GAP161 and CDDP was mediated through activation of apoptotic signal pathway. Meanwhile, GAP161 downregulated the level of RasGTP and inhibited the activation of Ras (Fig. 5f). CDDP‐induced activation of ERK and NF‐κB played an important role in chemoresistance.20, 22 As shown in Figure 5(f), CDDP induced ERK phosphorylation and aberrantly activated NF‐κB, but had no obvious effect on Akt activity. In agreement with GAP161 treatment, the combination of CDDP and GAP161 clearly inhibited the activation of ERK and Akt, and significantly downregulated the phosphorylation and expression of NF‐κB.
GAP161 suppress tumor growth and sensitized CDDP in vivo
We next conducted an in vivo experiment to examine the potential effect of GAP161 on tumor growth. HCT116 xenograft tumors (50–100 mm3) were treated intraperitoneally with various concentrations of GAP161 once per day. As shown in Figure 6(a), GAP161 significantly reduced tumor volume in a dose‐dependent and time‐dependent manner. On day 24, the average tumor weight in the GAP161‐treated group is lower than that in the PBS group (Fig. 6b).
Figure 6.
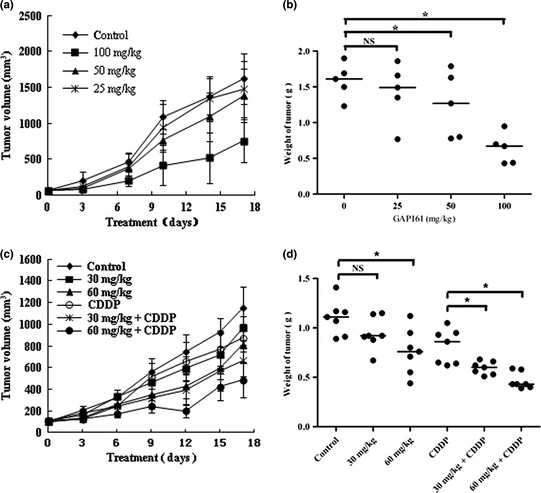
GAP161 suppresses the growth of xenograft human colon tumors and sensitized HCT116 xenograft tumors to CDDP. (a) Growth of tumor volumes in different GAP161 treatment groups (n = 5 per group). Tumor volumes are measured every 3 days after treatment. Zero stands for volume before first injection. (b) Tumor weight of each GAP161 treatment group. (c) The growth curve of HCT116 tumors (n = 7 per group) subjected to GAP161 alone or combined with CDDP treatments. (d) A graph showing relative tumor weight following various treatments. Each dot represents an individual mouse, with a line demarcating the median for each cohort. The results were statistically analyzed with Student's t‐test (*P < 0.05).
To investigate the effects of GAP161 on tumor growth in vivo when combined with CDDP, we conducted two types of experiments to examine the anti‐tumor effect of GAP161 and the synergistic effect to CDDP, particularly under the doses of CDDP with poor or marginal effects. In the first experiment, female BALB/c mice were injected subcutaneously with 1.5 million mouse colon carcinoma CT26 cells on the right flank. The following day and thereafter daily for GAP161 and every other day for CDDP, the mice were injected intraperitoneally with PBS, GAP161, CDDP alone, or a combination of GAP161 and CDDP. A combination of CDDP with GAP161 displayed markedly antitumor effects (Table 1).
Table 1.
Combination treatment of GAP161 and CDDP significantly suppressed the tumor growth of BALB/c mice bearing colon CT26 tumors
| Groups | Dosage (mg/kg) | No. of mice (begin/end) | Body weight change (g) | Tumor weight (g) (mean ± SD) | %TGI† |
|---|---|---|---|---|---|
| Control | — | 10/10 | +1.34 | 2.47 ± 0.31 | — |
| CDDP | 1 | 10/10 | −0.76 | 1.53 ± 0.39* | 38 |
| GAP161 | 20 | 10/10 | −0.47 | 2.27 ± 0.42 | 8 |
| 40 | 10/10 | −0.99 | 1.91 ± 0.43* | 23 | |
| GAP161 + CDDP | 20 + 1 | 10/10 | −0.42 | 0.97 ± 0.32*,** | 61 |
| 40 + 1 | 10/10 | −1.43 | 0.81 ± 0.25*,** | 67 |
*P < 0.05 (vs control group); **P < 0.05 (vs CDDP group). †The %TGI was calculated using the final mean excised tumor weight for each treatment group rather than the final tumor weights estimated by dimensional measurement.
In the second experiment, we transplanted HCT116 cells into nude mice and treated them with GAP161and CDDP. As shown in Figure 6(c), GAP161 could inhibit the growth of HCT116 tumors and markedly enhanced CDDP‐induced growth inhibition. On day 24, the tumor weight of GAP161‐treated group is lower than that of the PBS group, and the tumor weights of the combination‐treated groups were significantly lower than those of the CDDP group (Fig. 6d). Outward signs of drug toxicity were minimal, and there was no significant weight‐loss in the treated groups (data not shown). According to the tumor weight, 0.5 mg/kg of CDDP alone showed a slight inhibitory effect, with 22.5% TGI as compared with the PBS control. In addition, 30 and 60 mg/kg of GAP161 showed modest therapeutic efficacy, with 17.1% and 31.5% TGI, respectively. The combination of GAP161 and CDDP displayed markedly antitumor effects, with 45.95% and 61.26% TGI (P < 0.05 vs CDDP), respectively.
Moreover, GAP161 reduced the expression of G3BP and inhibited the activation of ERK, Akt and NF‐κB, and promoted the cleavage of caspase‐3 in HCT116 xenograft tumors (Fig. 7a). Inconsistent with the in vitro effect, GAP161 decreased the binding of RasGAP to G3BP in vivo (Fig. 7b). H&E staining and TUNEL staining revealed large fractions of apoptotic cells in the tumors treated with GAP161 or the combination of GAP161 and CDDP, but not in those treated with CDDP or PBS (Fig. 7c). In addition, immunohistochemistry assay showed that G3BP and NF‐κB were downregulated in GAP161‐treated tumors (Fig. 7c). These data showed that GAP161 could effectively inhibit the tumor growth in vivo through induction of apoptosis.
Figure 7.
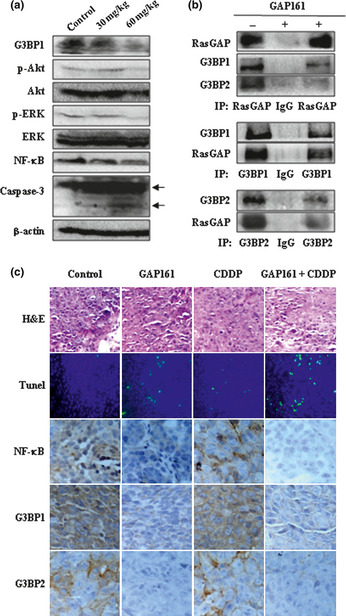
GAP161 downregulate G3BP and impairs Ras signal pathway to induce apoptosis in tumor tissue. (a) Immunoblot analysis of HCT116 xenograft tumors from mice treated for 3 days with 30 and 60 mg/kg GAP161. (b) Cell lysates from HCT116 tumors treated with 60 mg/kg GAP161 for 3 days or not were used for immunoprecipitation (IP). (c) Effects of GAP161 alone or combined with CDDP on paraffin‐embedded HCT116 tumor sections. Tumor histology was analyzed by H&E staining (×200). Apoptosis was analyzed by TUNEL staining (green, ×200) and nucleus stained with Hoechst (blue). The immunohistochemical analysis of NF‐κB and G3BP as described in Materials and Methods (×400), respectively.
Discussion
G3BP were first isolated in a screen for proteins that bind to SH3 domain of RasGAP,29 and the NTF2‐like domain was responsible for its binding to the SH3 domain of RasGAP.30 Parker et al.31 developed a monoclonal antibody directed against the NTF2‐like domain of G3BP that showed a specific effect in the therapy for the G3BP‐related tumors. According to the G3BP NTF2‐like domain and RasGAP SH3 domain interaction model established by Cui et al.18 we designed a new peptide GAP161 through amino acid substitution, which, theoretically, had strong interaction with G3BP. To improve the penetrating ability, we therefore synthesized GAP161 covalently bound to the amino acids carrier peptide HIV‐TAT48–57. Glycine residues were inserted between the TAT and GAP161 as spacers to allow flexibility. GAP161directly interacted with G3BP proteins, blocked their binding to RasGAP, and downregulated the cellular level of G3BP proteins. Consistent with this, GAP161 impaired Ras activity, strongly inhibited HCT116 cell growth, and significantly sensitized HCT116 cells to CDDP both in vitro and in vivo.
RasGAP is an important protein in the modulation of Ras signal transduction. RasGAP and G3BP interaction occurs only in growing cells and both proteins are recruited to activate Ras,6, 29 suggesting that G3BP could participate in Ras signal pathway through RasGAP. In this study, the binding of RasGAP to G3BP was decreased in GAP161‐treated cells, demonstrating that GAP161 competed with RasGAP to bind G3BP. At the same time, the level of RasGTP in GAP161‐treated cells was much lower than that of the control and the CDDP group. GAP161 competitively bound to G3BP and released RasGAP to inactivate Ras from its active GTP‐bound form to its inactive GDP‐bound form by enhancing the endogenous GTPase activity of Ras.
In consistent with the inactivation of Ras in GAP161‐treated cells, the downstream targets of Ras signaling were inhibited, including MEK, ERK and Akt. In contrast, CDDP abnormally induced ERK phosphorylation, which may contribute to antagonizing apoptosis.20, 22 Previously, we have demonstrated that MEK inhibitor U0126 and PI3K inhibitor LY294002 could sensitize HCT116 cells to CDDP.32 When HCT116 cells were treated with GAP161 plus CDDP, CDDP‐induced ERK activation was blocked. In addition, NF‐κB was significantly downregulated in GAP161‐treated cells. Taken together, the multiple‐target inhibitions by GAP161 might contribute to the synergism.
Overexpressed G3BP induce numerous downstream signaling events, including the activation of Ras signaling, MAPK activatio and phosphorylation of multiple signaling molecules.6, 33, 34 Interestingly, GAP161 significantly decreased G3BP protein levels. This effect was probably not caused by influencing the stability of the G3BP transcription because there was no significant change in the steady‐state levels of G3BP mRNA. G3BP could participate in ubiquitin proteasome system (USP10) signaling pathways but G3BP itself was not a substrate of USP10.8 Similarly, proteasome inhibitor MG132 could not reverse GAP161‐induced downregulation of G3BP (data not shown). So far, whether GAP161 specifically represses the post‐transcription or initiates the degradation of G3BP remains unknown. Therefore, it is important to carry out further investigations to identify the detailed regulatory network.
To mimic GAP161‐induced downregulation of G3BP, the knockdown of G3BP by RNA interfering also suppressed HCT116 cell growth and Ras signaling molecules (Fig. 4d). Furthermore, the knockdown of G3BP1 modestly decreased NF‐κB, while the knockdown of G3BP2 significantly downregulated NF‐κB. Moreover, MEK inhibitor U0126 and PI3K inhibitor LY294002 did not affect G3BP protein levels in HCT116 cells, indicating that G3BP was upstream of MEK and Akt. These data suggested that not only G3BP1 but also G3BP2 played an important role in cell proliferation and that GAP161‐induced downregulation of G3BP inhibited HCT116 cell growth and sensitized HCT116 cells to CDDP.
In summary, GAP161 targeting to the NTF2 domain of G3BP showed antitumor activity and potentiated CDDP‐induced cytotoxicity of the HCT116 cells both in vitro and in vivo. GAP161 blocked RasGAP binding to G3BP and downregulated G3BP protein to inhibit cell growth and sensitize HCT116 cells to CDDP. The knockdown of G3BP also suppressed HCT116 cell growth and potentiated CDDP‐induced cell death. These findings suggest that downregulation of G3BP1 and G3BP2 is a promising strategy for cancer treatment.
Disclosure Statement
The authors declare no conflict of interest.
Acknowledgments
We thank Yongjie Xu (Wright State University School of Medicine) for helpful discussion and suggestions. This research was supported by grants from the National Basic Research Program of China (No. 2009CB521807) and the National S&T Major Special Project on Major New Drug Innovation (No. 2009ZX09301‐003).
References
- 1. Barnes CJ, Li F, Mandal M, Yang Z, Sahin AA, Kumar R. Heregulin induces expression, ATPase activity, and nuclear localization of G3BP, a Ras signaling component, in human breast tumors. Cancer Res 2002; 62: 1251–5. [PubMed] [Google Scholar]
- 2. French J, Stirling R, Walsh M, Kennedy HD. The expression of Ras‐GTPase activating protein SH3 domain‐binding proteins, G3BPs, in human breast cancers. Histochem J 2002; 34: 223–31. [DOI] [PubMed] [Google Scholar]
- 3. Guitard E, Parker F, Millon R, Abecassis J, Tocque B. G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett 2001; 162: 213–21. [DOI] [PubMed] [Google Scholar]
- 4. Taniuchi K, Nishimori I, Hollingsworth MA. Intracellular CD24 inhibits cell invasion by posttranscriptional regulation of BART through interaction with G3BP. Cancer Res 2011; 71: 895–905. [DOI] [PubMed] [Google Scholar]
- 5. Taniuchi K, Nishimori I, Hollingsworth MA. The N‐terminal domain of G3BP enhances cell motility and invasion by posttranscriptional regulation of BART. Mol Cancer Res 2011; 9: 856–66. [DOI] [PubMed] [Google Scholar]
- 6. Gallouzi IE, Parker F, Chebli K et al A novel phosphorylation‐dependent RNase activity of GAP‐SH3 binding protein: a potential link between signal transduction and RNA stability. Mol Cell Biol 1998; 18: 3956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prigent M, Barlat I, Langen H, Dargemont C. IkappaBalpha and IkappaBalpha/NF‐kappa B complexes are retained in the cytoplasm through interaction with a novel partner, RasGAP SH3‐binding protein 2. J Biol Chem 2000; 275: 36441–9. [DOI] [PubMed] [Google Scholar]
- 8. Soncini C, Berdo I, Draetta G. Ras‐GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene 2001; 20: 3869–79. [DOI] [PubMed] [Google Scholar]
- 9. Huang Y, Wernyj RP, Norton DD, Precht P, Seminario MC, Wange RL. Modulation of specific protein expression levels by PTEN: identification of AKAP121, DHFR, G3BP, Rap1, and RCC1 as potential targets of PTEN. Oncogene 2005; 24: 3819–29. [DOI] [PubMed] [Google Scholar]
- 10. Kim MM, Wiederschain D, Kennedy D, Hansen E, Yuan ZM. Modulation of p53 and MDM2 activity by novel interaction with Ras‐GAP binding proteins (G3BP). Oncogene 2007; 26: 4209–15. [DOI] [PubMed] [Google Scholar]
- 11. Tourriere H, Chebli K, Zekri L et al The RasGAP‐associated endoribonuclease G3BP assembles stress granules. J Cell Biol 2003; 160: 823–31. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Arimoto K, Fukuda H, Imajoh‐Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress‐responsive MAPK pathways. Nat Cell Biol 2008; 10: 1324–32. [DOI] [PubMed] [Google Scholar]
- 13. Campbell SL, Khosravi‐Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene 1998; 17: 1395–413. [DOI] [PubMed] [Google Scholar]
- 14. Leblanc V, Tocque B, Delumeau I. Ras‐GAP controls Rho‐mediated cytoskeletal reorganization through its SH3 domain. Mol Cell Biol 1998; 18: 5567–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007; 129: 865–77. [DOI] [PubMed] [Google Scholar]
- 16. Michod D, Yang JY, Chen J, Bonny C, Widmann C. A RasGAP‐derived cell permeable peptide potently enhances genotoxin‐induced cytotoxicity in tumor cells. Oncogene 2004; 23: 8971–8. [DOI] [PubMed] [Google Scholar]
- 17. Yang JY, Widmann C. Antiapoptotic signaling generated by caspase‐induced cleavage of RasGAP. Mol Cell Biol 2001; 21: 5346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cui W, Wei Z, Chen Q et al Structure‐based design of peptides against G3BP with cytotoxicity on tumor cells. J Chem Inf Model 2010; 50: 380–7. [DOI] [PubMed] [Google Scholar]
- 19. Chen W, Wang X, Bai L, Liang X, Zhuang J, Lin Y. Blockage of NF‐kappaB by IKKbeta‐ or RelA‐siRNA rather than the NF‐kappaB super‐suppressor IkappaBalpha mutant potentiates adriamycin‐induced cytotoxicity in lung cancer cells. J Cell Biochem 2008; 105: 554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin‐induced activation of mitogen‐activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal‐regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res 1999; 5: 1007–14. [PubMed] [Google Scholar]
- 21. Venkatraman M, Anto RJ, Nair A, Varghese M, Karunagaran D. Biological and chemical inhibitors of NF‐kappaB sensitize SiHa cells to cisplatin‐induced apoptosis. Mol Carcinog 2005; 44: 51–9. [DOI] [PubMed] [Google Scholar]
- 22. Yoon H, Min JK, Lee JW, Kim DG, Hong HJ. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal‐regulated kinase (ERK)1/2. Biochem Biophys Res Commun 2011; 405: 333–7. [DOI] [PubMed] [Google Scholar]
- 23. Jiang X, Takahashi N, Ando K, Otsuka T, Tetsuka T, Okamoto T. NF‐kappa B p65 transactivation domain is involved in the NF‐kappa B‐inducing kinase pathway. Biochem Biophys Res Commun 2003; 301: 583–90. [DOI] [PubMed] [Google Scholar]
- 24. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF‐kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci 2005; 30: 43–52. [DOI] [PubMed] [Google Scholar]
- 25. Sun HX, He HW, Zhang SH et al Suppression of N‐Ras by shRNA‐expressing plasmid increases sensitivity of HepG2 cells to vincristine‐induced growth inhibition. Cancer Gene Ther 2009; 16: 693–702. [DOI] [PubMed] [Google Scholar]
- 26. Ren K, Jin H, Bian C et al MR‐1 modulates proliferation and migration of human hepatoma HepG2 cells through myosin light chains‐2 (MLC2)/focal adhesion kinase (FAK)/Akt signaling pathway. J Biol Chem 2008; 283: 35598–605. [DOI] [PubMed] [Google Scholar]
- 27. Dolcet X, Llobet D, Pallares J, Matias‐Guiu X. NF‐kB in development and progression of human cancer. Virchows Arch 2005; 446: 475–82. [DOI] [PubMed] [Google Scholar]
- 28. He HN, Wang X, Zheng XL et al Concurrent blockade of the NF‐kappaB and Akt pathways potently sensitizes cancer cells to chemotherapeutic‐induced cytotoxicity. Cancer Lett 2010; 295: 38–43. [DOI] [PubMed] [Google Scholar]
- 29. Parker F, Maurier F, Delumeau I et al A Ras‐GTPase‐activating protein SH3‐domain‐binding protein. Mol Cell Biol 1996; 16: 2561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kennedy D, French J, Guitard E, Ru K, Tocque B, Mattick J. Characterization of G3BPs: tissue specific expression, chromosomal localisation and rasGAP(120) binding studies. J Cell Biochem 2001; 84: 173–87. [DOI] [PubMed] [Google Scholar]
- 31. Parker F, Kenigsberg M, Duchesne M, Barlat I. Monoclonal antibodies directed against the G3BP protein, and uses. US patent 7,001,980. 2006; 21 Feb. [Google Scholar]
- 32. Zhang H, Zhang S, He H et al RasGAP‐derived peptide 38GAP potentiates the cytotoxicity of cisplatin through inhibitions of Akt, ERK and NF‐kappaB in colon carcinoma HCT116 cells. Cancer Lett 2011; 308: 62–70. [DOI] [PubMed] [Google Scholar]
- 33. Chen G, Goeddel DV. TNF‐R1 signaling: a beautiful pathway. Science 2002; 296: 1634–5. [DOI] [PubMed] [Google Scholar]
- 34. Malumbres M, Pellicer A. RAS pathways to cell cycle control and cell transformation. Front Biosci 1998; 3: d887–912. [DOI] [PubMed] [Google Scholar]