

Abstract
Patients with triple‐negative breast cancers (TNBCs) typically have a poor prognosis because such cancers have no effective therapeutic targets, such as estrogen receptors for endocrine therapy or human epidermal growth factor receptor 2 (HER2) receptors for anti‐HER2 therapy. As the phosphatidylinositol 3′ kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) cascade is activated in TNBCs, mTOR is a potential molecular target for anticancer therapy. In this study, we investigated the antitumor activities of everolimus, an oral mTOR inhibitor, in nine TNBC cell lines. Everolimus effectively inhibited cell growth at concentrations under 100 nM (IC 50) in five cell lines and even in the 1‐nM range in three of the five cell lines. To identify specific characteristics that could be used as predictive markers of efficacy, we evaluated the expressions of proteins in the mTOR cascade, basal markers, and cancer stem cell markers using western blotting, fluorescent in situ hybridization (FISH), or immunohistochemistry. All five of the sensitive cell lines were categorized as a basal‐like subtype positive for either epidermal growth factor receptor (EGFR) or CK5/6, although resistant cell lines were not of this subtype and tended to exhibit the characteristics of cancer stem cells, with decreased E‐cadherin and the increased expression of Snail or Twist. In vivo assays demonstrated antitumor activity in a mouse xenograft model of basal‐like breast cancer, rather than non‐basal breast cancer. These results suggest that everolimus has favorable activity against basal‐like subtypes of TNBCs. Epidermal growth factor receptor and CK5/6 are positive predictive markers of the TNBC response to everolimus, while cancer stem cell markers are negative predictive markers.
Triple‐negative breast cancers (TNBCs) are defined as estrogen receptor (ER)‐negative, progesterone receptor (PGR)‐negative, and human epidermal growth factor receptor 2 (HER2)‐negative tumors; these tumors account for 11–23% of all breast cancers.1, 2, 3 Triple‐negative breast cancers follow a more aggressive clinical course than other forms of breast cancers and have a poor prognosis.1 As TNBCs have no indications for endocrine therapy or HER2 inhibitors, which are the main treatment options for breast cancers, novel molecular‐targeted therapies against TNBCs are crucially needed.
Triple‐negative breast cancers are a heterogeneous population containing a subgroup that is extremely sensitive to chemotherapy, while another subgroup is resistant to such therapy.2, 4, 5, 6 For example, familial breast cancers with the BRCA1/2 germline mutation are frequently included in the TNBC category that is chemosensitive.5 In contrast, metaplastic carcinoma of the breast, which often lacks ER, PGR, and HER2 expressions, is quite chemoresistant.6 Gene expression profiling can be used to separate breast cancers into five distinct molecular subtypes: luminal A (ER or PGR positive and HER2 negative); luminal B (ER or PGR positive and HER2 positive); HER2 overexpressing (ER or PGR negative and HER2 positive); normal breast‐like; and basal‐like.7, 8, 9, 10 Recently, Herschkowitz et al.11 reported a novel subgroup of TNBCs – the claudin‐low subgroup, which is characterized by low gene expressions of the tight junction proteins claudin 3, 4, 7, and E‐cadherin – which is clearly different from the basal‐like subtype. The claudin‐low subtype has been shown to have cancer‐stem‐cell‐like features because it exhibits a high CD44/CD24 expression ratio12 and the upregulation of snail and twist,13 which have been described as specific markers for cancer stem cells.14, 15 Clearly, TNBC subtypes must be classified to facilitate the development of effective therapies for individuals and to improve therapeutic outcomes.
Everolimus (RAD001) is an inhibitor of serine‐threonine kinase mammalian target of rapamycin (mTOR) and has shown broad antitumor activities in preclinical models.16, 17 Everolimus has been approved for the treatment of refractory renal cell carcinoma,18 progressive neuroendocrine tumors of pancreatic origin (PNET),19 and subependymal Giant cell astrocytoma associated with tuberous sclerosis.20 In addition, several clinical trials have reported the effectiveness of everolimus used in combination with trastuzumab or hormone therapy against HER2‐overexpressing or hormone‐receptor‐overexpressing breast cancers, respectively.21, 22 However, the effect of everolimus against TNBCs has not yet been examined. The loss of function of phosphatase and tensin homolog deleted in chromosome 10 (PTEN) has been reported with varying frequencies in breast cancers23, 24 and has been shown to occur frequently in TNBCs.23, 25 As PTEN dysfunction leads to the activation of the phosphatidylinositol 3′ kinase (PI3K)/Akt/mTOR signaling pathway, mTOR is a potential molecular target for the treatment of TNBCs.
In this study, we investigated the antitumor activities of everolimus in TNBC cell lines in vitro and in vivo and identified predictive markers of the response of TNBCs to everolimus.
Material and Methods
Cell lines and reagents
The following TNBC cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) for use in this study: MDA‐MB‐157, MDA‐MB‐231, MDA‐MB‐436, MDA‐MB‐468, Hs578T, BT20, BT549, HCC38, and HCC1937. All the cell lines were cultured in modified Eagle's medium essential (MEME) or RPMI medium supplemented with 10% FBS at 37°C and in humidified 5% CO2. Everolimus was a generous gift of Novartis Pharma AG (Basel, Switzerland). GDC0914 bismesylate and perifosine were purchased from Selleck Chemicals (Houston, TX, USA). Antibodies against epidermal growth factor receptor (EGFR) (2232; Cell Signaling Technology, Inc, Beverly, MA, USA), phospho‐EGFR (p‐EGFR; Tyr 1069) (2234; Cell Signaling), PTEN (9552; Cell Signaling), Akt (9272; Cell Signaling), phospho‐Akt (p‐Akt; Ser473) (9271; Cell Signaling), mTOR (2972; Cell Signaling), phospho‐mTOR (p‐mTOR; Ser2448) (2971; Cell Signaling), S6 ribosomal protein (2212; Cell Signaling), phospho‐S6 ribosomal protein (p–S6; Ser235/236) (2211; Cell Signaling), 4EBP1 (9452; Cell Signaling), phospho‐4EBP1 (p‐4EBP1; Ser65) (9451; Cell Signaling), E‐cadherin (4065; Cell Signaling), Snail (ab17732; Abcam, Cambridge, UK), Twist (Twist2C1a; Bio Matrix Research, Chiba, Japan), and β‐actin (4967; Cell Signaling) were also purchased.
Cell proliferation assays
Cell proliferation assays were performed using a Cell Counting Kit‐8 assay (CCK‐8; Dojindo, Kumamoto, Japan) according to the product protocol. Briefly, cells were plated into 96‐well, flat‐bottomed plates at 2–3 × 103 cells/180 μL/well. After overnight incubation, triplicate wells were treated with varying concentrations of everolimus ranging from 0.1 to 500 nM for 96 h. The existing medium was removed and replaced with 110 μL of fresh medium containing 10 μL of CCK‐8 reagent and allowed to incubate for 4 h. Absorbance was measured for each well at a wavelength of 450 nm. The percent survival and IC50‐values were calculated as described previously.26
Western blotting
Cultured cells were washed with cold PBS and lysed in M‐PER buffer (Pierce, Rockford, IL, USA). The protein concentration of the supernatant was measured using the bicinchoninic acid (BCA) protein assay (Pierce). The membrane was probed with the first antibody and then with horseradish‐peroxidase‐conjugated secondary antibody. The bands were visualized using enhanced chemiluminescence (ECL Plus Western Blotting Detection Kit; Amersham, Piscataway, NJ, USA).
Immunohistochemistry
Cells were cultured in chamber slides for 48 h. The cultured cells were washed with PBS and fixed with 100% ethanol. The slides were then treated with 3% hydrogen peroxide for 30 min. The slides were incubated with primary antibodies against cytokeratin (CK) 5/6 protein (1:40; Dako Cytomation, Glostrup, Denmark) for 60 min at room temperature. Immunoreactions were detected using the EnVison Plus system (Dako).
Fluorescent in situ hybridization analysis
All the cell lines were cultured in appropriate media supplemented with 10% FBS; the FISH analyses were outsourced to SRL (Tokyo, Japan).
DNA sequencing
Sequencing was performed to detect the following mutations: in EGFR,27 deletions in exon 19 (del 19) and L858R in exon 21; in PI3KCA,28, 29 E542K and E545K in exon 9 and H1047R in exon 20; and in AKT1,30 E17K in exon 4. Briefly, the total RNAs were extracted from each cell line using ISOGEN (Nippon Gene, Tokyo, Japan) according to the manufacturer's instructions. First‐strand cDNA was synthesized from 1 μg of total RNA using the High‐Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). The first‐strand cDNA was amplified by PCR using specific primers for EGFR. Genomic DNA was extracted from each cell line using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany), and exon regions were amplified via PCR using specific primers for PI3KCA and AKT1. DNA sequencing of the PCR products was performed using the dideoxy chain termination method and an ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Small interfering RNA treatment
Individual small interfering RNA (siRNA) duplexes specific to human PTEN (Invitrogen, Carlsbad, CA, USA) or a control siRNA that does not target any sequence in the human genome (non‐target control; Invitrogen) was transfected into MDA‐MB‐231 and BT20 cells according to the product protocol. Briefly, for each well transfection, we prepared RNAi duplex–Lipofectamine RNAiMax Transfection Reagent (Invitrogen) complexes with final a concentration of 10 nM and diluted the cells in complete growth medium without antibiotics. We added 3 × 103 cells to each well with RNAi duplex–Lipofectamine RNAiMax complexes. After 24 h of incubation, the medium was removed and replaced with fresh medium with 10% FBS and antibiotics containing various concentrations of everolimus for the cell proliferation assay.
Transfection of wild‐type EGFR
Constructs of wild‐type EGFR (EGFRwt) and empty vectors were generously contributed by Dr Kazuto Nishio (Osaka, Japan). The pVSV‐G vector (BD Biosciences Clontech, Mountain View, CA, USA) for the constitution of the viral envelope and the pQCXIX constructs were co‐transfected into HEK293 cells (BD Biosciences Clontech) using a FuGENE6 transfection reagent (Roche Diagnostics, Basel, Switzerland). Forty‐eight hours after transfection, the culture medium was collected, and the viral particles were concentrated by centrifugation. MDA‐MB‐231 and MDA‐MB‐436 target cells were infected using a virus‐containing medium according to standard procedures and were used for the cell proliferation assay.
Xenograft studies
Experiments were performed in accordance with the United Kingdom Coordinating Committee on Cancer Research Guidelines for the welfare of animals in experimental neoplasia (second edition).
Suspensions of MDA‐MB‐231 and MDA‐MB‐468 cells (5 × 106) were injected subcutaneously into the backs of 5‐week‐old BALB/cAJcl‐nu/nu mice (CLEA Japan, Tokyo, Japan). After 5 weeks (tumors > 120 mm3), the mice were randomly allocated into groups of five animals to receive everolimus (10 mg/kg per day, three times per week) or the vehicle only by oral gavage for 3 weeks. The tumor diameter and body weight of each mouse were measured three times weekly. The tumor diameters were measured using calipers three times per week to evaluate the effects of treatment, and the tumor volume was determined using the following equation: tumor volume = ab 2/2 (mm3) (where a is the largest diameter of the tumor and b is the shortest diameter). Day “x” denotes the day on which the effect of the drugs was estimated and day “0” denotes the first day of treatment. All the mice were killed on day 22 after measuring their tumors.
Results
Sensitivity to everolimus in TNBC cell lines
We screened nine TNBC cell lines for sensitivity to everolimus. The IC50 values for everolimus in the nine cell lines ranged from 0.7 nM to over 200 nM (Fig. 1). As also shown in Figure 1, everolimus effectively inhibited growth in five of the nine cell lines at an IC50 under 100 nM. Among them, MDA‐MB‐468, Hs578T, and BT549 were highly sensitive to everolimus, with an IC50 of around 1 nM. We examined the induction of apoptosis in everolimus‐sensitive cell lines using three different assays. However, we did not observe any significant apoptosis events (data not shown). In addition, we examined the growth‐inhibitory effect of GDC0914 (a PI3K inhibitor) and perifosine (an Akt inhibitor) using MDA‐MB‐468 and BT549, two everolimus‐sensitive cell lines, and MDA‐MB‐231 and MDA‐MB‐157, two everolimus‐resistant cell lines. We found no significant differences in sensitivity to either inhibitor between everolimus‐sensitive cell lines and resistant cell lines with a sub‐μM IC50 concentration (data not shown).
Figure 1.
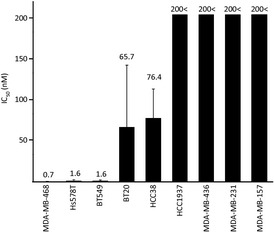
IC 50 values for everolimus of nine triple‐negative breast cancer (TNBC) cell lines. Each cell line was treated with the indicated concentrations of everolimus for 96 h. Viable cell numbers were relatively quantified using the CCK‐8 assay and were expressed as a percent of the untreated control.
Baseline expressions of proteins in the mTOR cascade
We measured the protein expressions of PTEN, p‐AKT, Akt, p‐mTOR, mTOR, p‐S6, S6, p‐4EBP1, and 4EBP1 using a western blot analysis in the nine TNBC cell lines (Fig. 2a). No differences in the expression levels of p‐mTOR and mTOR, which are the targets of everolimus, were seen among the TNBC cell lines. As also shown in Figure 2(a), PTEN was not detected in five cell lines, among which MDA‐MB‐468, BT549, HCC1937, and MDA‐MB‐436 have been reported to harbor a somatic mutation of PTEN.31 Among the five cell lines with a loss of PTEN, MDA‐MB‐468, BT549, and HCC38 were sensitive to everolimus; however, HCC1937 and MDA‐MB‐436 were resistant to everolimus.
Figure 2.
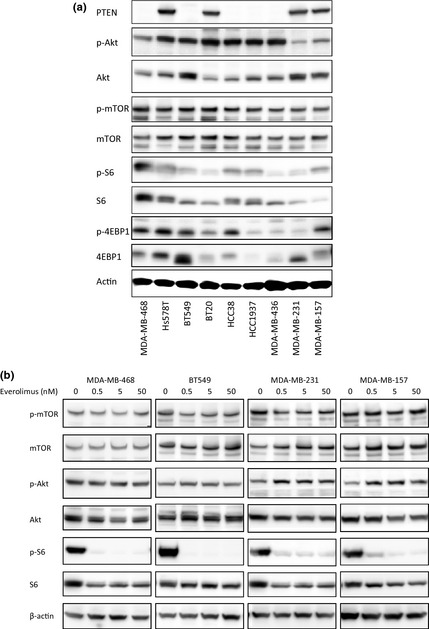
Baseline expressions of mammalian target of rapamycin (mTOR) cascade proteins and mTOR cascade modulation by everolimus in nine triple‐negative breast cancer (TNBC) cell lines. (a) Baseline protein expressions of PTEN, p‐Akt, Akt, p‐mTOR, mTOR, p‐S6, S6, p‐4EBP1, 4EBP1, and β‐actin in nine TNBC cell lines. Ten micrograms of protein were prepared from the indicated cell lines at 60–70% confluence. The cell lines are arranged in decreasing order of sensitivity to everolimus, from left to right. β‐Actin was used as a loading control. (b) Cells were untreated or treated with 0.5, 5, or 50 nM of everolimus for 1 h. Five micrograms of protein were prepared from the indicated cell lines. Equivalent reductions of p‐S6 and S6 were observed with only 0.5 nM in the two sensitive cell lines (MDA‐MB‐468 and BT549) and the two resistant cell lines (MDA‐MB‐231 and MDA‐MB‐157).
mTOR cascade modulation by everolimus
To observe the effect of everolimus, we measured the protein expressions of p‐S6 and S6 before and after everolimus treatment in MDA‐MB‐468 and BT549, two everolimus‐sensitive cell lines, and MDA‐MB‐231 and MDA‐MB‐157, two everolimus‐resistant cell lines. Equivalent reductions of p‐S6 and S6 in response to only 0.5 nM of everolimus were observed in all four cell lines (Fig. 2b), indicating that everolimus acted on all the cell lines. We also measured the protein expressions of p‐Akt and Akt and found an elevation in p‐Akt after treatment with everolimus in BT549, MDA‐MB‐231, and MDA‐MB‐157 cells; however, this result was not observed in MDA‐MB‐468 cells.
Basal markers and cancer stem cell markers
Nielsen et al.32 defined basal‐like breast cancers as those showing a positive expression of EGFR or CK5/6 in TNBCs. In our results, EGFR was overexpressed in MDA‐MB‐468, Hs578T, BT549, and BT20 cells (Fig. 3a), while MDA‐MB‐468 and BT20 cells exhibited the gene amplification of EGFR (Fig. 3b). No mutations in the EGFR gene (del E746–A750, L858R) were detected in any of the cell lines (data not shown). CK5/6 was positive in MDA‐MB‐468, BT20, and HCC38 cells based on an immunocytochemical analysis (Fig. 3c). The status of basal‐cell‐like markers in the nine TNBC cell lines is shown in Table 1. According to the definition by Nielsen et al.,32 we were able to categorize all five of the sensitive cell lines – MDA‐MB‐468, Hs578T, BT549, BT20, and HCC38 – as basal‐like breast cancer. In contrast, the four resistant cell lines were not characterized as basal‐like breast cancer. We also measured the expressions of the cancer stem cell marker proteins, E‐cadherin, Snail, and Twist in all of the cell lines. E‐cadherin was decreased in Hs578T, BT549, MDA‐MB‐436, MDA‐MB‐231, and MDA‐MB‐157. The expression of Snail gradually increased in the more resistant cells (HCC1937, MDA‐MB‐436, MDA‐MB‐231, and MDA‐MB‐157). Twist was overexpressed in MDA‐MB‐436 and MDA‐MB‐157 cells. In summary, the resistant cell lines tended to show characteristics of cancer stem cells, with decreased E‐cadherin expression and the increased expression of Snail or Twist.
Figure 3.
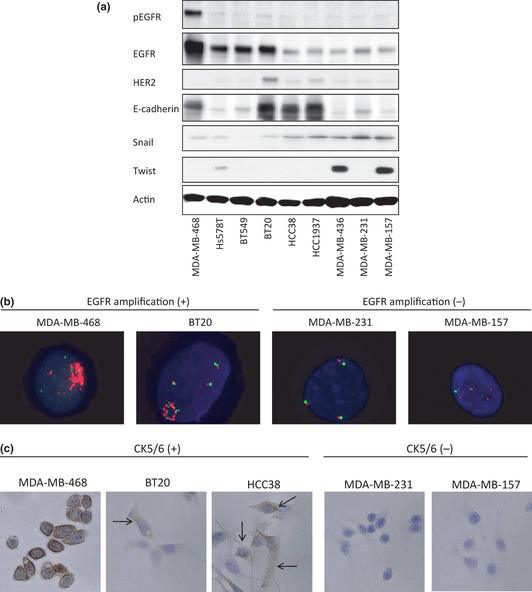
Determination of breast cancer subtypes using basal markers and stem cell‐like characteristics. (a) Protein expressions of p‐EGFR, EGFR, HER2, E‐cadherin, Snail, Twist, and β‐actin in nine triple‐negative breast cancer (TNBC) cell lines. Ten micrograms of protein were prepared from the indicated cell lines at 60–70% confluence. The cell lines are arranged in order of decreasing sensitivity to everolimus, from left to right. β‐Actin was used as a loading control. (b) Epidermal growth factor receptor (EGFR) gene fluorescence in situ hybridization (FISH) analysis. MDA‐MB‐468 and BT20 showed the gene amplification of EGFR. The other seven TNBC cell lines did not exhibit the gene amplification of EGFR. Two positive cell lines and two negative cell lines are shown. (c) Immunohistochemical analysis of CK5/6. MDA‐MB‐468, BT20, and HCC38 were positive for CK5/6, and the other six cell lines were negative. Three positive cell lines and two negative cell lines are shown. Cell membranes were stained by the CK 5/6 antibody in all MDA‐MB‐468 cells and in some BT20 and HCC38 cells (indicated by arrows).
Table 1.
The status of basal‐cell‐like markers in the triple‐negative breast cancer (TNBC) cell lines
| MDA‐MB‐468 | Hs578t | BT549 | BT20 | HCC38 | HCC1937 | MDA‐MB‐436 | MDA‐MB‐231 | MDA‐MB‐157 | |
|---|---|---|---|---|---|---|---|---|---|
| EGFR protein | +++ | ++ | ++ | ++ | ± | ± | ± | ± | ± |
| EGFR amplification | +++ | − | − | ++ | − | − | − | − | − |
| EGFR mutationa | − | − | − | − | − | − | − | − | − |
| CK5/6 protein | ++ | − | − | + | + | − | − | − | − |
| PIK3CA mutationb | − | − | − | H1047R | − | − | − | − | − |
| AKT1 mutationc | − | − | − | − | − | − | − | − | − |
EGFR mutations: deletions in exon 19 (del 19) and L858R in exon 21.
PI3KCA mutations: E542K and E545K in exon 9 and H1047R in exon 20.
AKT1 mutation: E17K in exon 4.
Effect of PTEN knockdown or EGFR overexpression on everolimus sensitivity
We used siRNA oligonucleotides to silence the expression of PTEN in the BT20 and MDA‐MB‐231 cell lines to test whether PTEN expression confers everolimus sensitivity. Before the cell proliferation assay, we determined that the siRNA oligonucleotides against PTEN selectively reduced the mRNA expression levels by 80% or more after 48 h and that the PTEN protein levels were reduced after 72 h, compared with a nonspecific control siRNA. However, the sensitivity of these cells to everolimus did not change with the loss of PTEN expression. The results for MDA‐MB‐231 are shown in Figure 4(a).
Figure 4.
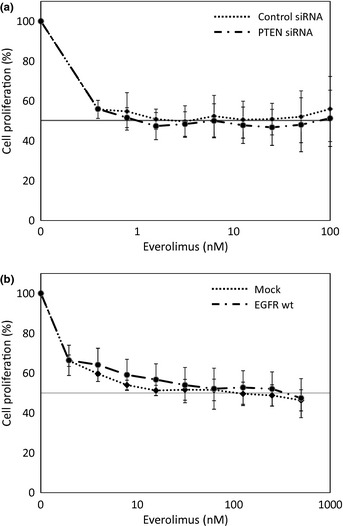
Effect of PTEN or epidermal growth factor receptor (EGFR) modulation on everolimus sensitivity. (a) MDA‐MB‐231 and BT20 cells were transfected with siRNA specific to human PTEN or nonspecific control siRNA. After 24 h, the cells were treated with the indicated concentrations of everolimus for 72 h. The results for MDA‐MB‐231 are shown. (b) MDA‐MB‐231 and MDA‐MB‐436 cells were transfected with retrovirus containing either an empty vector or an EGFRwt vector and then were treated with the indicated concentrations of everolimus for 96 h. The results for MDA‐MB‐231 are shown.
We transfected construct expressing EGFRwt into the EGFR‐silenced cell lines MDA‐MB‐231 and MDA‐MB‐436 to determine the effect of EGFR expression on everolimus sensitivity. However, the overexpression of EGFR did not affect the sensitivity to everolimus. The results for MDA‐MB‐231 are shown in Figure 4(b).
In vivo antitumor effects
To determine whether everolimus is also effective against basal‐like breast cancer in vivo, the growth inhibitory effect was evaluated against MDA‐MB‐468, basal‐like breast cancer cell line, and MDA‐MB‐231, non‐basal‐like breast cancer cell line, tumor xenografts. Everolimus treatment (10 mg/kg day, three times per week for 3 weeks) significantly suppressed the tumor volumes of the MDA‐MB‐468 xenografts, with T/C values of 38.3% (P = 0.016) on day 22 (Fig. 5a). On the other hand, everolimus treatment did not significantly suppress the tumor volumes of the MDA‐MB‐231 xenografts, with T/C values of 58.7% (P = 0.35) on day 22 (Fig. 5b). Body weight loss after treatment was not observed in the MDA‐MB‐468 and MDA‐MB‐231 xenograft groups (data not shown).
Figure 5.
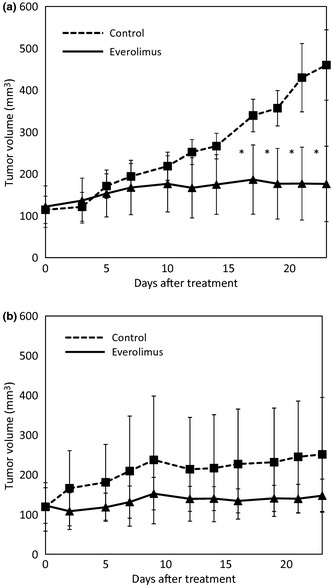
Effect of RAD001 on the growth of breast cancer cell lines in vivo. Athymic nude mice were inoculated with MDA‐MB‐468 cells (a) or MDA‐MB‐231 cells (b). When the tumors reached an average size of 120 mm3, mice were treated with placebo or 10 mg/kg per day RAD001, three times per week for 3 weeks. The tumors were measured twice weekly and tumor size was averaged for each treatment group. Points, mean; bars, standard deviation (SD); *P < 0.05, significantly different from placebo‐treated mice.
Discussion
Patients with TNBCs have relatively poor outcomes and cannot be treated with endocrine therapy or therapies targeted to HER2 receptors.1 The lack of tailored therapies is problematic for the treatment of TNBCs, and the development of novel therapies is crucial. In this study, everolimus effectively inhibited growth in some TNBC cell lines with a sub‐nM IC50 concentration in vitro. In previous reports, everolimus has shown limited growth‐inhibitory activities against several human cancer cell lines, compared with TNBC cell lines.33, 34 Our results suggest that everolimus is a promising therapy for TNBCs.
The classification of TNBC subgroups is necessary for the future development of therapies. In this study, we found that TNBC cell lines classified as basal‐like breast cancer were highly sensitive to everolimus, while cell lines characterized as cancer stem‐cell‐like were less sensitive to everolimus. Similar to the results of the in vitro assay, we found that treatment with everolimus significantly inhibited tumor growth in basal‐like breast cancers in vivo. In a previous report, EGFR expression was associated with a poor prognosis and was a significant independent negative prognostic factor in a multivariate analysis.32 Voduc et al.35 reported that the risk of local and regional relapse in basal‐like breast cancer was higher than those in other breast cancer subtypes. Our results suggest that everolimus is a promising therapy targeting basal‐like TNBC.
Previous studies have suggested that the loss of PTEN may predict sensitivity to everolimus, since PTEN dysfunction leads to the activation of the PI3K/Akt/mTOR signaling pathway.36, 37 In the present study, three of the five cell lines that were sensitive to everolimus were PTEN‐deficient cells, and the other two sensitive cell lines exhibited normal levels of PTEN protein expression. Two of the four everolimus‐resistant cell lines were PTEN‐deficient cells. Furthermore, in the siRNA experiment, although silencing the expression of PTEN activated Akt in BT20 and MDA‐MB‐231 cells, the sensitivities of these cell lines to everolimus did not change. Our results indicate that PTEN deficiency does not predict the response to everolimus in TNBCs. Furthermore, in a clinical trial with glioblastoma patients, no correlation between PTEN deficiency and the response to everolimus was observed.38 Thus, these results suggest that mTOR was not controlled only by Akt, but also by multiple factors and signaling pathways. Furthermore, the role of PTEN in the response to everolimus may differ according to the type of cancer.
In this study, the expression of EGFR was correlated with the sensitivity to everolimus, and we considered the possibility that EGFR may be a key molecule in determining efficacy. EGFR overexpression in breast cancer has been reported in approximately 20–30% of all cases.39, 40 In TNBCs, EGFR expression was reported in 41–57% of cases and EGFR amplification was reported in 18%.32, 41 However, we found that EGFR transfection did not affect the sensitivity to everolimus, suggesting that there is another cascade that affects everolimus sensitivity. We observed an elevation in phosphor EGFR after everolimus treatment in a sensitive cell line, suggesting that a feedback mechanism might influence the sensitivity to everolimus. We examined the induction of apoptosis using three different assays; however, we did not observe any significant apoptosis events. The difference in the sensitivity of the TNBCs can likely be explained by some mechanism of action other than apoptosis. Further studies are needed to clarify these mechanisms and to elucidate the mechanism responsible for the sensitivity to everolimus in TNBCs. Furthermore, we suggest that combination strategies including mTOR inhibitor and PI3K or MEK inhibitor are needed in future clinical trials to overcome the multiple cascades or compensatory feedback systems resulting in cell survival.
Recent reports have indicated that the emergence of cancer stem cells occurs, in part, as a result of the epithelial‐mesenchymal transition (EMT).42, 43 The EMT is characterized by a decrease in epithelial‐specific gene expression, including E‐cadherin, and a gain in mesenchymal‐specific gene expression, including twist and snail.14, 15 Our data shows that cancer stem cell‐positive TNBC cells tend to be resistant to everolimus. These EMT‐rich TNBCs do not respond to traditional cytotoxic drugs or targeted therapies that act on signal transduction. Thus, other therapeutic strategies against these TNBCs, such as stem cell‐ or EMT‐targeted drugs, are urgently needed.
This study suggests that everolimus is a promising agent for the treatment of TNBCs, especially basal‐like breast cancers. Basal markers (EGFR and CK5/6) or cancer stem cell markers (E‐cadherin, snail, or twist) may be predictive markers of the response to everolimus in TNBCs.
Disclosure Statement
The authors have no conflicts of interest.
Acknowledgments
We thank Dr Hitoshi Tsuda for providing technical support for the immunohistochemical analysis. This study was supported in part by a Research Resident Fellowship from Third‐Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour, and Welfare of Japan (H22‐general‐007) and a grant from the Third‐Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour, and Welfare of Japan (H22‐general‐23).
(Cancer Sci, 2012; 103: 1665–1671)
References
- 1. Dent R, Trudeau M, Pritchard KI et al Triple‐negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13: 4429–34. [DOI] [PubMed] [Google Scholar]
- 2. Liedtke C, Mazouni C, Hess KR et al Response to neoadjuvant therapy and long‐term survival in patients with triple‐negative breast cancer. J Clin ncol 2008; 26: 1275–81. [DOI] [PubMed] [Google Scholar]
- 3. Thike AA, Cheok PY, Jara‐Lazaro AR, Tan B, Tan P, Tan PH. Triple‐negative breast cancer: clinicopathological characteristics and relationship with basal‐like breast cancer. Mod Pathol 2010; 23: 123–33. [DOI] [PubMed] [Google Scholar]
- 4. Colleoni M, Cole BF, Viale G et al Classical cyclophosphamide, methotrexate, and fluorouracil chemotherapy is more effective in triple‐negative, node‐negative breast cancer: results from two randomized trials of adjuvant chemoendocrine therapy for node‐negative breast cancer. J Clin Oncol 2010; 28: 2966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foulkes WD, Stefansson IM, Chappuis PO et al Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 2003; 95: 1482–5. [DOI] [PubMed] [Google Scholar]
- 6. Hennessy BT, Giordano S, Broglio K et al Biphasic metaplastic sarcomatoid carcinoma of the breast. Ann Oncol 2006; 17: 605–13. [DOI] [PubMed] [Google Scholar]
- 7. Perou CM, Sorlie T, Eisen MB et al Molecular portraits of human breast tumours. Nature 2000; 406: 747–52. [DOI] [PubMed] [Google Scholar]
- 8. Sorlie T, Perou CM, Tibshirani R et al Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001; 98: 10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van't Veer LJ, Dai H, van de Vijver MJ et al Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–6. [DOI] [PubMed] [Google Scholar]
- 10. Sotiriou C, Neo SY, McShane LM et al Breast cancer classification and prognosis based on gene expression profiles from a population‐based study. Proc Natl Acad Sci USA 2003; 100: 10393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herschkowitz JI, Simin K, Weigman VJ et al Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 2007; 8: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hennessy BT, Gonzalez‐Angulo AM, Stemke‐Hale K et al Characterization of a naturally occurring breast cancer subset enriched in epithelial‐to‐mesenchymal transition and stem cell characteristics. Cancer Res 2009; 69: 4116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taube JH, Herschkowitz JI, Komurov K et al Core epithelial‐to‐mesenchymal transition interactome gene‐expression signature is associated with claudin‐low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 2010; 107: 15449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cano A, Perez‐Moreno MA, Rodrigo I et al The transcription factor snail controls epithelial‐mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol 2000; 2: 76–83. [DOI] [PubMed] [Google Scholar]
- 15. Yang J, Mani SA, Donaher JL et al Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–39. [DOI] [PubMed] [Google Scholar]
- 16. Lu CH, Wyszomierski SL, Tseng LM et al Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res 2007; 13: 5883–8. [DOI] [PubMed] [Google Scholar]
- 17. Marinov M, Ziogas A, Pardo OE et al AKT/mTOR pathway activation and BCL‐2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res 2009; 15: 1277–87. [DOI] [PubMed] [Google Scholar]
- 18. Motzer RJ, Escudier B, Oudard S et al Efficacy of everolimus in advanced renal cell carcinoma: a double‐blind, randomised, placebo‐controlled phase III trial. Lancet 2008; 372: 449–56. [DOI] [PubMed] [Google Scholar]
- 19. Pavel ME, Hainsworth JD, Baudin E et al Everolimus plus octreotide long‐acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT‐2): a randomised, placebo‐controlled, phase 3 study. Lancet 2011; 378: 2005–12. [DOI] [PubMed] [Google Scholar]
- 20. Krueger DA, Care MM, Holland K et al Everolimus for subependymal giant‐cell astrocytomas in tuberous sclerosis. N Engl J Med 2010; 363: 1801–11. [DOI] [PubMed] [Google Scholar]
- 21. Baselga J, Semiglazov V, van Dam P et al Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor‐positive breast cancer. J Clin Oncol 2009; 27: 2630–7. [DOI] [PubMed] [Google Scholar]
- 22. Jerusalem G, Fasolo A, Dieras V et al Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre‐treated patients with HER2‐overexpressing metastatic breast cancer. Breast Cancer Res Treat 2011; 125: 447–55. [DOI] [PubMed] [Google Scholar]
- 23. Perren A, Weng LP, Boag AH et al Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999; 155: 1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol 2001; 14: 672–6. [DOI] [PubMed] [Google Scholar]
- 25. Saal LH, Holm K, Maurer M et al PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 2005; 65: 2554–9. [DOI] [PubMed] [Google Scholar]
- 26. Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non‐small cell lung cancer cell line resistant to gefitinib. Int J Cancer 2005; 116: 36–44. [DOI] [PubMed] [Google Scholar]
- 27. Rosell R, Moran T, Queralt C et al Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361: 958–67. [DOI] [PubMed] [Google Scholar]
- 28. Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004; 3: 1221–4. [DOI] [PubMed] [Google Scholar]
- 29. Samuels Y, Wang Z, Bardelli A et al High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 30. Carpten JD, Faber AL, Horn C et al A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007; 448: 439–44. [DOI] [PubMed] [Google Scholar]
- 31. Saal LH, Gruvberger‐Saal SK, Persson C et al Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet 2008; 40: 102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nielsen TO, Hsu FD, Jensen K et al Immunohistochemical and clinical characterization of the basal‐like subtype of invasive breast carcinoma. Clin Cancer Res 2004; 10: 5367–74. [DOI] [PubMed] [Google Scholar]
- 33. Mabuchi S, Kawase C, Altomare DA et al mTOR is a promising therapeutic target both in cisplatin‐sensitive and cisplatin‐resistant clear cell carcinoma of the ovary. Clin Cancer Res 2009; 15: 5404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Di Nicolantonio F, Arena S, Tabernero J et al Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest 2010; 120: 2858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol 2010; 28: 1684–91. [DOI] [PubMed] [Google Scholar]
- 36. Neshat MS, Mellinghoff IK, Tran C et al Enhanced sensitivity of PTEN‐deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA 2001; 98: 10314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi Y, Gera J, Hu L et al Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI‐779. Cancer Res 2002; 62: 5027–34. [PubMed] [Google Scholar]
- 38. Galanis E, Buckner JC, Maurer MJ et al Phase II trial of temsirolimus (CCI‐779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol 2005; 23: 5294–304. [DOI] [PubMed] [Google Scholar]
- 39. Gori S, Sidoni A, Colozza M et al EGFR, pMAPK, pAkt and PTEN status by immunohistochemistry: correlation with clinical outcome in HER2‐positive metastatic breast cancer patients treated with trastuzumab. Ann Oncol 2009; 20: 648–54. [DOI] [PubMed] [Google Scholar]
- 40. Korsching E, Jeffrey SS, Meinerz W, Decker T, Boecker W, Buerger H. Basal carcinoma of the breast revisited: an old entity with new interpretations. J Clin Pathol 2008; 61: 553–60. [DOI] [PubMed] [Google Scholar]
- 41. Ryden L, Jirstrom K, Haglund M, Stal O, Ferno M. Epidermal growth factor receptor and vascular endothelial growth factor receptor 2 are specific biomarkers in triple‐negative breast cancer. Results from a controlled randomized trial with long‐term follow‐up. Breast Cancer Res Treat 2010; 120: 491–8. [DOI] [PubMed] [Google Scholar]
- 42. Mani SA, Guo W, Liao MJ et al The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9: 265–73. [DOI] [PubMed] [Google Scholar]