

Abstract
The management of myelodysplastic syndrome (MDS) remains challenging. We performed a phase I/II study to evaluate the safety and efficacy of decitabine in patients with MDS in Japan. Patients with MDS with red cell transfusion dependence or 5–30% blasts in marrow and with an International Prognostic Scoring System score of intermediate‐1 or higher were eligible. Patients received intravenous decitabine at 15 or 20 mg/m2 daily for 5 days every 4 weeks. A total of 37 patients were enrolled. Three patients received 15 mg/m2 and experienced no dose limiting toxicity during the first cycle. Thirty‐four patients received 20 mg/m2. Grade 3 or greater non‐hematologic toxicities included cerebral infarction (n = 1), subdural hematoma (n = 1), elevated blood glucose (n = 1), and pulmonary hypertension (n = 1). At 20 mg/m2, complete response, partial response, and hematologic improvement were observed in 7 (20.6%), 2 (5.9%), and 7 (20.6%) patients, respectively. Complete cytogenetic response was observed in 30% of evaluable 20 patients. The median number of cycles to clinical response was 4 (range 4–8), and duration of remission was 474+ days (range 294–598+). The 2‐year rate of acute myeloid leukemia‐free survival was 52%. Correlative studies revealed hypomethylation in multiple genes in peripheral blood cells after treatment. Hypomethylation was generally more profound in CD15 + peripheral blood cells, which reflects myeloid cells, than in peripheral blood mononuclear cells. In summary, decitabine was safe and demonstrated efficacy in Japanese patients with high‐risk MDS. This trial was registered at ClinicalTrials.gov (NCT00796003).
Myelodysplastic syndrome (MDS) is a heterogeneous group of hematopoietic stem cell disorders presenting as cytopenias and dysplastic hematopoiesis with or without increased blast cells.1 The disease is associated with a dismal outcome due to progression of cytopenias or transformation to acute leukemia. Management of MDS varies depending on a patient's age, degree of cytopenias, performance status and estimated risk of disease progression1 as assessed by, for example, the International Prognostic Scoring System (IPSS) score.2
Development of MDS is a multistep event. One important mechanism involved is epigenetic change, such as promoter DNA methylation.3 Frequent methylation often confers a poor prognosis.4 In recent years, treatment with DNA methyltransferase inhibitors has been studied extensively in the management of MDS. Two drugs in this class, azacitidine5 and decitabine,6, 7, 8 have been approved in the management of MDS in multiple countries. However, very little data exist regarding treatment of Asian patients with decitabine.9 Therefore, we conducted a phase I/II study in Japan to assess the safety and efficacy of decitabine in Japanese patients with high‐risk MDS. We also performed correlative methylation analysis using 3‐fucosyl‐N‐acetyl‐lactosamine (CD15)‐positive myeloid cells selected from the peripheral blood of patients undergoing treatment with decitabine.
Materials and Methods
This open‐label multicenter phase I/II study of decitabine in patients with MDS was approved by the institutional review boards of each participating institution and was conducted in compliance with the International Conference on Harmonisation Good Clinical Practice guidelines (ClinicalTrials.gov identifier: NCT00796003).
Phase I: The primary objective was to assess the safety of intravenous decitabine at 15 and 20 mg/m2 administered over 1 h daily for 5 days every 4 weeks. Secondary objectives were pharmacokinetic and pharmacodynamic assessments during the first cycle of treatment.
Phase II: The primary objective was to evaluate patient response (rates of complete response [CR] and partial response [PR]). Secondary objectives were time to response, response duration, time to acute myelogenous leukemia (AML) or death, transfusion dependency, and cytogenetic response.
Patients
Eligibility criteria included the following: diagnosis of MDS based on French‐American‐British (FAB) morphologic classification, including refractory anemia (RA), RA with ringed sideroblasts (RARS), RA with excess blasts (RAEB), RAEB in transformation and chronic myelomonocytic leukemia (CMML); patients with RA or RARS were to have required red blood cell transfusion more than once every 4 weeks, while patients with CMML were to have a white blood cell count of <13 000/mm3; IPSS assessment of intermediate or high risk; age 20 years or older; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; and normal organ functions including creatinine ≤176.8 μM (2 mg/dL), bilirubin ≤ 25.7 μM (1.5 mg/dL) and aspartate aminotransferase and alanine aminotransferase levels ≤2 × the upper limit of normal.
Patients were ineligible if they had: bone marrow blast percentage ≥30% on local marrow review; prior chemotherapy with cytarabine ≥1 g/m2; or other serious comorbidities, active infectious disease, autoimmune cytopenia, hepatitis B surface antigen positivity, hepatitis C antibody positivity or HIV antibody positivity. Pregnant or lactating female patients were also ineligible.
Treatment
Two decitabine doses and schedules have been recommended in previous studies in MDS, namely 15 mg/m2 over 3 h every 8 h for 3 days every 6‐week cycle6 and 20 mg/m2 over 1 h daily for 5 days every 4‐week cycle.7, 8 Because the latter method has shown clinical efficacy and convenience in the outpatient setting, we chose the 5‐day dosing schedule for this study. We started treatment at 15 mg/m2 daily, escalating to 20 mg/m2 after safety was assessed.
Treatment was repeated every 4 weeks in the absence of dose limiting toxicity (DLT, defined below) or disease progression. In order to receive the next cycle of treatment, a patient was required to have normal organ function as defined above. If the patient did not meet these criteria on day 29, a maximum 2‐week delay was allowed. Treatment was also delayed if the patient had febrile neutropenia, grade 3 or 4 infection with neutropenia, or ≥grade 2 bleeding. If, by day 43, the patient did not meet the above criteria, the patient was taken off the study.
Antiemetics were not routinely given. The use of erythropoietin was not allowed. Granulocyte colony stimulating factor was allowed as clinically indicated, but response was recorded when patients were not receiving growth factor support. Dose reductions of the study drug were not allowed.
Toxicity and response evaluation
Toxicity was evaluated based on Common Terminology Criteria for Adverse Event (CTCAE) version 3.0. DLT was defined as: non‐hematologic toxicity ≥grade 3, excluding nausea and vomiting, and neutropenic fever and infection ≥grade 3 that did not improve despite a delay in the initiation of the next cycle of treatment by 2 weeks.
Bone marrow aspiration slides were centrally reviewed and final data analysis was conducted based on central review. Response was evaluated based on International Working Group (IWG) 200010 and IWG 200611 response criteria in myelodysplasia. Complete cytogenetic response was defined as the disappearance of cytogenetic abnormalities; partial cytogenetic response was defined as a ≥50% reduction in cytogenetic abnormalities.10, 11 Response duration was calculated from first evidence of response until disease progression.10, 11 Survival was calculated from start of therapy to death from any cause. Time to AML was calculated from start of therapy to the first date of documented marrow blast percentage ≥30%.
Statistical considerations
In phase I, three patients were to be accrued at 15 mg/m2. If DLT was not observed in any patients during the first cycle, six more patients were accrued at 20 mg/m2. If the 20 mg/m2 dose was not associated with DLT, this population was carried into phase II. Based on prior studies (response rate 17–35%6, 7, 8, 12), the expected response rate was 25%. The drug was considered ineffective if the response rate was ≤5%. With α = 0.05 and β = 0.2, 21 patients would be required to evaluate drug efficacy. Estimating that up to 20% of patients might be found ineligible after central pathologic review, a total of at least 26 patients (including six patients from phase I study at the same dose) were to be enrolled.
Correlative analysis
Pharmacokinetics
Plasma decitabine levels were analyzed on days 1 and 5 of treatment in patients in phase I. Blood was drawn at 0, 30, 60, 65, 75, 90, 120, 180 and 240 min after initiation of infusion. Samples were immediately stored at 4°C, and plasma decitabine concentrations were determined by liquid chromatography coupled with tandem mass spectrometry (lower limit of quantification: 1.0 ng/mL).13
Pharmacodynamics
Preparation of patient samples: In phase I, we conducted DNA methylation analysis using peripheral blood samples from all nine patients. Samples were obtained before treatment (at baseline) and on days 5, 12, and 28 of the first cycle of decitabine treatment. Two separate cell populations were obtained from whole blood: peripheral blood mononuclear cells (PBMCs) were isolated with standard ficoll separation of whole blood and CD15‐positive peripheral blood cells (CD15 + PBCs) were isolated using CD15‐recognizing antibodies (Dynabeads CD15; Life Technologies, Carlsbad, CA, USA). CD15 + PBCs were chosen because it has been shown that the majority of PBMCs are lymphocytes, which may not be representative of myelodysplastic cells.14 CD15, however, is expressed on myeloid cells, including neutrophils, eosinophils and monocytes, and this selection enriched the myeloid cell population and eliminated lymphocytes.
Bisulfite‐pyrosequencing for DNA methylation analysis: DNA methylation levels were quantitatively measured using bisulfite‐pyrosequencing15, 16, 17 (Pyrosequencing AB, Uppsala, Sweden) for PGR, ESR1, CDH1, CDH13, and LINE1. The list of primers for bisulfite‐pyrosequencing is provided in Table S1. LINE1 is a repetitive component that we used as a surrogate for global methylation. The methylation levels at different CpG sites were averaged to represent the degree of methylation.
Methylated CpG island amplification and microarray analysis: The DNA methylation status of CD15 + PBC during the first cycle of treatment was further analyzed with methylated CpG island amplification and microarray (MCAM) technology as previously reported18 using microarrays from Agilent Technologies, which analyzes 6157 genes.
Results
Study enrollment
In phase I, the first cycle of treatment was not associated with DLT. Therefore, the study was expanded into phase II. Data from 34 patients treated with 20 mg/m2 decitabine were analyzed for safety and efficacy (Fig. 1). Patient characteristics and disease characteristics are summarized in Tables 1 and 2, respectively. All patients had MDS based on local hemato‐morphological review, among whom two were found to have AML upon central review of marrow (blast count ≥ 30%).
Figure 1.
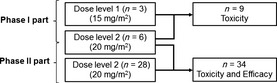
Study schema. A total of 37 patients were treated in this study. In the phase I part, three received 15 mg/m2 and six received 20 mg/m2. In the phase II part, 28 additional patients received 20 mg/m2. Toxicity was analyzed in all patients. Efficacy was analyzed in all patients who received 20 mg/m2.
Table 1.
Baseline characteristics of patients
| Patient characteristics | Phase | I | I (n = 6) and II (n = 28) | Total | |
|---|---|---|---|---|---|
| Dose | 15 mg/m2 | 20 mg/m2 | 20 mg/m2 | ||
| n | 3 | 6 | 34 | 37 | |
| Sex | Male | 3 (100.0%) | 4 (66.7%) | 26 (76.5%) | 29 (78.4%) |
| Female | 0 | 2 (33.3%) | 8 (23.5%) | 8 (21.6%) | |
| Age | <65 | 0 | 3 (50.0%) | 10 (29.4%) | 10 (27.0%) |
| 65–74 | 3 (100.0%) | 1 (16.7%) | 17 (50.0%) | 20 (54.1%) | |
| >75 | 0 | 2 (33.3%) | 7 (20.6%) | 7 (18.9%) | |
| Median | 68 (67–71) | 66.5 (54–77) | 69 (52–81) | 69 (52–81) | |
| PS | 0 | 1 (33.3%) | 5 (83.3%) | 22 (64.7%) | 23 (62.2%) |
| 1 | 2 (66.7%) | 1 (16.7%) | 12 (35.3%) | 14 (37.8%) | |
| Type | De novo | 2 (66.7%) | 5 (83.3%) | 29 (85.3%) | 31 (83.8%) |
| Secondary | 1 (33.3%) | 1 (16.7%) | 5 (14.7%) | 6 (16.2%) | |
| Hb (g/dL) | Median (range) | 7.1 (5.4–8.7) | 7.6 (7.1–8.5) | 8.1 (4.7–15.2) | 8.0 (4.7–15.2) |
| Neu (/μL) | Median (range) | 434 (312–1243) | 887 (173–1910) | 898 (143–7416) | 807 (143–7416) |
| Plt | Median (range) | 1.2 (1.1–3.6) | 7.6 (3.1–22.4) | 5.25 (0.3–74.2) | 4.9 (0.3–74.2) |
| Serum EPO (mU/mL) | Median (range) | 594 (132–728) | 615.5 (31.6–1840) | 369 (1538–13 600) | 393 (15.8–13 600) |
| Time from diagnosis | Median | 1.1 (0.1–4.1) | 1.1 (0.1–4.2) 5 | 0.55 (0.1–12.7) | 0.6 (0.1–12.7) |
| Prior chemotherapy for MDS | Yes | 0 | 1 (16.7%) | 6 (17.6%) | 6 (16.2%) |
| RBC transfusion dependent | Yes | 2 | 4 (66.7%) | 25 (73.5%) | 27 (73.0%) |
| Platelet transfusion dependent | Yes | 0 | 0 | 5 (14.7%) | 5 (13.5%) |
| Previous G‐CSF | Yes | 0 | 1 (16.7%) | 1 (2.9%) | 1 (2.7%) |
| Type of MDS and IPSS | De novo – Low | 0 | 0 | 1 (2.9%) | 1 (2.7%) |
| De novo – Intermediate‐1 | 1 (33.3%) | 1 (16.7%) | 9 (26.5%) | 10 (27.0%) | |
| De novo – Intermediate‐2 | 0 | 2 (33.3%) | 7 (20.6%) | 7 (18.9%) | |
| De novo – High | 0 | 1 (16.7%) | 11 (32.4%) | 11 (29.7%) | |
| Secondary MDS | 1 (33.3%) | 1 (16.7%) | 5 (14.7%) | 6 (16.2%) | |
| AML | 1 (33.3%) | 1 (16.7%) | 1 (2.9%) | 2 (5.4%) | |
EPO, erythropoietin; G‐CSF, granulocyte‐colony stimulating factor; IPSS, International Prognostic Scoring System; MDS, myelodysplastic syndrome; RBC, red blood cell.
Table 2.
Baseline characteristics of bone marrow
| Bone marrow characteristics | Phase | I | I (n = 6) and II (n = 28) | Total | |
|---|---|---|---|---|---|
| Dose | 15 mg/m2 | 20 mg/m2 | 20 mg/m2 | ||
| n | 3 | 6 | 34 | 37 | |
| Marrow Blast percent | <5 | 0 (0.0%) | 4 (66.7%) | 12 (35.3%) | 12 (32.4%) |
| 5–10 | 2 (66.7%) | 0 (0.0%) | 4 (11.8%) | 6 (16.2%) | |
| 11–20 | 0 (0.0%) | 0 (0.0%) | 11 (32.4%) | 11 (29.7%) | |
| 21–30 | 0 (0.0%) | 1 (16.7%) | 6 (17.6%) | 6 (16.2%) | |
| >30 | 1 (33.3%) | 1 (16.7%) | 1 (2.9%) | 2 (5.4%) | |
| Median | 9.2 (7.4–37.1) | 4.2 (2.2–52.5) | 11.1 (1.0–52.5) | 11 (1–52.5) | |
| Bone marrow cellularity | Hyper | 0 (0.0%) | 3 (50.0%) | 13 (38.2%) | 13 (35.1%) |
| Normo | 1 (33.3%) | 2 (33.3%) | 9 (26.5%) | 10 (27.0%) | |
| Hypo | 2 (66.7%) | 1 (16.7%) | 12 (35.3%) | 14 (37.8%) | |
| FAB classification | RA | 0 (0.0%) | 3 (50.0%) | 11 (32.4%) | 11 (29.7%) |
| RARS | 0 (0.0%) | 1 (16.7%) | 1 (2.9%) | 1 (2.7%) | |
| RAEB | 2 (66.7%) | 0 (0.0%) | 14 (41.2%) | 16 (43.2%) | |
| RAEB‐T | 0 (0.0%) | 1 (16.7%) | 7 (20.6%) | 7 (18.9%) | |
| CMML | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| AML | 1 (33.3%) | 1 (16.7%) | 1 (2.9%) | 2 (5.4%) | |
| WHO classification | RA | 0 (0.0%) | 0 (0.0%) | 3 (8.8%) | 3 (8.1%) |
| RARS | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| RCMD | 0 (0.0%) | 3 (50.0%) | 7 (20.6%) | 7 (18.9%) | |
| RCMD‐RS | 0 (0.0%) | 1 (16.7%) | 1 (2.9%) | 1 (2.7%) | |
| RAEB‐1 | 2 (66.7%) | 0 (0.0%) | 4 (11.8%) | 6 (16.2%) | |
| RAEB‐2 | 0 (0.0%) | 0 (0.0%) | 10 (29.4%) | 10 (27.0%) | |
| MDS‐U | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Other | 0 (0.0%) | 0 (0.0%) | 1 (2.9%) | 1 (2.7%) | |
| AML | 1 (33.3%) | 2 (33.3%) | 8 (23.5%) | 9 (24.3%) | |
| Chromosome | Any chromosome abnormality | 1 (33.3%) | 2 (33.3%) | 17 (50.0%) | 18 (48.6%) |
| Good | 2 (66.7%) | 4 (66.7%) | 14 (41.2%) | 16 (43.2%) | |
| Intermediate | 0 (0.0%) | 0 (0.0%) | 3 (8.8%) | 3 (8.1%) | |
| Poor | 1 (33.3%) | 2 (33.3%) | 17 (50.0%) | 18 (48.6%) | |
| Chromosome 7 | 0 (0.0%) | 1 (16.7%) | 12 (35.3%) | 12 (32.4%) | |
| Complex | 1 (33.3%) | 2 (33.3%) | 12 (35.3%) | 13 (35.1%) | |
AML, acute myelogenous leukemia; CMML, chronic myelomonocytic leukemia; FAB, French‐American‐British; MDS, myelodysplastic syndrome; RA, refractory anemia; RARS, RA with ringed sideroblasts; RAEB, RA with excess blasts; RAEB‐T, RAEB in transformation; RCMD, refractory cytopenia with multilineage dysplasia; RCMD‐RS, RCMD with ringed sideroblast; WHO, World Health Organization.
Treatment delivery
A total of 298 cycles were given to 37 patients. A median of 6.0 (range 1–17) and 5.5 cycles (1–17) were delivered per patient in the whole group (15 and 20 mg/m2) and 20 mg/m2 group, respectively. Drug administration was delayed due to cytopenia and/or infection by longer than 7 days (i.e. interval >5 weeks) for 84 cycles (28%). Eight patients experienced DLT within two cycles of treatment and did not continue treatment. Reasons for discontinuation included pneumonia associated with neutropenia (n = 5), fungal pneumonia with pulmonary hypertension (n = 1), chronic subdural hematoma (n = 1), and elevated liver enzymes (n = 1). Seven patients were actively receiving decitabine upon trial closure in March 2011 and had received 14–22 cycles of treatment.
Toxicity
Toxicity data are summarized in Table 3. If one patient experienced the same toxicity multiple times, the highest grade is recorded.
Table 3.
Observed toxicities
| Grade | 15 mg/m2 (n = 3) | 20 mg/m2 (n = 34) | |||||
|---|---|---|---|---|---|---|---|
| 1 or 2 | 3 | 4 | 1 or 2 | 3 | 4 | ||
| Hematologic | Leukocytopenia | 0 | 0 | 3 (100%) | 2 (5.9%) | 9 (26.5%) | 23 (67.6%) |
| Neutropenia | 0 | 0 | 3 (100%) | 0 | 2 (5.9%) | 25 (73.5%) | |
| Thrombocytopenia | 0 | 0 | 3 (100%) | 1 (2.9%) | 6 (17.6%) | 21 (61.8%) | |
| Anemia | 0 | 1 (33.3%) | 2 (66.7%) | 1 (2.9%) | 13 (38.2%) | 16 (47.1%) | |
| Lymphocytopenia | 0 | 2 (66.7%) | 1 (33.3%) | 11 (32.4%) | 11 (32.4%) | 6 (17.6%) | |
| Infection | Febrile neutropenia | 0 | 0 | 1 (33.3%)a | 0 | 8 (23.5%) | 2 (5.9%)a |
| Other infection | 1 (33.3%) | 1 (33.3%) | 0 | 6 (17.6%) | 10 (29.4%) | 0 | |
| General | Insomnia | 1 (33.3%) | 0 | 0 | 0 | 0 | 0 |
| Abdominal pain | 1 (33.3%) | 0 | 0 | 0 | 0 | 0 | |
| Dermatologic | Erythema multiforme | 1 (33.3%) | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal | Elevated transaminase | 1 (33.3%) | 0 | 0 | 8 (23.5%) | 1 (2.9%) | 0 |
| Elevated bilirubin | 1 (33.3%) | 0 | 0 | 0 | 0 | 0 | |
| Elevated alkaline phosphatase | 1 (33.3%) | 0 | 0 | 0 | 0 | 0 | |
| Endocrine | Hyperglycemia | 0 | 0 | 0 | 9 (26.5%) | 1 (2.9%) | 0 |
| Cardiovascular | Cerebral infarction | 0 | 0 | 0 | 0 | 1 (2.9%) | 0 |
| Pulmonary hypertension | 0 | 0 | 0 | 0 | 1 (2.9%) | 0 | |
| Hemorrhage | Subdural hematoma | 0 | 0 | 0 | 0 | 1 (2.9%) | 0 |
Including grade 5 pneumonia (n = 1 each).
15 mg/m2 group
None of the three patients enrolled in the phase I study experienced DLT during the first cycle of treatment, and all continued treatment at this dose. All three patients experienced grade 3 or 4 hematologic toxicities and all received granulocyte‐colony stimulating factor (G‐CSF). One patient developed pneumonia on day 20 of the 9th course, which progressed rapidly, and this patient died 10 days later. There were no other severe non‐hematologic toxicities reported.
20 mg/m2 group
Thirty‐four patients received the 20 mg/m2 dose. As shown in Table 3, myelosuppression was very common, a similar finding to that of previous studies. A total of 17 patients received G‐CSF. One patient experienced prolonged grade 4 neutropenia and was taken off the study. Another patient developed pneumonia on day 13 of cycle 2 and died 3 weeks later. Non‐hematologic toxicities ≥grade 3 included cerebral infarction (n = 1, grade 3), subdural hematoma (n = 1, grade 3), elevated blood glucose (n = 1, grade 3), and pulmonary hypertension (n = 1, grade 3).
Response
Response data are summarized in Table 4. Out of 37 patients, CR, PR and hematologic improvement (HI) defined by IWG2000 criteria were observed in 7 (18.9%), 3 (8.1%) and 6 (16.2%), respectively. By IWG2006 criteria, CR, PR, marrow CR and HI were observed in 7 (18.9%), 3 (8.1%) and 2 (5.4%) and 4 (10.8%), respectively. When analysis was limited to the 34 patients who received treatment at 20 mg/m2, CR and PR were observed in 7 (20.6%) and 2 (5.9%), respectively, both by IWG2000 and IWG2006 criteria. In patients who achieved response (CR + PR, n = 9) at 20 mg/m2, median time to remission was 130 days (range 67–220), or four cycles (range 4–8), and duration of remission was 474+ days (range 294–598+).
Table 4.
Response to decitabine treatment
| Doses | 15 mg/m2 (n = 3) | 20 mg/m2 (n = 34) | Total (n = 37) | |
|---|---|---|---|---|
| IWG 2000 | CR | 0 (0.0%) | 7 (20.6%) | 7 (18.9%) |
| PR | 1 (33.3%) | 2 (5.9%) | 3 (8.1%) | |
| HI | 1 (33.3%) | 5 (14.7%) | 6 (16.2%) | |
| SD | 1 (33.3%) | 4 (11.8%) | 5 (13.5%) | |
| PD | 0 (0.0%) | 3 (8.8%) | 3 (8.1%) | |
| NE | 0 (0.0%) | 13 (38.2%) | 13 (35.1%) | |
| CR + PR (%, 95% CI) | 1 (33.3% [0.8–90.6%]) | 9 (26.5% [12.9–44.4%]) | 10 (27.0% [13.8–44.1%]) | |
| CR + PR + HI (%, 95% CI) | 2 (66.7% [9.4–99.2%]) | 14 (41.2% [24.6–59.3%]) | 16 (43.2% [27.1–60.5%]) | |
| IWG2006 | CR | 0 (0.0%) | 7 (20.6%) | 7 (18.9%) |
| PR | 1 (33.3%) | 2 (5.9%) | 3 (8.1%) | |
| mCR | 1 (33.3%) | 1 (2.9%) | 2 (5.4%) | |
| HI | 0 (0.0%) | 4 (11.8%) | 4 (10.8%) | |
| SD | 1 (33.3%) | 4 (11.8%) | 5 (13.5%) | |
| PD | 0 (0.0%) | 3 (8.8%) | 3 (8.1%) | |
| NE | 0 (0.0%) | 13 (38.2%) | 13 (35.1%) | |
| CR + PR (%, 95% CI) | 1 (33.3% [0.8–90.6%]) | 9 (26.5% [12.9–44.4%]) | 10 (27.0% [13.8–44.1%]) | |
| CR + PR + mCR (%, 95% CI) | 2 (66.7% [9.4–99.2%]) | 10 (29.4% [15.1–47.5%]) | 12 (32.4% [18.0–49.8%]) | |
| CR + PR + mCR+HI (%, 95% CI) | 2 (66.7% [9.4–99.2%]) | 14 (41.2% [24.6–59.3%]) | 16 (43.2% [27.1–60.5%]) |
CR, complete response; HI, hematologic improvement; mCR, marrow complete response; NE, not evaluated; PD, progressive disease; PR, partial response; SD, stable disease.
When the analysis was limited to patients with marrow blast percentage <20% (n = 28), the CR rate was 17.9% and CR + PR rate was 25.0% by both IWG2000 and IWG2006 criteria (Table S1).
Cytogenetic response was evaluable in 20 patients who had karyotype abnormalities upon study entry. Six patients (30%) achieved complete cytogenetic response, while one patient (5%) achieved partial cytogenetic response. Cytogenetic changes are shown in detail in Table 5.
Table 5.
Cytogenetic response in patients who originally had cytogenetic abnormalities
| Patient No. | Karyotype | Chromosome abnormality | Cytogenetic response |
|---|---|---|---|
| 11 | Poor | Chromosome 7 abnormalities, complex | Major/Complete |
| 16 | Poor | Chromosome 7 abnormalities, complex | Major/Complete |
| 18 | Intermediate | Trisomy 8 | Major/Complete |
| 20 | Poor | Complex | Major/Complete |
| 24 | Good | 20q‐ | Major/Complete |
| 25 | Poor | Complex | Minor/Partial |
| 28 | Poor | Chromosome 7 abnormalities, complex | Major/Complete |
Transfusion independence
Transfusion independence over time is shown in Figure 2. Percentages were calculated using all enrolled patients as the denominator (n = 34).
Figure 2.
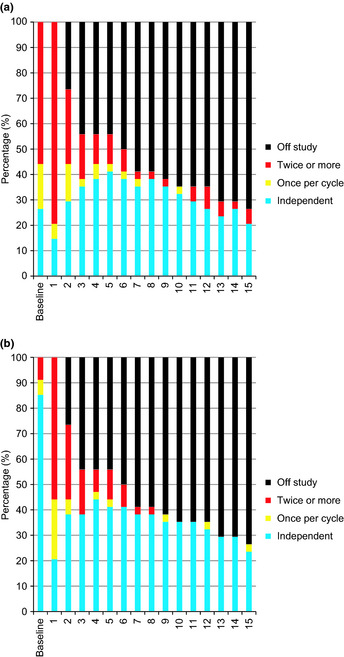
Transfusion independence. Transfusion independence was calculated using all enrolled patients as the denominator (n = 34). (a) Red cell transfusion independence. The transfusion independence rate increased from 26% (baseline) to 41% after five cycles. (b) Platelet transfusion independence. The color indicates the number of transfusions required per cycle. At baseline 85% of patients were transfusion independent. The majority of patients who continued on treatment past cycle 6 did not require platelet transfusion after that cycle.
Red cell transfusion independence
At baseline, 26% of patients were red cell transfusion‐independent. Although this number decreased to 15% of patients after the first cycle, it increased again after two cycles, hitting a peak of 41% after five cycles. Transfusion independence decreased again after 10 cycles, but many who were transfusion‐independent continued treatment. Details are shown in Figure 2(a).
Platelet transfusion independence
At baseline, 85% of patients were platelet transfusion‐independent, with that number falling to 21% after the first cycle. However, transfusion independence increased as patients continued on treatment, and most patients who were able to continue treatment through cycle 6 became platelet transfusion‐independent after that cycle. Details are shown in Figure 2(b).
Time to acute myelogenous leukemia or death and overall survival
Kaplan–Meier estimates of time to AML or death are plotted in Figure 3. Median time to AML or death has not been reached. The 2‐year rate of AML‐free survival is 52%. Kaplan–Meier estimates of overall survival are shown in Figure 4. Median survival has not been reached. The 2‐year survival rate is 56%. These data are comparable to previously reported data in similar patient populations receiving the same treatment in the United States and Europe.6, 7, 8
Figure 3.
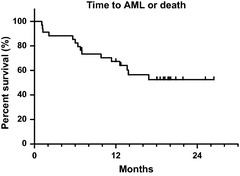
Time to acute myeloid leukemia or death. Median time to acute myeloid leukemia or death has not been reached. The 2‐year rate was 52%.
Figure 4.
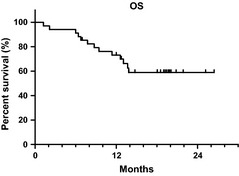
Overall survival. Median survival has not been reached. The 2‐year rate was 56%.
Pharmacokinetics
Plasma concentrations of decitabine were analyzed on days 1 and 5 in three patients receiving 15 mg/m2 and five patients receiving 20 mg/m2 (Fig. 5). Plasma decitabine became undetectable within 240 min. No significant difference in plasma decitabine concentrations between days 1 and 5 was observed. In addition, plasma levels at each time point and the calculated area under the curve were not apparently different between the 15 and 20 mg/m2 groups (data not shown), although this could be due to the limited number of evaluable subjects and considerable individual variation.
Figure 5.
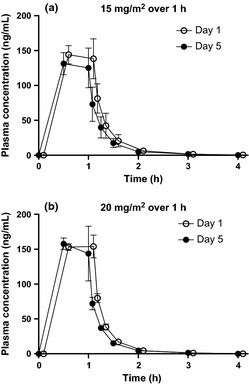
Plasma concentrations of decitabine – time profile (Mean ± standard error of mean). Mean plasma concentration is plotted with standard error of mean. The data are from: (a) three patients receiving 15 mg/m2, and (b) five patients receiving 20 mg/m2 (one patient receiving 20 mg/m2 in phase I chose not to participate in the pharmacokinetic analysis).
Pharmacodynamics
Changes in DNA methylation in peripheral blood mononuclear cells and CD15 + peripheral blood cells
Changes in methylation of five selected genes in PBMCs and CD15 + PBCs are summarized in Figure 6(a). In general, the degree of methylation at baseline was higher in PBMCs than in CD15 + PBCs, although the difference was not statistically significant. Average baseline methylation in PBMCs and CD15 + PBCs was 24% and 14%, respectively, for PGR (P = 0.007), 12% and 8% for ESR1 (P = 0.03), 14% and 9% for CDH1 (P = 0.002), and 15% and 11% for CDH13 (P = 0.28). The hypomethylating effect of decitabine seemed most prominent on day 12, and was more prominent in CD15 + PBCs than in PBMCs, particularly in LINE1 (P < 0.0001). With this result, and given that CD15 + PBCs represent affected myeloid cells more accurately than PBMCs, we chose to analyze the DNA methylation status of CD15 + PBCs. Changes in methylation status after treatment were more prominent in patients receiving 20 mg/m2 than in those receiving 15 mg/m2 (Fig. 6b).
Figure 6.
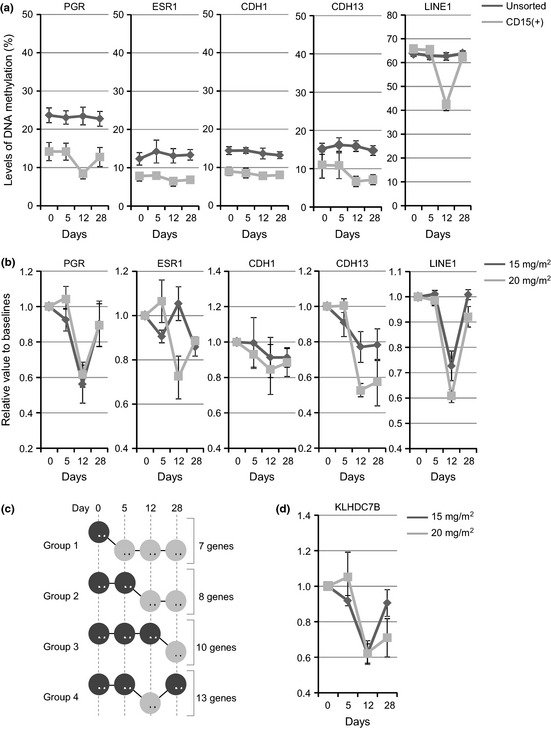
Changes in the methylation status in five selected genes using peripheral blood mononuclear cells or CD15 + peripheral blood cells (Mean ± standard error of mean). (a) Average methylation changes (0, baseline, and 5, 12 and 28 days after treatment) were compared between peripheral blood mononuclear cells (black lines) and CD15‐positive blood cells (gray lines). The y‐axis shows methylation level (%). (b) Average methylation changes (0, baseline, and 5, 12 and 28 days after treatment) were compared between patients treated with 15 mg/m2 (black lines) and 20 mg/m2 (gray lines). The y‐axis shows relative changes to methylation levels at baseline. (c) DNA demethylation status after decitabine treatment was classified into four groups. (d) Genes in Group 4 show prominent demethylation around 12 days after decitabine treatment. Methylation status of a representative newly identified gene, KLHDC7B, is shown. Methylation changes (at 0, baseline, and 5, 12 and 28 days after treatment) were compared between patients treated with 15 mg/m2 (black lines) and 20 mg/m2 (gray lines). The y‐axis shows relative changes compared to methylation levels at baseline.
Methylated CpG island amplification and microarray analysis
A variable number of genes were found to be methylated at baseline (average: 548 genes; range: 140–1029 genes) after genome‐wide DNA methylation profiling using MCAM.19 While a majority of these genes sustained detectable levels of DNA methylation after decitabine treatment, 25 genes showed significant hypomethylation. Persistent hypomethylation was observed starting on day 5 (Group 1), day 12 (Group 2), and day 28 (Group 3) in seven, eight, and 10 genes, respectively (Fig. 6c, Table S3). Notably, prominent hypomethylation on day 12 followed by recovery of methylation, as observed most dramatically in LINE1, was observed in 13 genes (Group 4). Thus, 21 genes (55%) showed prominent DNA demethylation on day 12. Using MCAM analysis, we identified a kelch domain‐containing protein, 7B (KLHDC7B), which warrants further investigation as a surrogate marker for demethylation after decitabine treatment (Fig. 6d). As we did not observe major clinical response in phase I, correlation between methylation and clinical outcome could not be analyzed.
Discussion
Decitabine was well tolerated and induced durable response in this Japanese population. Decitabine has previously been investigated in multiple clinical trials (Table 6). The treatment regimen studied here was investigated in a phase II study at MD Anderson20 and in a multi‐institutional study.8 Compared to treatment with 15 mg/m2 over 3 h every 8 h for 3 days, treatment with 20 mg/m2 over 1 h daily for 5 days is more convenient, can be given in an outpatient setting and has comparable efficacy.
Table 6.
Summary of large studies of decitabine and azacitidine
| Study | Phase | Drug | Dose | Outcome |
|---|---|---|---|---|
| CALGB24 | III | Azacitidine |
Best supportive care (n = 92) Crossover allowed |
CR 0%, PR 0% (IWG2000) Median OS 14 months |
|
75 mg/m2 per day × 7 days (n = 99) CR + 2 more cycles |
CR 6.1%, PR 10.1% Median OS 20 months (P = 0.10) A land mark analysis eliminating the effect of cross over showed benefit of azacitidine P = 0.03 |
|||
| International23 | III | Azacitidine | Physician's choice (n = 179) |
CR rate 16% (IWG2000) Median OS 15.0 months |
|
75 mg/m2 per day × 7 days (n = 179) At least four cycles |
CR rate 18% (P = 0.80) Median OS 24.4 months (P < 0.01) |
|||
| US multi institutional6 | III | Decitabine | Best supportive care (n = 81) |
CR 0%, PR 0% (IWG2000) Median leukemia free survival 7.8 months |
|
15 mg/m2 Q 8 h, nine doses (n = 89) Max eight cycles |
CR 9%, PR 8% Median leukemia free survival 12.1 months (P = 0.16) |
|||
| EORTC12 | III | Decitabine | Best supportive care (n = 114) |
CR 0%, PR 0% (IWG2000) Median OS 8.5 months |
|
15 mg/m2 Q 8 h, nine doses (n = 119) Max eight cycles |
CR 13%, PR 6% Median OS 10.1 months (P = 0.38) |
|||
| MDACC7 | II | Decitabine | 20 mg/m2 daily for 5 days and two other arms 100 mg/m2 per course (n = 95) |
CR 34%, mCR 24%, PR 1%, HI 13% (IWG2006) Median OS 19 months |
| ADOPT8 | II | Decitabine | 20 mg/m2 daily for 5 days (n = 99) |
CR 17%, mCR 15%, PR 0%, HI 18% (IWG2006) Median OS 19.4% |
| Japanese Azacitidine | II | Azacitidine | 75 mg/m2 per day × 7 days (n = 53) | CR 15%, mCR 13%, PR 0%, HI 55% (IWG2006) |
| Our study | II | Decitabine | 20 mg/m2 daily for 5 days (n = 34) |
CR 21%, mCR 3%, PR 6% (IWG2006) Median OS not reached |
ADOPT, Alternative Dosing for Outpatient Treatment Trial; CALGB, Cancer and Leukemia Group B; CR, complete remission; EORTC, European Organization for Research and Treatment of Cancer; HI, hematologic improvement; IWG, Response criteria by International Working Group; mCR, marrow complete remission; MDACC, MD Anderson Cancer Center; OS, overall survival; PR, partial remission.
The only grade 3/4 toxicities observed in >10% of patients were hematologic and infectious toxicities, which are characteristic of the disease itself, although treatment with decitabine certainly requires careful blood count monitoring and infection surveillance. Overall, the observed toxicity profile was comparable to that of azacitidine reported in a phase I/II study conducted in Japan.21 In particular, the incidences of grade 3/4 febrile neutropenia in the present study and the Japanese azacitidine study were similar at 29.4% and 30.2%, respectively.21
Randomized phase III studies of decitabine versus best supportive care have not shown significant survival benefit for decitabine.6, 12 This could be due to the fact that decitabine at 15 mg/m2 over 3 h every 8 h for 3 days, not the dose and schedule studied here, was used in the US and European Organization for Research and Treatment of Cancer (EORTC) studies. In addition, only a limited percentage of patients in these studies continued treatment after experiencing clinical benefit.6, 12 Continuation of treatment in patients who are responding is beneficial, but previous randomized phase III studies set a maximum number of treatment cycles, which affected responders in particular.
There are several differences in the mechanisms of action of azacitidine and decitabine. First, azacitidine is a ribose‐based nucleoside and is mostly incorporated into RNA. A small proportion of azacitidine diphosphate is reduced by ribonucleotide reductase to decitabine diphosphate and is eventually incorporated into DNA, which can result in the inhibition of DNA methyltransferase. In contrast, decitabine is incorporated into DNA after phosphorylation and does not require reduction. Second, azacitidine is incorporated into RNA and inhibits DNMT2. This can inhibit methylation of tRNA(Asp)‐cytosine 38 and may exert toxicity.22 Decitabine does not have this effect. Finally, the mechanisms of resistance may be different, as illustrated by the difference in the rate‐determining step of phosphorylation of each; deoxycytidine kinase limits the rate of phosphorylation for decitabine while uridine‐cytidine kinase limits that of azacitidine. Decreases in the expression or activity of these enzymes may be associated with drug resistance, and thus certain cells may be more sensitive to one drug than the other.
Our study confirmed that hypomethylation is observed after decitabine therapy. Robust methylation analysis also revealed that certain sets of genes were hypomethylated after decitabine therapy, although the significance of these changes should be further evaluated. In this study, methylation status in CD15 + PBCs, rather than PBMCs, was analyzed for the first time. The changes in methylation were generally more prominent in CD15 + PBSs than in PBMCs. As this cell population better reflects myeloid cell behavior, we propose this method be used in future analysis of in vivo methylation changes in patients treated with a hypomethylating agent.
In summary, our study has shown the safety and clinical activity of decitabine in Japanese patients with high‐risk MDS. Responses and toxicities are comparable to those reported in previous studies. Correlative analysis revealed hypomethylation in a number of genes in vivo after decitabine treatment, which warrants further investigation in the setting of epigenetic therapy.
Disclosure Statement
Y. Kobayashi and M. Ogura have received a consultant fee from Janssen Pharmaceutical KK. The other authors reported no conflicts of interest.
Supporting information
Table S1. List of the primers for bisulfite pyrosequencing analysis.
Table S2. Response to decitabine treatment in patients whose marrow blast was <20%.
Table S3. List of hypomethylated genes in response to decitabine treatment.
Acknowledgments
We thank all the patients and families who participated in this trial. We also thank all institutional investigators and their clinical research staff. This study was supported by Janssen Pharmaceutical K.K.
(Cancer Sci, doi: 10.1111/j.1349-7006.2012.02386.x, 2012)
References
- 1. Garcia‐Manero G. Myelodysplastic syndromes: 2011 update on diagnosis, risk‐stratification, and management. Am J Hematol 2011; 86: 490–8. [DOI] [PubMed] [Google Scholar]
- 2. Greenberg P, Cox C, LeBeau MM et al International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–88. [PubMed] [Google Scholar]
- 3. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 4. Shen L, Kantarjian H, Guo Y et al DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 2010; 28: 605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaminskas E, Farrell A, Abraham S et al Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res 2005; 11: 3604–8. [DOI] [PubMed] [Google Scholar]
- 6. Kantarjian H, Issa JP, Rosenfeld CS et al Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006; 106: 1794–803. [DOI] [PubMed] [Google Scholar]
- 7. Kantarjian H, Oki Y, Garcia‐Manero G et al Results of a randomized study of 3 schedules of low‐dose decitabine in higher‐risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007; 109: 52–7. [DOI] [PubMed] [Google Scholar]
- 8. Steensma DP, Baer MR, Slack JL et al Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol 2009; 27: 3842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee JH, Jang JH, Park J et al A prospective multicenter observational study of decitabine treatment in Korean patients with myelodysplastic syndrome. Haematologica 2011; 96: 1441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheson BD, Bennett JM, Kantarjian H et al Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 2000; 96: 3671–4. [PubMed] [Google Scholar]
- 11. Cheson BD, Greenberg PL, Bennett JM et al Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–25. [DOI] [PubMed] [Google Scholar]
- 12. Lubbert M, Suciu S, Baila L et al Low‐dose decitabine versus best supportive care in elderly patients with intermediate‐ or high‐risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol 2011; 29: 1987–96. [DOI] [PubMed] [Google Scholar]
- 13. Cashen AF, Shah AK, Todt L et al Pharmacokinetics of decitabine administered as a 3‐h infusion to patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Cancer Chemother Pharmacol 2008; 61: 759–66. [DOI] [PubMed] [Google Scholar]
- 14. Raza A, Raza FZ, Galili N. Low‐dose decitabine and high‐risk MDS. Blood 2006; 108: 4291. [DOI] [PubMed] [Google Scholar]
- 15. Gao W, Kondo Y, Shen L et al Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis 2008; 29: 1901–10. [DOI] [PubMed] [Google Scholar]
- 16. Colella S, Shen L, Baggerly KA et al Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques 2003; 35: 146–50. [DOI] [PubMed] [Google Scholar]
- 17. Yang AS, Estecio MR, Doshi K et al A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res 2004; 32: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Estecio MR, Gallegos J, Vallot C et al Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res 2010; 20: 1369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Estecio MR, Yan PS, Ibrahim AE et al High‐throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 2007; 17: 1529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kantarjian HM, O'Brien S, Cortes J et al Results of decitabine (5‐aza‐2′deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer 2003; 98: 522–8. [DOI] [PubMed] [Google Scholar]
- 21. Uchida T, Ogawa Y, Kobayashi Y et al Phase I and II study of azacitidine in Japanese patients with myelodysplastic syndromes. Cancer Sci 2011; 102: 1680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res 2009; 69: 8127–32. [DOI] [PubMed] [Google Scholar]
- 23. Fenaux P, Mufti GJ, Hellstrom‐Lindberg E et al Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher‐risk myelodysplastic syndromes: a randomised, open‐label, phase III study. Lancet Oncol 2009; 10: 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silverman LR, Demakos EP, Peterson BL et al Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002; 20: 2429–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the primers for bisulfite pyrosequencing analysis.
Table S2. Response to decitabine treatment in patients whose marrow blast was <20%.
Table S3. List of hypomethylated genes in response to decitabine treatment.