

Abstract
In mammals, hematopoietic homeostasis is maintained by a fine‐tuned balance among the self‐renewal, proliferation, differentiation and survival of hematopoietic stem cells and their progenies. Each process is also supported by the delicate balance of the expression of multiple genes specific to each process. GATA1 is a transcription factor that comprehensively regulates the genes that are important for the development of erythroid and megakaryocytic cells. Accumulating evidence supports the notion that defects in GATA1 function are intimately linked to hematopoietic disorders. In particular, the somatic mutation of the GATA1 gene, which leads to the production of N‐terminally truncated GATA1, contributes to the genesis of transient myeloproliferative disorder and acute megakaryoblastic leukemia in infants with Down syndrome. Similarly, a mutation in the GATA1 regulatory region that reduces GATA1 expression is involved in the onset of erythroid leukemia in mice. In both cases, the accumulation of immature progenitor cells caused by GATA1 dysregulation underlies the pathogenesis of the leukemia. This review provides a summary of multi‐step leukemogenesis with a focus on GATA1 dysfunction. (Cancer Sci, doi: 10.1111/cas.12007, 2012)
Gata1 Gene Regulation in Erythropoiesis
GATA1 was originally identified in 1989 as a transcription factor that binds to the β‐globin regulatory region, and it was later revealed to be a member of the GATA transcription factor family, whose members bind the consensus “WGATAR” binding motif.1, 2, 3 In hematopoietic tissues, GATA1, GATA2 and GATA3 are key regulators of cell/tissue specific development.2 The expression profiles of these genes are restricted to specific cell lineages and differentiation stages. GATA2 is abundant in hematopoietic stem cells and early multipotential progenitor cells; GATA1 is mainly expressed in erythroid‐ and megakaryocyte‐committed cells; and GATA3 is expressed in the T‐cell lineage and is important for Th2 type T‐cell development.4, 5
Importantly, GATA1 and GATA2 show partially overlapping expression profiles and cooperatively regulate their own expression.6 When immature progenitor cells are triggered toward erythroid commitment, GATA2 initiates GATA1 expression. Subsequently, GATA1 expression is activated by GATA1 and GATA2, and the level of GATA1 increases rapidly. GATA2 expression is repressed by GATA1 and gradually decreases during erythroid differentiation. This dynamic GATA factor switching underlies normal erythropoiesis.6
The mouse Gata1 gene consists of two alternative first exons and five commonly used coding exons (Fig. 1).3 The proximal first exon (referred to as the IE exon) is used for Gata1 gene expression in hematopoietic cells, whereas the distal first exon (referred to as the IT exon) is expressed in Sertoli cells in testis. A region of approximately 8.5 kb flanking the IE exon is sufficient to promote Gata1 gene expression.7 Therefore, this region has been named G1HRD (GATA1 hematopoietic regulatory domain).
Figure 1.
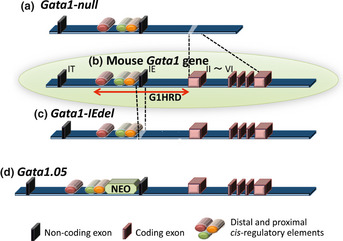
The structure of the mouse Gata1 gene. The mouse Gata1 gene is composed of two alternative non‐coding first exons and five constitutive coding exons (b; inside the green circle). G1HRD is composed of three cis‐regulatory regions (shown by red, green and orange bars) located upstream of the IE exon and the first intron. The Gata1‐null (a) and Gata1‐IEdel (b) alleles were constructed by deleting the five coding exons or the IE exon, respectively. The insertion of a Neo cassette between the cis‐regulatory elements and the IE exon reduces the gene expression of the Gata1.05 allele (d).
GATA1 hematopoietic regulatory domain has three cis‐regulatory elements within the 3.9‐kb upstream of the IE exon, and these elements are conserved between humans and mice.3, 8 One regulatory element is the G1HE (GATA1 hematopoietic enhancer), located between 3.9 and 2.6 kb upstream of the IE exon. The G1HE contains a bipartite DNA binding motif composed of an E‐box and GATA motif.9 A transgenic reporter mouse assay revealed that G1HRD activity was completely abolished by a mutation in the GATA motif, but not by a mutation in the E‐box,10 indicating that the direct binding of the GATA factor to this locus is required for Gata1 gene expression. The other two regulatory elements, located upstream of the IE exon, are a typical double GATA motif adjacent to the consensus CP2 binding sites and a CACCC motif. The combination of the GATA motif and the CP2 sites is conserved in the promoter region of multiple erythroid genes, and it is proposed to be essential for erythroid promoter activity.11, 12, 13 In addition, chromatin immunoprecipitation and sequence analysis revealed an enriched GATA motif and CACCC motif together in the regulatory region of a variety of erythroid genes,14 suggesting that the GATA factor promotes erythroid differentiation in combination with other transcription factors that may work cooperatively with GATA factors.
The importance of these three elements was further demonstrated in a transgenic reporter mouse assay using an artificially constructed reporter transgene.15 A 1‐kb artificial minigene containing these three elements can recapitulate GATA1 gene expression, and all three of the elements are required for this activity. Although mice with mutations in either the proximal or the distal GATA‐binding motif were viable,16, 17 the expression pattern of the Gata1 gene has been influenced. Indeed, a recent transgenic mouse experiment using a 200‐kb bacterial artificial chromosome sequence showed that the distal region containing the GATA binding motif has the potential to initiate Gata1 gene expression in early erythroid progenitor cells.18 Thus, spatiotemporal Gata1 gene regulation is a complex process that is organized by multiple molecules, including the GATA factors. We propose that the proper dynamic regulation of GATA1 expression is the key to erythroid homeostasis during erythroid development.
The Effect of Deregulated Gata1 Gene Expression in Erythropoiesis
The IE exon is used specifically in hematopoietic tissues. Therefore, mice lacking the IE exon die in utero due to yolk sac dyserythropoiesis, similar to mice that lack the entire Gata1 gene.19, 20 To investigate the function of GATA1 in adulthood, mouse strains carrying two different floxed alleles are used; one allele generates a Gata1‐null allele lacking five coding exons after treatment with cre recombinase, and the other allele generates a Gata1‐IEdel allele in which the IE non‐coding exon is specifically deleted (Fig. 1). As expected, the mice from both lines suffer from anemia and thrombocytopenia. Further investigation has revealed that the acquisition of the Gata1‐null mutation in adulthood leads to red cell aplasia with a hypotrophic red‐pulp area in the spleen,21 while mice with the deleted IE exon suffer from anemia with a massive accumulation of immature erythroid progenitor cells.20 These results indicate that the survival and/or proliferation of immature erythroid progenitor cells are disturbed in the mice lacking Gata1 coding exons but those are maintained in the mice without the IE exon. Five coding exons were retained in the Gata1‐IEdel mice; thus, aberrant Gata1 gene expression using alternative transcription start sites has been predicted. GATA1 is important for the regulation of a set of genes related to the proliferation, differentiation and cell survival of erythroid progenitor cells, and the well‐organized regulation of these genes is required for normal erythroid development. It has been proposed that inadequate Gata1 gene expression disturbs the balance of erythroid proliferation, survival and differentiation, leading to the aberrant accumulation of immature erythroid progenitor cells in Gata1‐IEdel mice.
Analysis of the Gata1‐IEdel mice provided new insight into the function of the IE exon. Based on RT‐PCR analyses, the amount of transcripts expressed in erythroid progenitor cells using the alternative first exons instead of the IE exon is comparable to endogenous expression levels. However, no Gata1 transcripts are observable in the megakaryocytic lineage.20 This finding indicates that the IE exon is required specifically in the megakaryocytic lineage, but its transcription start site function is dispensable in the erythroid lineage. Interestingly, no full‐length GATA1 protein is produced in the Gata1‐IEdel mice. Instead, inefficient translation using the AUG start codon at amino acid residue 84 of the full‐length GATA1 protein has occurred, leading to the low‐level production of an amino (N)‐terminally truncated GATA1 (GATA1‐S) protein.20 Recently, the importance of the carboxyl (C)‐terminal region of GATA1 as a transactivation domain was identified, which independently and cooperatively works with the N‐terminal transactivation domain of GATA1.22 Thus, the phenotype generated by the acquired deletion of the IE exon is due to an incomplete loss of GATA1 function.
Leukemogenesis in the Erythroid Lineage
The GATA1‐knockdown allele has been constructed by inserting a neomycin resistance cassette between the proximal regulatory region and the IE exon (Fig. 1).23 Strong promoter activity within the neomycin cassette interferes with the transcriptional regulation of the IE exon, leading to a reduction in Gata1 gene expression down to 5% of the endogenous level. This allele has been named Gata1.05. GATA1 is located on the X‐chromosome, and GATA1 deficiency in hemizygous male embryos confers lethal anemia resembling that of the Gata1‐null male embryos, which are established with a germ‐line deletion of the coding exons. In contrast, mice with a deletion of the distal cis‐regulatory element are able to survive to adulthood, as approximately 20% of the normal level of GATA1 expression is maintained in those mice.17 These findings indicate that an 80% reduction in the endogenous GATA1 level is permissible, but a reduction of 5% of the GATA1 level is lethal in embryos.
The Gata1.05 and Gata1‐null alleles have been maintained in heterozygous female mice. Intriguingly, Gata1.05 female mice frequently develop erythroleukemia between 3 and 6 months of age, whereas the onset of leukemia is completely abolished by the transgenic expression of wild‐type GATA1 under the transcriptional regulation of G1HRD.24 Immunohistochemistry has showed that the surfaces of the leukemic cells are positive for c‐Kit (a stem cell factor receptor) and CD71 (a transferrin receptor) antibodies but negative for an antibody against Ter119 (a molecule associated with glycophorin A25), which corresponds to the immature erythroid progenitor cells.3 In contrast, female mice harboring heterozygously the Gata1‐null allele have a normal life expectancy.
In both heterozygous females, erythroid progenitor cells are divided into two groups dependent on X‐chromosome inactivation. The erythroid progenitor cells with the activated wild‐type allele can develop normally into mature erythrocytes, because the GATA1 level is unaffected. In contrast, it is predicted that the development of the progenitor cells with the activated mutant allele would be altered due to the deficiency or absence of GATA1 (Fig. 2a). Therefore, leukemic event(s) would occur in the immature progenitor cells carrying the activated Gata1.05 allele, but not in the cells with the activated Gata1‐null allele.
Figure 2.
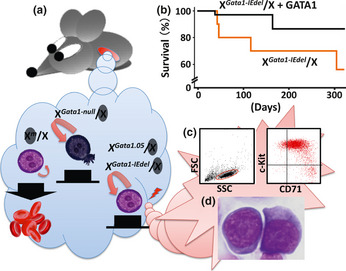
Multi‐step leukemogenesis caused by the dysregulation of Gata1 gene expression. (a) There are two types of erythroid progenitor cells in females heterozygous for the Gata1 gene mutation due to the random inactivation of the X chromosome. Erythroid progenitor cells with the activated wild‐type allele undergo terminal maturation, whereas those with the inactivated wild‐type allele fail to differentiate and continue to proliferate. Immature cells with the activated Gata1‐null allele die by apoptosis due to the absence of GATA1. In contrast, a low level of GATA1 expression elongates the lifespan of the progenitor cells expressing either the activated Gata1‐IEdel or Gata1.05 allele. Consequently, those cells may acquire additional genetic event(s). (b) Survival curves of Gata1‐IEdel heterozygous females with (black line; n = 35) or without (red line; n = 10) the G1HRD‐GATA1 transgene. (c) A typical flow cytometry dot plot showing leukemic cell population positive for c‐Kit and CD71 (right panel). Left panel depicts a graph for side‐scatter (SSC) versus forward‐scatter (FSC). Leukemic cells in peripheral blood are gated in a red circle. (d) Leukemic cells in bone marrow of Gata1‐IEdel heterozygous females are morphologically similar to proerythroblasts.
There are two possible explanations for the development of leukemia in the Gata1.05 females. One is that artificially strong promoter activity in the neomycin cassette may activate a latent oncogenic ability. Alternatively, the 5% residual expression of the Gata1 gene may contribute to the genesis of leukemia, which may not occur in the complete absence of GATA1. Recently, it was found that heterozygous female mice carrying the Gata1‐IEdel allele are also prone to develop leukemia (Shimizu R and Yamamoto M, unpublished observation). The leukemic cells in the Gata1‐IEdel female mice show similar phenotypic properties to those in the Gata1.05 female mice (Fig. 2). As in the Gata1.05 female mice, the onset of leukemia in the Gata1‐IEdel female mice is abolished by the transgenic expression of full‐length GATA1 (Fig. 2b). A low level of N‐terminal domain‐truncated GATA1 (i.e., GATA1‐S) is expressed in the erythroid progenitor cells bearing the activated Gata1‐IEdel allele, suggesting that inadequate GATA1 function due to low‐level expression of wild‐type GATA1 and expression of GATA1‐S may be involved in the leukemogenesis observed in Gata1.05 and Gata1‐IEdel female mice, respectively.
Based on the data from conditional Gata1 gene modification, progenitor cells with the activated mutant allele are found to develop differently in each line of the heterozygous females during erythroid differentiation due to remnant GATA1 expression.20 Progenitor cells in the Gata1‐null females are hypoplastic, whereas the relevant cells in the Gata1‐IEdel females, and possibly the Gata1.05 females, are hyperplastic and lack differentiation potential. This phenomenon has been further investigated using an in vitro differentiation assay with embryonic stem (ES) cells.26 Wild‐type ES cells had a restricted cell proliferation profile and differentiated into mature hemoglobinized erythroblasts, whereas both Gata1.05 ES cells and Gata1‐null ES cells remained immature and continued to proliferate. In sharp contrast to the Gata1.05 ES cells, Gata1‐null ES cells tended to die by apoptosis (Fig. 2). The expression of the Bcl2l1 gene, which encodes an anti‐apoptotic protein, was detected in the Gata1.05 ES cells, but not in the Gata1‐null ES cells.26 Coinciding with this finding, the number of TUNEL‐positive apoptotic cells was increased in the Gata1‐null females, but not in the Gata1.05 or wild‐type females.24 These findings indicate that the regulation of anti‐apoptotic function depends on the presence of GATA1. Progenitor cells with the activated Gata1‐null allele undergo apoptosis during erythroid differentiation.24, 26 In contrast, the level of GATA1 in the progenitor cells in of Gata1.05 and Gata1‐IEdel females is sufficient to support cell survival, but not to control proliferation and promote differentiation. Consequently, erythroid progenitor cells are protected from apoptotic elimination and can survive for long periods but remain immature. Such progenitor cells, which are unnaturally halted during the differentiation process, may have the opportunity to acquire additional genetic mutations that promote leukemic transformation (Fig. 2).
Leukemic Stem Cells in Multi‐Step Leukemogenesis
It has been proposed that a specific subset of leukemic cells, the so‐called leukemic stem cells (LSCs),27 are capable of self‐renewal and of producing clonogenic leukemic cells. Leukemic stem cells were first identified in the CD34+CD38− leukemic subpopulation of human acute myelogenous leukemia cells.28 Subsequently, a number of reports have provided evidence that LSCs accumulate in the CD34+CD38− subpopulation in cases of acute myelogenous leukemia and acute lymphocytic leukemia.29, 30, 31, 32 Normal hematopoietic stem cells (HSCs) also exhibit the Lin−CD34+CD38− phenotype,33 and thus, it has been proposed that HSCs are the most likely target for transformation into LSCs. In contrast, recent reports have clarified that additional subpopulations of LSCs may exist in some cases.34, 35, 36 Therefore, the precursors of LSCs may be committed progenitor cells that acquired the potential for self‐renewal.
The leukemic cells developed in the Gata1.05 females also caused leukemia when transplanted into immune‐deficient allogenic nude mice, whereas nude mice injected with the cells from the Gata1.05 females, who exhibited no clinical signs of leukemia, never developed leukemia.37 The exogenous leukemic cells expanded in the recipient nude mice serially regenerate leukemia in the next generation of recipient mice. The c‐Kit+CD71+Ter119− leukemic cells are divided into two populations that exhibit either weak (SP; side population) or strong (MP; major population) Hoechst dye staining.37 It is important to note that the majority of the cells in SP fraction are quiescent, whereas those in MP fraction proliferate rapidly. By analyzing the results of transplanting these two subpopulations, the capacity to transfer leukemia is revealed to reside exclusively in the quiescent SP fraction. Consistent with this finding, cancer stem cells from a variety of malignancies are enriched in the SP fraction.38, 39 SP phenotype is defined by the property to effectively exclude the Hoechst dye due to ATP‐binding cassette transporters, which are abundantly expressed in quiescent HSCs with long‐term reconstitution ability.40, 41, 42 Together with their quiescent characteristics, the excellent drug export system of LSCs may contribute to their resistance to chemo‐toxic agents.
The development of a leukemic nude mouse model has allowed the evaluation of LSC characteristics. Importantly, the nature of LSCs is found to be altered upon exposure to anti‐cancer drugs. When a patient encounters a hematopoietic emergency, such exposure to myelosuppressive agents, HSCs enter the cell cycle to expand their progenies. However, HSCs immediately return to the previous quiescent state and maintain their pool size after recovery.43 Although LSCs also enter the cell cycle in the recurrent phase of leukemia after chemotherapy, they never arrested in the quiescent state, and the LSC pool increased in size.37 Thus, therapeutic resistance and progressive characteristics in recurrent/refractory leukemias may arise in part from the activation of LSCs that have survived an inappropriate treatment (Fig. 3).
Figure 3.
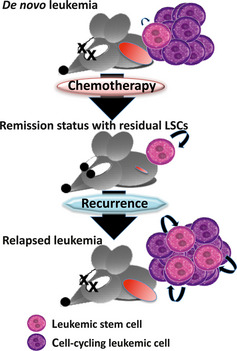
Pernicious changes in leukemic stem cells (LSCs) in relapsed leukemia. Most LSCs are quiescent, and the number of LSCs is maintained at a steady state in a de novo leukemia. After eliminating proliferating leukemic cells with inappropriate chemotherapy, the residual LSCs are stimulated to enter the cell cycle and produce siblings and progenies (remission status with residual LSCs). Upon relapsing to full‐blown leukemia, the LSCs fail to return to quiescence and are maintained at a higher number than before (relapsed leukemia).
Leukemogenesis in the Megakaryocytic Lineage: A Model for Multi‐Step Leukemogenesis
Down syndrome (DS) is a genetic disorder caused by an extra copy of chromosome 21. Children with DS share some common physical and mental features, although the severity of these symptoms varies among individuals. DS is the most common genetic risk factor for childhood leukemia. The incidence of acute megakaryoblastic leukemia (DS‐AMKL) in children with DS is approximately 500 times higher than that in the general population.44 Approximately 10% of babies with DS develop transient myeloproliferative disorder (DS‐TMD), which is characterized by the clonal expansion of immature megakaryocytes. This condition resolves spontaneously, although intensive care is required for some DS‐TMD patients who develop organ failure due to massive blast cell infiltration. A unique clinical characteristic of this disease is that approximately 20% of DS children with a history of DS‐TMD develop acute megakaryoblastic leukemia (AMKL) after several years of asymptomatic latency.44
In the first decade of this century, rapid progress has been made in elucidating the molecular mechanisms of DS‐TMD/AMKL. Almost all of the studied cases of DS‐TMD/AMKL involve a mutation in the GATA1 gene, leading to the production of GATA1‐S, which lacks the amino‐terminal 83 amino acids and is alternatively translated from the methionine codon at position 84 in the 3rd exon. Furthermore, when a GATA1 gene mutation is recognized in AMKL children without DS symptoms, an extra copy of chromosome 21 is always identified in the blast cells (consequently diagnosed as mosaic trisomy 21); except for only two reported cases.45, 46 Therefore, a model for multi‐step leukemogenesis has been proposed. Children with congenital DS possess the 1st hit (trisomy 21) for DS‐TMD/AMKL. The 2nd genetic hit is when a megakaryocytic progenitor cell in DS babies transforms into a DS‐TMD blast cell as a consequence of the acquired somatic GATA1 gene mutation. Finally, a DS‐TMD blast cell subsequently becomes a leukemic cell when an additional, currently unknown, genetic event occurs.47
Molecular Basis of Leukemogenesis in the Megakaryocytic Lineage
GATA1 has two functional finger domains in addition to its N‐terminal transactivation domain. One is a carbonyl‐terminal zinc‐finger (C‐finger), which is required for DNA binding and interactions with various co‐factors. The other is the N‐terminal zinc‐finger (N‐finger) domain, which is important for association with the GATA factor‐specific co‐factor FOG1 and works to stabilize GATA1‐DNA binding.12, 48 Transgenic complementation rescue analyses using transgenic mouse lines expressing mutant GATA1 under the regulation of G1HRD have clarified the independent and cooperative functions of these functional domains during erythroid development.49 One advantage of this approach is that the rescued mice express various levels of mutant GATA1, so that the dosage‐dependent effect of mutant GATA1 can be determined by evaluating the phenotypes in each line. For example, the transgenic expression of a high amount of mutant GATA1 with poor interaction with FOG1 in GATA1‐deficient mice phenocopies the human disease caused by an inherited mutation in the GATA1 gene.50, 51 The FOG1‐GATA1 interaction is important for the regulation of multiple membrane protein genes and to terminate the maturation of megakaryocytes. Thus, newborn mice expressing normal levels of GATA1 mutant lacking the FOG1 interaction die due to spherocytic hemolysis.52 An excessive level of mutant GATA1 can partially support the expression of membrane proteins. Consequently, rescued mice grow to adulthood and exhibit thrombocytopenia resembling that in human cases.
GATA1 supports megakaryocyte differentiation at multiple stages. The N‐terminal transactivation domain of GATA1 is required for the controlled growth of immature megakaryocytic progenitor cells, whereas the GATA1‐FOG1 interaction mediates the terminal maturation of megakaryocytes and proplatelet formation.53, 54 As a matter of course, GATA1‐deficiency leads to arrest of the differentiation and perturbation of the growth control in megakaryocytes.55 A transgenic complementation rescue analysis has been exploited to investigate the role of the GATA1‐S mutation on the pathogenesis of TMD. As expected, the number of immature megakaryocytes increased in the fetal livers.56 Importantly, although the megakaryocyte colony forming ability in rescued mice is equivalent to that of wild‐type mice, there are significantly more cells per colony, and those cells are morphologically immature. These findings support the hypothesis that GATA1‐S cannot regulate the proliferation of immature megakaryocytes, most likely after the megakaryocyte colony‐forming units are produced. Notably, this hyper‐proliferative megakaryocyte phenotype is observed in fetal livers, but not in neonatal spleens or adult bone marrow, in synchronizing with the switch of hematopoietic sites from livers in fetus to spleens and bone marrows in pups. Thus, the GATA1‐S mutation alone is sufficient to induce the abnormal proliferation of immature megakaryocytic progenitor cells in embryonic livers.
The phenotype observed in the rescued mice expressing GATA1‐S closely resembles that of newborns with DS‐TMD. The hyper‐proliferative megakaryocytes seen in livers of the rescued embryos are no longer identified in the spleens and bone marrows at the weaning stage.56 In this regard, previous reports showed that a GATA1 mutant lacking amino acids 3–63 enables the hyper‐proliferation of megakaryocytes of mid‐gestation embryos but lost this ability by the late‐gestation period.57 The difference between these two mouse studies is most likely due to the additional amino acid residues preserved in the latter mouse study. Specifically, GATA1‐S lacks a consensus retinoblastoma protein (pRb)‐binding motif (LxC/SxE, amino acids 81–85) that is conserved in humans and mice. Indeed, the interaction of GATA1 with pRb through this motif seems to be vital for proper erythropoiesis.58 pRb is known as a tumor suppressor protein that acts partly through the transcriptional repression of E2F‐regulated genes. It has been reported that GATA1‐S failed to repress E2F activation followed by the activation of mammalian target of rapamycin (mTOR) signaling in DS‐AMKL cells.59 These findings support the notion that the loss of the GATA1‐pRb‐E2F complex formation may in part potentiate cell cycle progression in the blasts of DS‐TMD, but direct verification of the function of these complexes remains to be established.
A high level of exogenous GATA1‐S expression rescues the defect in differentiation, but not in growth control in GATA1‐deficient megakaryocytes.54 Consistent with the finding, the embryos rescued by the overexpression of transgenic GATA1‐S showed thrombocytosis, corresponding to an increased number of immature megakaryocytes.56 In the embryos, megakaryocytic progenitor cells accumulated in the fetal livers are spontaneously eliminated when they lose the hyper‐proliferative potential (Fig. 4). Consequently, mice recover from thrombocytosis by the weaning stage.56 In contrast, mice expressing normal levels of GATA1‐S suffer from thrombocytopenia (Shimizu R and Yamamoto M, unpublished observation). These findings suggest that the differentiation process is weakened due to the lack of the GATA1 N‐terminal domain, but this disadvantage is compensated in part by abundant GATA1‐S expression. Recently, it was reported that the level of GATA1‐S protein produced in the blasts of DS‐TMD/AMKL patients varies, and leukemic transformation occurs more frequently in the cases with low GATA1‐S expression than in those with high GATA1‐S expression.60 Thus, an immature megakaryocyte progenitor cell lacking differentiation potential may survive for long periods in the hematopoietic organs of children with DS‐TMD. These long‐lived cells may have an increased chance to acquire subsequent mutation(s) and transform into genuine leukemic cells (Fig. 4).
Figure 4.
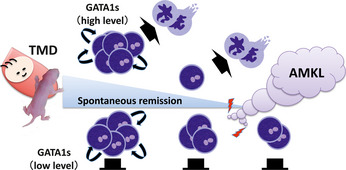
Multi‐step leukemogenesis caused by a structural mutation in the GATA1 protein. Because the contribution of the GATA1‐S mutation to the hyper‐proliferative phenotype of megakaryocytes is restricted to fetal hematopoiesis, the phenotypes observed in babies with Down syndrome with transient myeloproliferative disorder (DS‐TMD) and mice rescued with GATA1‐S disappear spontaneously. At high levels of expression, GATA1‐S has the potential to stimulate megakaryocytic differentiation and rapidly eliminate accumulated abnormal megakaryocytic progenitor cells. However, a low level of GATA1‐S fails to promote differentiation and instead extends the life spans of those progenitor cells bearing the GATA1‐S mutation. Consequently, those cells have an opportunity to acquire additional genetic hit(s) and eventually transform into a leukemic cell.
Conclusions
GATA1 is a key regulator of erythroid and megakaryocytic homeostasis. Specific abnormalities of GATA1 function are involved in leukemogenesis in both lineages. This review has described two types of leukemias that occur due to GATA1 dysfunction. One is an erythroleukemia in which the preleukemic status or the accumulation of immature erythroblasts occurs due to a qualitative deficit in GATA1. The other is a megakaryoblastic leukemia in which the preleukemic status or the accumulation of immature megakaryocytes is generated by qualitative defect in the GATA1 protein.61 Whereas the latter has been found in human DS cases, no human cases of acute erythroleukemia carrying a GATA1 gene mutation have been reported. We surmise that mutations in the regulatory locus of human GATA1 gene might trigger the onset of this type of leukemia. The function of GATA1 in the balance of differentiation, proliferation and cell survival provides important clues to the molecular pathogenesis of the leukemias. However, a simple GATA1 gene mutation is not sufficient to trigger leukemia, and subsequent genetic hit(s) to the preleukemic progenitor cells seems to be required. The GATA1‐related leukemias provide important insights into multi‐step leukemogenesis.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (RS and MY), the Asahi Glass Foundation (RS), the Mitsubishi Foundation (RS and MY), the Daiichi‐Sankyo Foundation of Life Science (RS), the Takeda Science Foundation (MY) and the Naito Memorial Foundation (MY).
(Cancer Sci, 2012; 103: 2039–2044)
References
- 1. Tsai SF, Martin DI, Zon LI, D'Andrea AD, Wong GG, Orkin SH. Cloning of cDNA for the major DNA‐binding protein of the erythroid lineage through expression in mammalian cells. Nature 1989; 339: 446–51. [DOI] [PubMed] [Google Scholar]
- 2. Yamamoto M, Ko LJ, Leonard MW, Beug H, Orkin SH, Engel JD. Activity and tissue‐specific expression of the transcription factor NF‐E1 multigene family. Genes Dev 1990; 4: 1650–62. [DOI] [PubMed] [Google Scholar]
- 3. Shimizu R, Yamamoto M. Gene expression regulation and domain function of hematopoietic GATA factors. Semin Cell Dev Biol 2005; 16: 129–36. [DOI] [PubMed] [Google Scholar]
- 4. Zheng W, Flavell RA. The transcription factor GATA‐3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997; 89: 587–96. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki M, Shimizu R, Yamamoto M. Transcriptional regulation by GATA1 and GATA2 during erythropoiesis. Int J Hematol 2011; 93: 150–5. [DOI] [PubMed] [Google Scholar]
- 6. Kaneko H, Shimizu R, Yamamoto M. GATA factor switching during erythroid differentiation. Curr Opin Hematol 2010; 17: 163–8. [DOI] [PubMed] [Google Scholar]
- 7. Onodera K, Takahashi S, Nishimura S et al GATA‐1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc Natl Acad Sci USA 1997; 94: 4487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valverde‐Garduno V, Guyot B, Anguita E, Hamlett I, Porcher C, Vyas P. Differences in the chromatin structure and cis‐element organization of the human and mouse GATA1 loci: implications for cis‐element identification. Blood 2004; 104: 3106–16. [DOI] [PubMed] [Google Scholar]
- 9. Wadman IA, Osada H, Grutz GG et al The LIM‐only protein Lmo2 is a bridging molecule assembling an erythroid, DNA‐binding complex which includes the TAL1, E47, GATA‐1 and Ldb1/NLI proteins. EMBO J 1997; 16: 3145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishimura S, Takahashi S, Kuroha T et al A GATA box in the GATA‐1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol Cell Biol 2000; 20: 713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicolis S, Bertini C, Ronchi A et al An erythroid specific enhancer upstream to the gene encoding the cell‐type specific transcription factor GATA‐1. Nucleic Acids Res 1991; 19: 5285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trainor CD, Omichinski JG, Vandergon TL, Gronenborn AM, Clore GM, Felsenfeld G. A palindromic regulatory site within vertebrate GATA‐1 promoters requires both zinc fingers of the GATA‐1 DNA‐binding domain for high‐ affinity interaction. Mol Cell Biol 1996; 16: 2238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bose F, Fugazza C, Casalgrandi M et al Functional interaction of CP2 with GATA‐1 in the regulation of erythroid promoters. Mol Cell Biol 2006; 26: 3942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tallack MR, Whitington T, Yuen WS et al A global role for KLF1 in erythropoiesis revealed by ChIP‐seq in primary erythroid cells. Genome Res 2010; 20: 1052–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohneda K, Yamamoto M. Roles of hematopoietic transcription factors GATA‐1 and GATA‐2 in the development of red blood cell lineage. Acta Haematol 2002; 108: 237–45. [DOI] [PubMed] [Google Scholar]
- 16. Yu C, Cantor AB, Yang H et al Targeted deletion of a high‐affinity GATA‐binding site in the GATA‐1 promoter leads to selective loss of the eosinophil lineage in vivo . J Exp Med 2002; 195: 1387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. A “knockdown” mutation created by cis‐element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA‐1. Proc Natl Acad Sci USA 1997; 94: 6781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki M, Moriguchi T, Ohneda K, Yamamoto M. Differential contribution of the Gata1 gene hematopoietic enhancer to erythroid differentiation. Mol Cell Biol 2009; 29: 1163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA‐1. Proc Natl Acad Sci USA 1996; 93: 12355–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi E, Shimizu R, Kikuchi Y, Takahashi S, Yamamoto M. Loss of the Gata1 gene IE exon leads to variant transcript expression and the production of a GATA1 protein lacking the N‐terminal domain. J Biol Chem 2010; 285: 773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutierrez L, Tsukamoto S, Suzuki M et al Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady‐state and stress erythropoiesis. Blood 2008; 111: 4375–85. [DOI] [PubMed] [Google Scholar]
- 22. Kaneko H, Kobayashi E, Yamamoto M, Shimizu R. Amino‐ and carboxyl‐terminal transactivation domains of GATA1 protein coordinate the hematopoietic program. J Biol Chem 2012; 287: 21439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takahashi S, Onodera K, Motohashi H et al Arrest in primitive erythroid cell development caused by promoter‐ specific disruption of the GATA‐1 gene. J Biol Chem 1997; 272: 12611–5. [DOI] [PubMed] [Google Scholar]
- 24. Shimizu R, Kuroha T, Ohneda O et al Leukemogenesis caused by incapacitated GATA‐1 function. Mol Cell Biol 2004a; 24: 10814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kina T, Ikuta K, Takayama E et al The monoclonal antibody TER‐119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br J Haematol 2000; 109: 280–7. [DOI] [PubMed] [Google Scholar]
- 26. Pan X, Ohneda O, Ohneda K et al Graded levels of GATA‐1 expression modulate survival, proliferation and differentiation of erythroid progenitors. J Biol Chem 2005; 280: 22385–94. [DOI] [PubMed] [Google Scholar]
- 27. Dick JE. Normal and leukemic human stem cells assayed in SCID mice. Semin Immunol 1996; 8: 197–206. [DOI] [PubMed] [Google Scholar]
- 28. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3: 730–7. [DOI] [PubMed] [Google Scholar]
- 29. Jordan CT, Upchurch D, Szilvassy SJ et al The interleukin‐3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000; 14: 1777–84. [DOI] [PubMed] [Google Scholar]
- 30. Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy‐1 (CD90) on acute myeloid leukemia cells with long‐term proliferative ability in vitro and in vivo. Blood 1997; 89: 3104–12. [PubMed] [Google Scholar]
- 31. Blair A, Hogge DE, Sutherland HJ. Most acute myeloid leukemia progenitor cells with long‐term proliferative ability in vitro and in vivo have the phenotype CD34(+)/CD71(−)/HLA‐DR. Blood 1998; 92: 4325–35. [PubMed] [Google Scholar]
- 32. Cobaleda C, Gutierrez‐Cianca N, Perez‐Losada J et al A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia‐positive acute lymphoblastic leukemia. Blood 2000; 95: 1007–13. [PubMed] [Google Scholar]
- 33. Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 2007; 1: 635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kong Y, Yoshida S, Saito Y et al CD34+CD38+CD19+ as well as CD34+CD38‐CD19+ cells are leukemia‐initiating cells with self‐renewal capacity in human B‐precursor ALL. Leukemia 2008; 22: 1207–13. [DOI] [PubMed] [Google Scholar]
- 35. Taussig DC, Miraki‐Moud F, Anjos‐Afonso F et al Anti‐CD38 antibody‐mediated clearance of human repopulating cells masks the heterogeneity of leukemia‐initiating cells. Blood 2008; 112: 568–75. [DOI] [PubMed] [Google Scholar]
- 36. Taussig DC, Vargaftig J, Miraki‐Moud F et al Leukemia‐initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood 2010; 115: 1976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abe K, Shimizu R, Pan X, Hamada H, Yoshikawa H, Yamamoto M. Stem cells of GATA1‐related leukemia undergo pernicious changes after 5‐fluorouracil treatment. Exp Hematol 2009; 37: 435–45. [DOI] [PubMed] [Google Scholar]
- 38. Wu C, Alman BA. Side population cells in human cancers. Cancer Lett 2008; 268: 1–9. [DOI] [PubMed] [Google Scholar]
- 39. Moserle L, Ghisi M, Amadori A, Indraccolo S. Side population and cancer stem cells: therapeutic implications. Cancer Lett 2010; 288: 1–9. [DOI] [PubMed] [Google Scholar]
- 40. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo . J Exp Med 1996; 183: 1797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guo Y, Lubbert M, Engelhardt M. CD34‐ hematopoietic stem cells: current concepts and controversies. Stem Cells 2003; 21: 15–20. [DOI] [PubMed] [Google Scholar]
- 42. Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003; 22: 7340–58. [DOI] [PubMed] [Google Scholar]
- 43. Venezia TA, Merchant AA, Ramos CA et al Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol 2004; 2: e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Isaacs H Jr. Fetal and neonatal leukemia. J Pediatr Hematol Oncol 2003; 25: 348–61. [DOI] [PubMed] [Google Scholar]
- 45. Hama A, Yagasaki H, Takahashi Y et al Acute megakaryoblastic leukaemia (AMKL) in children: a comparison of AMKL with and without Down syndrome. Br J Haematol 2008; 140: 552–61. [DOI] [PubMed] [Google Scholar]
- 46. Harigae H, Xu G, Sugawara T, Ishikawa I, Toki T, Ito E. The GATA1 mutation in an adult patient with acute megakaryoblastic leukemia not accompanying Down syndrome. Blood 2004; 103: 3242–3. [DOI] [PubMed] [Google Scholar]
- 47. Izraeli S, Rainis L, Hertzberg L, Smooha G, Birger Y. Trisomy of chromosome 21 in leukemogenesis. Blood Cells Mol Dis 2007; 39: 156–9. [DOI] [PubMed] [Google Scholar]
- 48. Fox AH, Kowalski K, King GF, Mackay JP, Crossley M. Key residues characteristic of GATA N‐fingers are recognized by FOG. J Biol Chem 1998; 273: 33595–603. [DOI] [PubMed] [Google Scholar]
- 49. Shimizu R, Takahashi S, Ohneda K, Engel JD, Yamamoto M. In vivo requirements for GATA‐1 functional domains during primitive and definitive erythropoiesis. EMBO J 2001; 20: 5250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shimizu R, Ohneda K, Engel JD, Trainor CD, Yamamoto M. Transgenic rescue of GATA‐1‐deficient mice with GATA‐1 lacking a FOG‐1 association site phenocopies patients with X‐linked thrombocytopenia. Blood 2004b; 103: 2560–7. [DOI] [PubMed] [Google Scholar]
- 51. Nichos KE, Crispino JD, Poncz M et al Familial dyserythropoietic anaemia and thromobocytopenia due to an inherited mutation in GATA1. Nat Genet 2000; 24: 266–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hasegawa A, Shimizu R, Mohandas N, Yamamoto M. Mature erythrocyte membrane homeostasis is compromised by loss of the GATA1‐FOG1 interaction. Blood 2012; 119: 2615–23. [DOI] [PubMed] [Google Scholar]
- 53. Muntean AG, Crispino JD. Differential requirements for the activation domain and FOG‐interaction surface of GATA‐1 in megakaryocyte gene expression and development. Blood 2005; 106: 1223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuhl C, Atzberger A, Iborra F, Nieswandt B, Porcher C, Vyas P. GATA1‐mediated megakaryocyte differentiation and growth control can be uncoupled and mapped to different domains in GATA1. Mol Cell Biol 2005; 25: 8592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA‐1 deficiency in megakaryocytes and platelets. Blood 1999; 93: 2867–75. [PubMed] [Google Scholar]
- 56. Shimizu R, Kobayashi E, Engel JD, Yamamoto M. Induction of hyperproliferative fetal megakaryopoiesis by an N‐terminally truncated GATA1 mutant. Genes Cells 2009; 14: 1119–31. [DOI] [PubMed] [Google Scholar]
- 57. Li Z, Godinho FJ, Klusmann JH, Garriga‐Canut M, Yu C, Orkin SH. Developmental stage‐selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet 2005; 37: 613–9. [DOI] [PubMed] [Google Scholar]
- 58. Kadri Z, Shimizu R, Ohneda O et al Direct binding of pRb/E2F‐2 to GATA‐1 regulates maturation and terminal cell division during erythropoiesis. PLoS Biol 2009; 7: e1000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klusmann JH, Godinho FJ, Heitmann K et al Developmental stage‐specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev 2010; 24: 1659–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kanezaki R, Toki T, Terui K et al Down syndrome and GATA1 mutations in transient abnormal myeloproliferative disorder: mutation classes correlate with progression to myeloid leukemia. Blood 2010; 116: 4631–8. [DOI] [PubMed] [Google Scholar]
- 61. Shimizu R, Engel JD, Yamamoto M. GATA1‐related leukaemias. Nat Rev Cancer 2008; 8: 279–87. [DOI] [PubMed] [Google Scholar]