

Abstract
Arm protein lost in epithelial cancers, on chromosome X (ALEX; also known as armadillo repeat containing, X‐linked [ARMCX]) is a novel subgroup within the armadillo (ARM) family, which has several ARM repeat domains. The biological function of classical ARM family members such as β‐catenin is well understood, but that of the ALEX/ARMCX family members is largely unknown. Here we evaluate the effects of ALEX1 overexpression on in vitro colony formation ability and expression of ALEX1 mRNA in human colorectal tumor. Overexpression of ALEX1 suppressed the anchorage‐dependent and ‐independent colony formation of human colorectal carcinoma cell lines by the study of stable clones of HCT116 cells expressing ALEX1 protein. Bisulfite genomic sequencing revealed that the promoter region of ALEX1 gene was highly methylated in both HCT116 and SW480 cells in comparison with PANC‐1 and MCF‐7 cells, which express endogenous ALEX1 mRNA, indicating the capability of promoter methylation to silence ALEX1 gene in HCT116 and SW480 cells. Our current findings suggest that overexpression of ALEX1 play a negative role in human colorectal tumorigenesis. (Cancer Sci 2012; 103: 1267–1271)
Armadillo (ARM) repeat proteins, characterized by the presence of a repeating 42 amino acid motif, form a large family implicated in a variety of processes such as cell adhesion, embryogenesis and tumorigenesis.1, 2 Armadillo repeats mediate protein–protein interaction with diverse binding partners involved in cell junction assembly, nuclear transport and transcription activation.3, 4, 5 β‐Catenin (CTNND1 gene), a classical member of ARM family, is a multifunctional protein that plays essential roles both at adherence junctions and in Wnt signaling through interaction with E‐cadherin and T cell factor/lymphoid enhancer factor (TCF/LEF) family transcription factors, respectively.6, 7 Tumor suppressor adenomatous polyposis coli (APC) is also an ARM family member and acts synergistically with casein kinase Ι, glycogen synthase kinase‐3β and AXIN to regulate Wnt signaling via β‐catenin degradation.8, 9, 10 Approximately 80% of the sporadic colorectal carcinomas have inactivating mutations in APC and degradation‐resistant mutations in β‐catenin occur in around 50% of the remaining colorectal carcinomas. These mutations can cause aberrant nuclear accumulation of β‐catenin, leading to the transcriptional activation of the Wnt target genes such as oncogenic c‐MYC and CCND1.8, 9, 11, 12, 13
Arm protein lost in epithelial cancers, on chromosome X (ALEX; also known as armadillo repeat containing, X‐linked [ARMCX]) is a novel subgroup within the ARM family. The ALEX/ARMCX gene family consists of six genes including three predicted genes (ALEX1‐6).14, 15, 16 Bioinformatics analyses suggest that the ALEX1, ALEX2 and ALEX3 are each encoded by a single exon and contain an N‐terminal transmembrane domain, some ARM repeat domains, and a DUF634 (domain of unknown function 634).14, 15, 16 However, little is known about the ALEX/ARMCX genes. The only report regarding the biological function of the ALEX/ARMCX proteins demonstrates that the ALEX3 directly interacts with the sex determining region Y (Sry)‐box 10 (SOX10) transcription factor via the ARM repeat domains and alters its subcellular localization and transcriptional activity.15 In addition, gene expression analysis revealed that both ALEX1 and ALEX2 mRNA is expressed in a variety of adult human tissues, including colon, but dramatically reduced or even undetectable in several human carcinoma cell lines and tissues.14 In mouse embryos, ALEX2 mRNA is expressed in the developing testis, forebrain and somites, and in dorsal root ganglia and ribs.16 In vivo RNAi screening for potential tumor suppressor genes using immortalized embryonic hepatocytes lacking p53 and overexpressing MYC revealed that knockdown of ALEX1 and ALEX2 individually accelerated hepatocarcinogenesis in mice.17 From these results, the members of the ALEX/ARMCX gene family are suspected to function as a tumor suppressor and a regulatory factor of embryonic development. However, the role and the expression profile of ALEX1 gene in colorectal tumor are not well examined.
Here we evaluate the effects of ALEX1 overexpression on colony formation ability of human colorectal carcinoma cell lines and investigate the capability of promoter methylation to silence ALEX1 gene using no endogenous ALEX1 mRNA expressing cell lines.
Materials and Methods
Cell culture
HCT116, SW480, and MCF‐7 cells were maintained in DMEM (Invitrogen, Tokyo, Japan) and PANC‐1 cells were maintained in RPMI1640 (Invitrogen) supplemented with 10% heat‐inactivated FBS (Invitrogen) in a 5% CO2 atmosphere at 37°C.18
Conventional RT‐PCR
Total RNA from cells was extracted using TRIzol Reagent (Invitrogen) and RNeasy mini kit (Qiagen, Tokyo, Japan), respectively, and then treated with RQ1 RNase‐Free DNase (Promega, Tokyo, Japan). First strand cDNA was synthesized from 1 μg of DNase‐treated total RNA using Transcriptor Reverse Transcriptase (Roche Diagnostics, Tokyo, Japan) in a total volume of 20 μL reaction mixtures. The PCR reactions contained 20 ng of cDNA, 1× PCR buffer (TaKaRa Bio, Shiga, Japan), 0.2 mM dNTP each, 0.5 units of Ex Taq Polymerase (TaKaRa), and 0.1 μM each primers and were subjected to the following amplification scheme: One cycle at 95°C for 2 min, 25 (for GAPDH) or 35 (for ALEX1) cycles at 95°C for 30 s, 55°C for 1 min, and 72°C for 30 s, and final extension at 72°C for 5 min after the last cycle.
Western blot analysis
Cells were washed once with PBS and then suspended in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% deoxycholic acid, 1% Nonidet P‐40) containing protease inhibitor cocktail (Sigma‐Aldrich, Tokyo, Japan). Protein concentrations were quantified using the Quick Start Bradford protein assay (Bio‐Rad Laboratories, Tokyo, Japan) according to the manufacturer's instructions. Equal protein level of whole lysates were resolved by SDS‐PAGE and transferred onto Immobilon‐P transfer membranes (Nihon Millipore, Tokyo, Japan). Anti‐human ALEX1/ARMCX1 polyclonel antibody (Abnova, Taipei, Taiwan) and anti‐β‐actin monoclonal antibody (Abcom, Tokyo, Japan) were used at a dilution of 1:1000 and 1:10 000, respectively.
Bisulfite genomic sequencing
Cells and tissues were digested with 0.1 mg/mL Proteinase K in lysis buffer (10 mM Tris–HCl, pH 7.6, 1 mM EDTA, 100 mM NaCl, 0.3% SDS) at 60°C overnight, and then genomic DNA was purified by the phenol chloroform extraction and ethanol precipitation. Bisulfite treatment of the genomic DNA was carried out with the EpiTect bisulfite kit (Qiagen) according to the manufacturer's instructions. The amplified fragments with the specific primers to the regulatory region, 5′‐GTTGGTAAAGAGGAAAATGAGTG‐3′ and 5′‐CCCTCCCTAATTCAAAACCCT‐3′, and exonic CpG island, 5′‐GGGTGGAAGAAAGGAATTAGTG‐3′ and 5′‐TCACAATCTCAACCCCAATCTC‐3′, of ALEX1 gene were cloned into the pGEM‐T easy vector (Promega), and subsequently sequenced with the BigDye Terminator Cycle Sequencing system (Applied Biosystems). Bisulfite conversion efficiency of non‐CpG cytosines were over 98% for all individual clones for each sample.
Plasmid constructs and transfection
Human ALEX1 gene (accession No. NM_016608) was amplified by PCR and inserted into the XhoI site of pCAGIPuro plasmid (kindly provided by Dr H. Niwa, RIKEN), designated as pCAGIPuro/ALEX1. The pCAGIPuro/EGFP plasmid (kindly provided by Dr H. Niwa, RIKEN) encoding enhanced GFP (EGFP) was used as a control. Plasmid transfections were performed by LipofectAMINE 2000 or LipofectAMINE LTX (Invitrogen) according to the manufacturer's instructions.
Colony formation assay
One day before transfection, each cell line was plated in a 24‐well plate at 1 × 105 cells/well. After 24 h post‐transfection, the cells were replated into 100‐mm dishes and cultured in the presence of 5 μg/mL puromycin (Invitrogen, Grand Island, NY, USA) for 4 weeks. The colonies were fixed with methanol, stained with 0.5% crystal violet solution, and counted. All assays were done in triplicate and repeated three times.
Soft agar colony formation assay
A total of 1000 cells of each clone in 1 mL DMEM containing 0.3% agar (Difco Laboratories, Detroit, MI, USA), 10% FBS, and 1 μg/mL puromycin, were plated per well onto six‐well plates coated with 1 mL DMEM containing 0.6% agar, 10% FBS, and 1 μg/mL puromycin. After 2 weeks, colonies were stained with 0.5% crystal violet solution and counted. The experiment was performed in triplicate and repeated three times.
Statistical analysis
Statistics were performed using Mann–Whitney's U‐test. A P‐value < 0.05 was considered to be statistically significant.
Results
Overexpression of ALEX1 suppresses the anchorage‐dependent and ‐independent colony formation of human colorectal carcinoma cell lines
To examine the effect of overexpression of ALEX1 on cancer cell proliferation, we performed a colony formation assay of two colorectal carcinoma cell lines HCT116 and SW480 and breast carcinoma cell line MCF‐7. Quantitative real‐time RT‐PCR and Western blot analysis showed that ALEX1 expression was lost in HCT116 and SW480 cells (Fig. 1A,B). In contrast to HCT116 and SW480 cells transfected with control pCAGIPuro/EGFP plasmid successfully formed colonies, those transfected with pCAGIPuro/ALEX1 plasmid failed to form colonies (Fig. 1C,D), indicating that overexpression of ALEX1 in colorectal carcinoma cells is capable of impairing colony formation. A similar result was observed using MCF‐7 cells in which the ALEX1 protein was endogenously expressed (Fig. 1A,B,E), suggesting that suppression of cancer cell proliferation requires high levels of ALEX1 protein expression.
Figure 1.
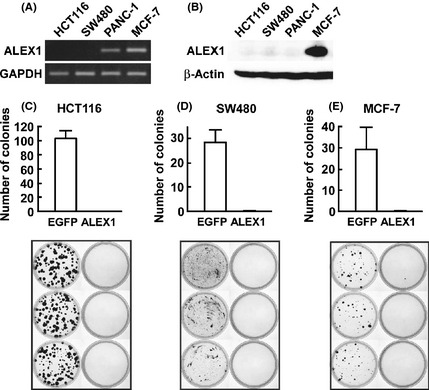
Effect of ALEX1 overexpression on the anchorage‐dependent colony formation of HCT116, SW480, and MCF‐7 cells. (A) Expression of ALEX1 mRNA in HCT116, SW480, PANC‐1 and MCF‐7 cells was examined by reverse transcription‐polymerase chain reaction (RT‐PCR). GAPDH mRNA expression was used as an internal control. (B) Western blot analysis for ALEX1 was performed with cell lysate from HCT116, SW480, PANC‐1 and MCF‐7 cells. β‐Actin protein served as an internal control. Colony formation assay on HCT116 (C), SW480 (D) and MCF‐7 (E) cells transfected with pCAGIPuro/enhanced green fluorescent protein (EGFP) or pCAGIPuro/ALEX1 plasmids, and selected with 5 μg/mL puromycin for 4 weeks. Bar graphs at the upper panel represent the mean number of visible colonies obtained for each cell line from three independent experiments, and error bars represent standard deviation (SD).
To further investigate the role of ALEX1 in the colorectal carcinoma cell line, we generated stable clones of HCT116 cells expressing ALEX1 protein, designated as HCT116/ALEX1, by limiting dilution after selection in growth medium supplemented with lower concentrations of puromycin (1 μg/mL) than that used in the colony formation assay (5 μg/mL). As control cells, HCT116/empty and HCT116/EGFP clones that express puromycin resistance protein alone and EGFP, respectively, were generated. Western blot analysis showed that ALEX1 protein was expressed in the four HCT116/ALEX1 clones, but not in the two HCT116/empty clones and the two HCT116/EGFP clones (Fig. 2A). All stable clones attached to the bottom of the culture dish and grew similarly (data not shown). However, anchorage‐independent growth, a hallmark of malignant transformation, was reduced in the four HCT116/ALEX1 clones (range of average colony number of 37–56) in comparison to the two HCT116/empty and HCT116/EGFP clones (range of average colony number of 120–243 and 170–233, respectively) (Fig. 2B). These findings support the possibility that ALEX1 functions as a tumor suppressor in colorectal carcinoma cells.
Figure 2.
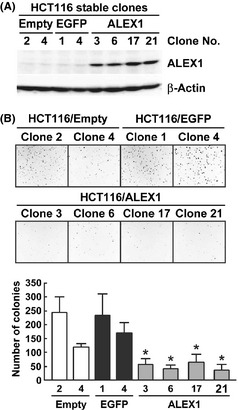
Effect of ALEX1 overexpression on the anchorage‐independent colony formation of HCT116 cells. (A) Western blot analysis for ALEX1 was performed with cell lysate from each HCT116/empty, HCT116/enhanced green fluorescent protein (EGFP), and HCT116/ALEX1 clones. β‐Actin protein served as an internal control. (B) Soft‐ager colony formation assay with the each stably transfected HCT116 clones. Bar graph at the lower panel represents the mean number of visible colonies obtained for each clones from three independent experiments, and error bars represent SD. *P < 0.05 versus each HCT116/empty and HCT116/EGFP clones.
ALEX1 is silenced by DNA methylation in colorectal carcinoma cell lines
Several tumor suppressor genes have been shown to be silenced via promoter hypermethylation in numerous colorectal tumors.19, 20, 21, 22 Therefore, DNA methylation is considered to be one mechanism of tumor suppressor gene inactivation, which can function identically to inactivating mutations. Although our previous report demonstrated that the exogenous human ALEX1 promoter was active in HCT116 and SW480 by luciferase reporter analysis,18 endogenous ALEX1 expression was not detected at the mRNA and protein levels in these cells (Fig. 1A,B). These data prompted us to investigate whether the expression of the ALEX1 gene was silenced by DNA methylation in colorectal carcinoma cell lines. HCT116 and SW480 cells treatment with DNA methyltransferase (DNMT) inhibitor, 5‐aza‐2′‐deoxycytidine (5‐aza‐dC), for 72 h, resulted in the reactivation of the ALEX1 gene (Fig. 3A). Furthermore, bisulfite genomic sequencing revealed that the promoter region of the ALEX1 gene was highly methylated in both HCT116 and SW480 cells in comparison to those in PANC‐1 and MCF‐7 cells, which express endogenous ALEX1 mRNA (Figs 1A,3B), whereas an exonic CpG island in the ALEX1 gene was hypermethylated in all four cell lines regardless of the ALEX1 mRNA expression levels (Fig. 3B). These results indicate the capability of promoter methylation to silence ALEX1 gene in HCT116 and SW480 cells.
Figure 3.
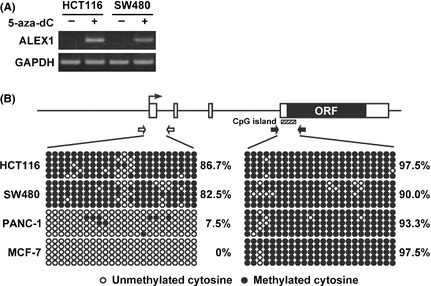
Silencing of ALEX1 gene through promoter hypermethylation in HCT116 and SW480 cells. (A) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis of ALEX1 gene in HCT116 and SW480 cells untreated (−) or treated with 0.5 μM 5‐aza‐dC for 3 days (+). (B) Bisulfite sequencing analysis of ALEX1 gene in HCT116, SW480, PANC‐1 and MCF‐7 cell lines. Open and filled boxes represent the exons of the ALEX1 gene and the open reading frame (ORF) encoding the ALEX1 protein, respectively. The bent arrow indicates the transcription start site of the ALEX1 gene. Locations of CpG island (shaded box) and the primers used for promoter region (white arrows) and CpG island (black arrows) are represented above. Open and filled circles represent unmethylated and methylated cytosine, respectively, and each row represents a single clone.
Discussion
Members of the ARM family of proteins have shown to exert diverse functions, such as signal transduction, cell adhesion, development, and tumorigenesis, through interactions of their ARM repeat domain with several binding partners. Meanwhile, the biological function of members of the ALEX/ARMCX family, a novel subgroup of the ARM family, is largely unknown. Here we clearly showed that overexpression of ALEX1 suppressed both the anchorage‐dependent and ‐independent colony formation of human colorectal carcinoma cell lines (Figs 1, 2). On the other hand, knockdown of ALEX1 accelerated hepatocarcinogenesis in mice.17 Together with the reduced expression of ALEX1 gene in several tumors, these lines of evidence support the hypothesis that ALEX1 protein functions as a tumor suppressor. Intriguingly, the B variant of specific splicing variant involved in hepatocarcinogenesis (SVH‐B; also known as armadillo repeat containing 10 [ARMC10]), a closely related ARM family member, was identified as an upregulated gene in the human hepatocellular carcinoma by representational difference analysis.23 The overexpression of SVH‐B accelerates cell growth and tumorigenicity in the normal liver cell lines and suppresses the transcriptional activity of tumor suppressor p53.23, 24 Accordingly, ALEX‐related genes may also be capable of playing different roles in tumorigenesis as well as APC and CTNNB1 function as a tumor suppressor gene and oncogene, respectively.
The first study on ALEX family reported that ALEX1 and ALEX2 mRNA were decreased in some carcinoma cell lines and tissues. Here we carried out the quantitative expression analysis of the ALEX1 mRNA in matched tissue pairs of normal colorectal mucosa and colorectal tumor tissues, and showed that the ALEX1 mRNA was frequently reduced in colorectal tumor. In addition, genome‐wide expression profiling of several carcinomas derived from lung, bladder, prostate and uterus using microarray have been accumulated and shown reduction of ALEX1 mRNA in these cancer tissues.25, 26 Thus, the low or absent ALEX1 expression seems to be a common feature of a variety of different cancers. It is noteworthy that ALEX1 was detected at the mRNA level in pancreatic carcinoma cell line PANC‐1, but not detected at the protein level, suggesting that posttranscriptional and/or posttranslational mechanisms for ALEX1 regulation exist.
Aberrant DNA methylation within the promoter of tumor suppressor genes results in the transcriptional silencing of these genes and is believed to contribute to colorectal cancer progression. For example, Wnt antagonists such as secreted frizzled‐related proteins, DICKKOPF‐1 and Wnt‐inhibitory factor‐1, which are the negative feedback regulators of Wnt signaling, are frequently silenced through promoter hypermethylation in colorectal tumors.19, 20, 21, 22 We previously revealed that the expression of ALEX1 mRNA is upregulated by continuous activation of Wnt/β‐catenin signaling.18 In this study, we demonstrated that the ALEX1 promoter was hypermethylated and reactivated by DNMT inhibitor in HCT116 and SW480 cells in which Wnt signaling is active, suggesting that DNA methylation serves as one mechanism for the reduction of ALEX1 gene expression induced by aberrant activation of Wnt signaling although other mechanism(s) such as a downregulation by oncogenic protein may contribute to ALEX1 gene reduction. Meanwhile, recent reports have indicated that a distinct subset of colorectal cancers showed a high frequency of DNA hypermethylation in multiple genes, which has been termed the CpG island methylator phenotype (CIMP).27, 28 The CIMP has been shown to associate with microsatellite instability and BRAF mutations in serrated colorectal polyps.29, 30, 31, 32 In our preliminary analysis of DNA methylation status with available five tissue pairs of normal colorectal mucosa and tumor with non‐serrated histology from male patients, in which the ALEX1 mRNA is decreased, the ALEX1 promoter was hypomethylated in all normal mucosa and four out of five tumor tissues, but hypermethylated in one out of five tumors (data not shown).
Expression of ALEX1 and ALEX2 mRNA is lost or significantly reduced in human lung, prostate, colon, pancreas, and ovarian carcinomas and also in cell lines established from different human carcinomas. These genes are, however, normally expressed in cell lines derived from other types of tumors, e.g., sarcomas, neuroblastomas, and gliomas, which will speculate that ALEX genes may play a role in suppression of tumors originating from epithelial tissue, i.e., carcinomas. The prognostic significance of ALEX1 in solid neoplasms has been suggested in several kinds of tumor.14 We also have examined the correlations between ALEX1 gene expression in tumor and postoperative prognosis in 49 primary colorectal tumors undergoing complete surgical resection. Patients showing ALEX1 expressions (n = 17) revealed a significantly better prognosis than those without ALEX1 (n = 34) (P = 0.045: data not shown). These results possibly imply that silencing of the ALEX1 gene through promoter hypermethylation possibly leads to the tumor growth in colorectal cancer with the CIMP and Wnt signaling. Therefore examination of ALEX1 expression might be helpful for predicting the prognosis of patients with curative resected colorectal cancer.
In summary, the current results indicate that overexpression of a non‐classical ARM protein family member ALEX1 suppresses colony formation of human carcinoma cells. Moreover, ALEX1 was frequently reduced in human colorectal tumor. The possibility that DNA methylation serves as one mechanism for the reduction of ALEX1 gene expression in human colorectal tumor cell is suggested, although the role of DNA methylation in colorectal tumorigenesis remains to be determined. These findings suggest that ALEX1 play a negative role in human colorectal tumorigenesis.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
The authors thank Hitoshi Niwa of the RIKEN Center for Developmental Biology for the generous gift of the pCAGIPuro and pCAGIPuro/EGFP plasmids. The authors also thank members of the Division of Functional Genomics and Systems Medicine for helpful discussion and advice. This work was supported by a Grant‐in‐Aid for Young Scientists (B) No. 19790953 (H. Iseki) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Hatzfeld M. The armadillo family of structural proteins. Int Rev Cytol 1999; 186: 179–224. [DOI] [PubMed] [Google Scholar]
- 2. Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell 1994; 76: 789–91. [DOI] [PubMed] [Google Scholar]
- 3. Ozawa M, Terada H, Pedraza C. The fourth armadillo repeat of plakoglobin (γ‐catenin) is required for its high affinity binding to the cytoplasmic domains of E‐cadherin and desmosomal cadherin Dsg2, and the tumor suppressor APC protein. J Biochem 1995; 118: 1077–82. [DOI] [PubMed] [Google Scholar]
- 4. Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E‐cadherin form similar but independent complexes with α‐catenin, β‐catenin, and plakoglobin. J Biol Chem 1995; 270: 5549–55. [DOI] [PubMed] [Google Scholar]
- 5. Troyanovsky RB, Chitaev NA, Troyanovsky SM. Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci 1996; 109 (Pt 13): 3069–78. [DOI] [PubMed] [Google Scholar]
- 6. Bass‐Zubek AE, Godsel LM, Delmar M, Green KJ. Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr Opin Cell Biol 2009; 21: 708–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu W, Kimelman D. Mechanistic insights from structural studies of β‐catenin and its binding partners. J Cell Sci 2007; 120: 3337–44. [DOI] [PubMed] [Google Scholar]
- 8. Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β‐catenin levels by the adenomatous polyposis coli (APC) tumor‐suppressor protein. Proc Natl Acad Sci USA 1995; 92: 3046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goss KH, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. J Clin Oncol 2000; 18: 1967–79. [DOI] [PubMed] [Google Scholar]
- 10. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC‐β‐catenin complex and regulation of complex assembly. Science 1996; 272: 1023–6. [DOI] [PubMed] [Google Scholar]
- 11. Iwao K, Nakamori S, Kameyama M et al Activation of the β‐catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res 1998; 58: 1021–6. [PubMed] [Google Scholar]
- 12. Wong SC, Lo ES, Lee KC, Chan JK, Hsiao WL. Prognostic and diagnostic significance of β‐catenin nuclear immunostaining in colorectal cancer. Clin Cancer Res 2004; 10: 1401–8. [DOI] [PubMed] [Google Scholar]
- 13. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β‐catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58: 1130–4. [PubMed] [Google Scholar]
- 14. Kurochkin IV, Yonemitsu N, Funahashi SI, Nomura H. ALEX1, a novel human armadillo repeat protein that is expressed differentially in normal tissues and carcinomas. Biochem Biophys Res Commun 2001; 280: 340–7. [DOI] [PubMed] [Google Scholar]
- 15. Mou Z, Tapper AR, Gardner PD. The armadillo repeat‐containing protein, ARMCX3, physically and functionally interacts with the developmental regulatory factor Sox10. J Biol Chem 2009; 284: 13629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith CA, McClive PJ, Sinclair AH. Temporal and spatial expression profile of the novel armadillo‐related gene, ALEX2, during testicular differentiation in the mouse embryo. Dev Dyn 2005; 233: 188–93. [DOI] [PubMed] [Google Scholar]
- 17. Zender L, Xue W, Zuber J et al An oncogenomics‐based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008; 135: 852–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iseki H, Takeda A, Andoh T et al Human Arm protein lost in epithelial cancers, on chromosome X 1 (ALEX1) gene is transcriptionally regulated by CREB and Wnt/β‐catenin signaling. Cancer Sci 2010; 101: 1361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aguilera O, Fraga MF, Ballestar E et al Epigenetic inactivation of the Wnt antagonist DICKKOPF‐1 (DKK‐1) gene in human colorectal cancer. Oncogene 2006; 25: 4116–21. [DOI] [PubMed] [Google Scholar]
- 20. He B, Reguart N, You L et al Blockade of Wnt‐1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005; 24: 3054–8. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki H, Watkins DN, Jair KW et al Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet 2004; 36: 417–22. [DOI] [PubMed] [Google Scholar]
- 22. Taniguchi H, Yamamoto H, Hirata T et al Frequent epigenetic inactivation of Wnt inhibitory factor‐1 in human gastrointestinal cancers. Oncogene 2005; 24: 7946–52. [DOI] [PubMed] [Google Scholar]
- 23. Huang R, Xing Z, Luan Z, Wu T, Wu X, Hu G. A specific splicing variant of SVH, a novel human armadillo repeat protein, is up‐regulated in hepatocellular carcinomas. Cancer Res 2003; 63: 3775–82. [PubMed] [Google Scholar]
- 24. Zhou X, Yang G, Huang R, Chen X, Hu G. SVH‐B interacts directly with p53 and suppresses the transcriptional activity of p53. FEBS Lett 2007; 581: 4943–8. [DOI] [PubMed] [Google Scholar]
- 25. Rohrbeck A, Borlak J. Cancer genomics identifies regulatory gene networks associated with the transition from dysplasia to advanced lung adenocarcinomas induced by c‐Raf‐1. PLoS ONE 2009; 4: e7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramaswamy S, Tamayo P, Rifkin R et al Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA 2001; 98: 15149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teodoridis JM, Hardie C, Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett 2008; 268: 177–86. [DOI] [PubMed] [Google Scholar]
- 28. Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4: 988–93. [DOI] [PubMed] [Google Scholar]
- 29. Velho S, Moutinho C, Cirnes L et al BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 2008; 8: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Brien MJ, Yang S, Mack C et al Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006; 30: 1491–501. [DOI] [PubMed] [Google Scholar]
- 31. Kim YH, Kakar S, Cun L, Deng G, Kim YS. Distinct CpG island methylation profiles and BRAF mutation status in serrated and adenomatous colorectal polyps. Int J Cancer 2008; 123: 2587–93. [DOI] [PubMed] [Google Scholar]
- 32. Kambara T, Simms LA, Whitehall VL et al BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004; 53: 1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]