

Abstract
Post‐transcriptional modifications, such as 5′ end capping, 3′ end polyadenylation and splicing, are necessary for the precise regulation of gene expression and transcriptome integrity. Therefore, it is not surprising that abnormalities of these post‐transcriptional modifications prompt numerous diseases, including cancer. In fact, many studies revealed that misregulation of mRNA processing, especially splicing, are observed in a variety of cancer cells. In this review we describe how changes within RNA splicing regulatory elements or mutations in the processing factors alter the expression of tumor suppressors or oncogenes with pathological consequences. In addition, we show how several small molecules that bind to spliceosomal components and splicing regulators inhibit or modulate splicing activity. These compounds have anticancer activity and further development of small molecule modulators has potential in next generation cancer therapy.
mRNA Splicing
In eukaryotes, nascent transcripts (precursor messenger RNA [pre‐mRNA]) are subjected to post‐transcriptional modifications, including capping of the 5′ end, intron removal by splicing, as well as cleavage and polyadenylation at the 3′ end, to become mature mRNA that are templates for translation.1 These post‐transcriptional modifications are important for efficient gene expression and for the integrity of the transcriptome, hence aberrations in these modifications might perturb gene expression and interfere with vital cellular functions, enabling pathogenesis including carcinogenesis or tumor progression.2
Pre‐mRNA consist of protein coding regions, exons and intervening sequences, introns.3 The splicing process joins the exon sequences while removing the introns. These reactions are coordinated and catalyzed by the spliceosome, a large, multi‐component ribonuclear complex, consisting of five sub‐component small nuclear ribonucleoprotein particles (snRNPs), named U1, U2, U4, U5 and U6 (Fig. 1). The splicing reactions start with recognition of the intron's 5′ end, the 5′ splice site (5′ ss), by U1 snRNP, followed by SF1 binding to the branch point sequence and interaction of U2AF with the 3′ end of the intron, the 3′ splice site (3′ ss), to form complex E. Complex E turns over to complex A when U2 snRNP replaces SF1. Complex B results from U4/U6•U5 tri‐snRNP binding to complex A. In the last step, conformational changes enable two transesterification reactions by which the intron sequence is excised and the adjacent exons joined.
Figure 1.
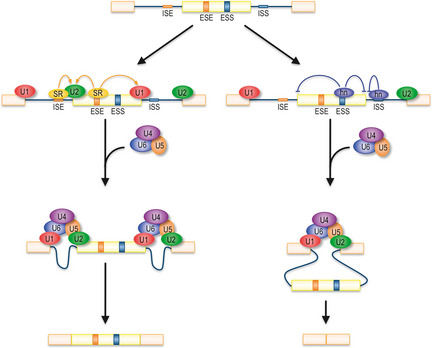
Schematic representation of mRNA splicing. U1 small nuclear ribonucleoprotein particle (snRNP) and U2 snRNP recognize 5′ splice site (5′ ss) and branch point sequence, respectively. U4/U6•U5 snRNP joining is followed by conformational changes to become the catalytically active spliceosome. Binding of SR proteins to exonic or intronic splicing enhancers (ESE and ISE, respectively) stimulates splicing efficiency, while binding of hnRNP to splicing silencers (ESS and ISS) suppresses splicing and causes exon skipping.
This process requires high precision and fidelity as gene expression depends on the integrity of the transcript. A change by only one nucleotide would introduce a frame‐shift, which would not only alter the amino acid sequence of the protein but likely introduce a premature termination codon (PTC). Similarly, intron retention by failure to splice will even more likely yield mRNA with a PTC, as intron sequences appear enriched in stop codons. Such mRNA with a PTC are degraded by nonsense mediated decay (NMD) or translated into truncated proteins.4, 5 Consequently, frequent failure in recognition of the splice site would completely disable transcriptional regulation and meaningful gene expression.
Alternative Splicing
Alternative splicing is a mechanism to produce multiple isoforms from a single gene through the use of alternative splice sites. Alternative splicing not only increases protein variety, but the resultant gene products might have considerably different functions. In some cases, alternative splice isoforms might even oppose each other's effect. Production of splice isoforms depends on the tissue at hand, as well as the organism's developmental stage.6, 7 Multicellular eukaryotes, like mammals, take advantage of this mechanism to expand protein diversity from a limited number of genes. In fact, more than 90% of genes are subjected to alternative splicing in humans.8, 9 As noted, alternative splicing greatly contributes to biological complexity, while failure to properly express the correct isoform can have deleterious consequences and aid tumor formation (see below).
Alternative splicing is regulated by cis elements within the RNA and trans RNA binding factors. Well‐studied trans factors are serine/arginine‐rich (SR) proteins and heterogeneous nuclear RNPs (hnRNPs)10 (Fig. 1). SR proteins, binding to exonic splicing enhancers (ESE) and intronic splicing enhancers (ISE), recruit spliceosomal components and promote splicing and exon inclusion.11 In contrast, hnRNPs mainly bind to exonic and intronic splicing silencer sequences (ESS and ISS) and inhibit splicing or promote exon skipping by inhibiting the positive splicing factors.12 Therefore, mutations in splicing enhancer/silencer elements and RNA binding trans factors might result in the expression of undesirable isoforms, which in turn can induce or aid carcinogenesis. We describe some examples in which a change of splicing pattern leads to cancer development below.
Splicing Factors Implicated in Cancer Development
Serine/arginine‐rich splicing factor 1 (SRSF1) is a well‐known SR protein regulating both constitutive and alternative splicing.11 Recently, it was reported that SRSF1 is upregulated in various human cancers and overexpression of SRSF1 transforms immortal cells, suggesting that SRSF1 has oncogenic activity.13 Actually, SRSF1 regulates splicing patterns of several important oncogenes and tumor suppressors, resulting in inhibition of apoptosis and increase of the rate of cell growth.
Tumor suppressor BIN1 interacts with the product of the MYC proto‐oncogene and suppresses oncogenic activity of MYC.14 Tumor‐suppressing activity of BIN1 is controlled by alternative splicing. Inclusion of exon 12A diminishes BIN1 binding to MYC and hence also decreases its antitumor activity (Fig. 2). Overexpression of this isoform has been observed in melanoma cells.15 Inclusion of exon 12A is increased by SRSF1 overexpression and decreased by knockdown of SRSF1, suggesting that SRSF1 contributes to cancer progression by changing the splicing pattern of the BIN1 mRNA.13
Figure 2.
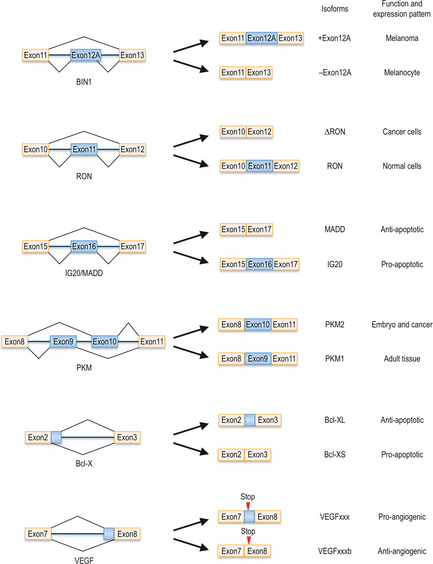
Alternative splicing isoforms discussed in this review. Example of proteins whose alternative splicing patterns greatly influence their activity and carcinogenic potential. Each isoform's function and expression pattern is noted on the right side of each diagram.
RPS6KB1 is another SRSF1 regulated gene. It encodes a kinase of the small subunit ribosomal protein S6 and regulates cell growth and apoptosis.16 RPS6KB1 has two isoforms (isoform‐1 and isoform‐2) and expression of isoform‐2 is stimulated by SRSF1 overexpression.13 Overexpression of isoform‐2 transforms NIH3T3 cells and the SRSF1 expression level is correlated with the isoform‐2/isoform‐1 ratio in human lung cancer cells, suggesting that a splicing pattern change in RPS6KB1 by SRSF1 also promotes cancer development.
RON is a tyrosine kinase receptor for the macrophage stimulating protein (MSP) and is involved in cell dissociation, mobility and invasion.17, 18 An alternative spliced isoform of RON without exon 11, named ΔRON, is constitutively active even in the absence of its ligand19 (Fig. 2). ΔRON is highly expressed in several cancers, suggesting that the splicing pattern change of RON regulates tumor progression.20 The splicing pattern of RON is regulated by SRSF1, which directly binds to an ESE of RON to stimulate exon 11 skipping and enhances cell motility, consistent with upregulation of SRSF1 in cancer cells.13, 20
The hnRNP protein family, which functions as a splicing silencer, is also implicated in cancer development. One of its family members, hnRNP H, regulates alternative splicing of the IG20/MADD gene encoding several alternative splicing isoforms including IG20 that triggers apoptosis, and another isoform MADD, the MAP kinase activating death domain protein, that is expressed at high levels in cancer cells, contributing to their survival by inactivating caspase‐821, 22, 23 (Fig. 2). HnRNP H binds to exon 16 of IG20/MADD and induces exon 16 skipping to produce the MADD isoform.21 Conversely, knockdown of hnRNP H results in exon 16 inclusion and IG20 isoform expression, leading to reduction of cell survival in both U373 glioma and HeLa cells. Interestingly, hnRNP H also controls alternative splicing of RON. The RON gene has several binding motifs for hnRNP H in exon 11 and binding of hnRNP H promotes exon 11 skipping and production of ΔRON that induces invasion and migration.21 HnRNP H is actually overexpressed in glioma cells and might function as an oncogene by modulating splicing of both IG20/MADD and RON.
Another hnRNP family member, hnRNP A2/B1, is overexpressed in several tumors, such as glioblastoma, lung cancer and breast cancer.24, 25, 26 Its expression level is correlated with poor prognosis. Knockdown of hnRNP A2/B1 induces apoptosis only in cancer cells, but not in normal cells.27 HnRNP A2/B1 modulates the splicing patterns of numerous genes including pyruvate kinase M (PKM), which controls glucose metabolism.28 PKM has two isoforms, PKM1 and PKM2, expressed in adult tissues and in embryonic cells, respectively (Fig. 2). The embryonic isoform PKM2, which is re‐expressed in cancer cells, promotes aerobic glycolysis and tumor growth, whereas PMK1 activates oxidative phosphorylation and reduces tumorigenicity.29 HnRNP A2/B1 binds to the flanking region of exon 9 of the PKM gene and promotes exon 9 skipping and exon 10 inclusion, leading to PKM2 production.28 These results suggest that hnRNP A2/B1 regulates tumorigenesis through the alternative splicing of PKM1/2. However, since Bluemlein et al.30 reported most recently that they did not observe a transition from PKM1 to PKM2 during tumorigenesis, further study is required to elucidate whether hnRNP A2/B1‐dependent alternative splicing of PKM does indeed enhance carcinogenicity.
Recently, Yoshida et al. reported that mutations in core components of the splicing machinery, namely SF3b1 and U2AF35, are found in patients with myelodysplasia, a myeloid neoplasm characterized by deregulated dysplastic blood cell production and a predisposition for acute myeloid leukemia.31, 32 Furthermore, a mutation in U2AF35, which is observed in patient samples, causes a reduction in the rate of cell growth and seems to induce apoptosis, which are features of myelodysplasia. While it is possible that abnormal splicing causes this hematological disorder, it currently remains uncertain how it could be involved in disease progression and tumorigenesis.
Splicing Pattern Changes by Alternative Splicing Affect Cancer Development
Beyond IG20/MADD or PKM1/2, the function of several other genes is regulated by alternative splicing, with each splice isoform fulfilling distinct or even opposing functions.6 Further examples include the apoptosis regulator Bcl‐X and the vascular endothelial growth factor (VEGF).
Bcl‐X
Bcl‐X is a well‐known regulator of apoptosis.33 Bcl‐X pre‐mRNA has two alternative 5′ ss in intron 2 (Fig. 2). If the upstream 5′ ss is used, the pro‐apoptotic isoform Bcl‐XS is produced. However, splicing at the downstream 5′ ss results in generation of the anti‐apoptotic isoform Bcl‐XL.34, 35, 36 Bcl‐XL is overexpressed in a wide variety of cancer cells,37, 38 whereas Bcl‐XS is usually downregulated.39 The splicing pattern of Bcl‐X is controlled by many splicing regulators.6, 40 One such factor is RBM25, also referred to as hRED120. It acts as a RNA binding protein, which interacts with RNA processing factors both physically and genetically.41 Co‐suppression of RBM25 with splicing and 3′ end processing factors results in aberrant growth or developmental defects in Caenorhabditis elegans. RBM25 binds to the Bcl‐X pre‐mRNA and recruits U1 snRNP to the upstream 5′ ss of intron 2 of the Bcl‐X gene through interaction with hLuc7A, a U1 snRNP binding protein, resulting in production of the short Bcl‐XS isoform.42, 43
Another RNA binding protein playing a role in alternative splicing is hnRNP K, which also influences the splicing pattern of the Bcl‐X gene. HnRNP K binds to a splicing silencer element located in the vicinity of the upstream 5′ ss of the Bcl‐X gene and represses the production of the Bcl‐XS isoform.40
Sam68, which was identified as a target of the oncogenic protein kinase Src‐family, is yet another splicing regulator of Bcl‐X.44, 45, 46 Inactivation of Sam68 causes neoplasmic transformation, while overexpression of Sam68 leads to cell cycle arrest and apoptosis. This suggests that Sam68 is a potential tumor suppressor.47, 48 Sam68 binds to Bcl‐X mRNA and promotes splicing at the upstream 5′ ss to increase the relative amount of isoform Bcl‐XS. This effect is reversed by tyrosine phosporylation of Sam68 through Src‐like kinase Fyn, indicating that dephosphorylated Sam68 induces apoptosis through alternative 5′ ss choice of Bcl‐X,46 although Sam68 also contributes to tumor progression in other contexts.49
Vascular endothelial growth factor
Vascular endothelial growth factor is a key regulator of angiogenesis, a prominent feature of cancer.50, 51 The VEGF family consists of several members, with each isoform designated as VEGFxxx, with xxx indicating the number of amino acids in the mature protein. Their lengths depend on whether alternative forms of exons 6 and 7 become incorporated into the mature mRNA. For a long time, it was not sufficiently appreciated that the VEGF gene gives rise to an entire family of growth regulators, some displaying rather different properties. VEGF isoforms fall into two main categories, VEGFxxx and VEGFxxxb.52, 53 The VEGF gene contains two possible splice sites in exon 853 (Fig. 2). If the proximal splice site is selected, the product will belong to the angiogenic VEGFxxx family. If the distal site is chosen, the sequence of the six most C‐terminal amino acids will be changed. In this case, the product belongs to the VEGFxxxb family and will have anti‐angiogenic properties, exactly the opposite of VEGFxxx. This splice site selection is controlled by SR proteins.54 Overexpression of SRSF1 and SRp40 causes VEGFxxx isoform production. In contrast, SRp55 enhances distal splice site selection, resulting in production of anti‐angiogenic VEGFxxxb isoforms.
Expression of VEGFxxxb isoforms is downregulated in renal cell carcinoma, prostate cancer and malignant melanoma.53, 55, 56 Not surprisingly, VEGFxxx is overexpressed in many cancer cells.57 VEGFxxx binding to VEGF receptor 2 (VEGFR2) induces dimerization of the receptor and activates several signal transduction cascades.58 It appears that the different C‐terminus of VEGFxxxb is unable to bind the neurophilin 1 co‐receptor required for the full activation of VEGF‐induced signaling.59 This difference of binding mode can explain opposing functions of each VEGF isoform.
Modulating Splicing by Small Molecules for Cancer Therapy
Thus far we have outlined the importance of accurate splicing in eukaryotes and described some of the pathologies arising from aberrant splicing or from loss of control over its regulation. With the discovery of small molecules that interfere with splicing, the possibility of using splice inhibition for therapeutic purposes arose.
Spliceostatin A
Spliceostatin A (SSA) is a methyl‐ketal derivative of FR901464, a metabolite from the bacterium Pseudomonas sp. No. 2663.5, 60 FR901464, which was originally isolated as a transcriptional activator, has potent cytotoxic activity against a number of different human cancer cell lines and the ability to prolong the life of tumor‐bearing mice.60, 61 The other distinguishing feature of FR901464 is that it causes cell cycle arrest at the G1 and G2/M phases. Further study revealed that FR901464 treatment causes production of a C‐terminal truncated form of the cyclin‐dependent kinase (CDK) inhibitor and tumor suppressor, p27, designated p27*. It became clear that under SSA treatment p27 pre‐mRNA accumulated and became translated, resulting in production of a shortened protein. As p27* was constitutively active and more stable than the original p27 protein, it acted as a negative regulator of cell growth. This finding suggests that splicing inhibition could allow targeted therapy and restore cell cycle control in transformed cells.
Spliceostatin A binds to the SF3b complex, which is a sub‐component of the U2 snRNP that recognizes the branch point sequence5, 62 and inhibits splicing both in vivo and in vitro. Recent studies revealed that SSA destabilizes interaction between pre‐mRNA and SF3b1, the largest subunit of the SF3b complex, resulting in reduced fidelity of branch point recognition63 (Fig. 3). In addition to production of p27*, SSA decreases VEGF in both mRNA and protein levels and inhibits tumor angiogenesis in vivo,64 suggesting that downregulation of VEGF is another reason why SSA displays antitumor activity.
Figure 3.

Small molecules inhibiting splicing. Spliceostatin A and pladienolide B inhibit stable binding of the U2 small nuclear ribonucleoprotein particle (snRNP) to precursor messenger RNA (pre‐mRNA). Isoginkgetin interferes with U4/U6•U5 binding. TG003 decreases the phosphorylation level of SR proteins by inhibiting the SR protein kinase CLK. ESE, exonic splicing enhancer; ESS, exonic splicing silencer; ISE, intronic splicing enhancer; ISS, intronic splicing silencer.
Pladienolide
Pladienolide is another potent antitumor natural compound, isolated from Streptomyces platensis.65 Pladienolide shares many common features with SSA. It decreases gene expression controlled by the VEGF promoter, arrests the cell cycle at G1 and G2/M phases and exerts potent antitumor activity in several human cancer xenograft models in mice.65, 66 Like SSA, the molecular target of pladienolide is SF3b1 and consequently pladienolide inhibits splicing in a similar manner.67, 68 In pladienolide‐treated cells, conformational changes and ATP‐dependent remodeling events of the U2 snRNP are inhibited and this inhibition weakens the binding between U2 snRNP and pre‐mRNA69 (Fig. 3). A synthetic derivative of pladienolide, E7107, has entered phase I clinical trials against thyroid cancer and has led to stable disease or delayed disease progression in a subset of patients.70
Other compounds
Spliceostatin A, pladienolide and GEX1A directly bind to the spliceosome and exert their antitumor activity through direct inhibition of splicing.5, 67, 71 Isoginkgetin is another general splicing inhibitor with antitumor activity.72, 73 It prevents recruitment of U4/U6•U5 tri‐snRNP and inhibits splicing, suggesting that the spliceosome is a novel anticancer drug target (Fig. 3). TG003 is not a general splicing inhibitor but modulates alternative splicing patterns through dephosphorylation of SR proteins, including SRSF1, by inhibiting SR protein kinases CLK1 and CLK474 (Fig. 3). Because SRSF1 is a proto‐oncogene (see above), this compound also has potential as an anticancer drug.
Perspective
As shown here, missplicing of cancer‐related genes can drive tumor development. Modulation of splicing patterns and restoring the balance between two alternative splicing forms using splicing modulators/inhibitors might be an effective method for cancer therapy. Several small molecules binding to splicing factors are in phase I clinical trials and the results will be available soon. Although it is challenging to control specific splicing events, the difficulty can be overcome by screening for novel small molecules and identifying subsets of tumors that might be susceptible to splice‐inhibition therapy. Further studies will surely uncover the detailed mode of action of further splicing modulators. Because the discovery of splicing inhibitors is still recent, the development of chemical modulation of pre‐mRNA processing has just begun.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Japan Society for the Promotion of Science.
(Cancer Sci, 2012; 103: 1611–1616)
References
- 1. Moore MJ, Proudfoot NJ. Pre‐mRNA processing reaches back to transcription and ahead to translation. Cell 2009; 136: 688–700. [DOI] [PubMed] [Google Scholar]
- 2. Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell 2009; 136: 777–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136: 701–18. [DOI] [PubMed] [Google Scholar]
- 4. Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J 2010; 430: 365–77. [DOI] [PubMed] [Google Scholar]
- 5. Kaida D, Motoyoshi H, Tashiro E et al Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre‐mRNA. Nat Chem Biol 2007; 3: 576–83. [DOI] [PubMed] [Google Scholar]
- 6. David CJ, Manley JL. Alternative pre‐mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010; 24: 2343–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 2011; 12: 715–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high‐throughput sequencing. Nat Genet 2008; 40: 1413–5. [DOI] [PubMed] [Google Scholar]
- 9. Wang ET, Sandberg R, Luo S et al Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456: 470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dreyfuss G, Kim VN, Kataoka N. Messenger‐RNA‐binding proteins and the messages they carry. Nat Rev Mol Cell Biol 2002; 3: 195–205. [DOI] [PubMed] [Google Scholar]
- 11. Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 2009; 417: 15–27. [DOI] [PubMed] [Google Scholar]
- 12. Martinez‐Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol 2007; 623: 123–47. [DOI] [PubMed] [Google Scholar]
- 13. Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto‐oncogene. Nat Struct Mol Biol 2007; 14: 185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakamuro D, Elliott KJ, Wechsler‐Reya R, Prendergast GC. BIN1 is a novel MYC‐interacting protein with features of a tumour suppressor. Nat Genet 1996; 14: 69–77. [DOI] [PubMed] [Google Scholar]
- 15. Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain‐specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci USA 1999; 96: 9689–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005; 123: 569–80. [DOI] [PubMed] [Google Scholar]
- 17. Comoglio PM, Trusolino L. Invasive growth: from development to metastasis. J Clin Invest 2002; 109: 857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaudino G, Follenzi A, Naldini L et al RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J 1994; 13: 3524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collesi C, Santoro MM, Gaudino G, Comoglio PM. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol 1996; 16: 5518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghigna C, Giordano S, Shen H et al Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005; 20: 881–90. [DOI] [PubMed] [Google Scholar]
- 21. Lefave CV, Squatrito M, Vorlova S et al Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J 2011; 30: 4084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mulherkar N, Prasad KV, Prabhakar BS. MADD/DENN splice variant of the IG20 gene is a negative regulator of caspase‐8 activation. Knockdown enhances TRAIL‐induced apoptosis of cancer cells. J Biol Chem 2007; 282: 11715–21. [DOI] [PubMed] [Google Scholar]
- 23. Ramaswamy M, Efimova EV, Martinez O, Mulherkar NU, Singh SP, Prabhakar BS. IG20 (MADD splice variant‐5), a proapoptotic protein, interacts with DR4/DR5 and enhances TRAIL‐induced apoptosis by increasing recruitment of FADD and caspase‐8 to the DISC. Oncogene 2004; 23: 6083–94. [DOI] [PubMed] [Google Scholar]
- 24. Golan‐Gerstl R, Cohen M, Shilo A et al Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 2011; 71: 4464–72. [DOI] [PubMed] [Google Scholar]
- 25. Tockman MS, Mulshine JL, Piantadosi S et al Prospective detection of preclinical lung cancer: results from two studies of heterogeneous nuclear ribonucleoprotein A2/B1 overexpression. Clin Cancer Res 1997; 3: 2237–46. [PubMed] [Google Scholar]
- 26. Zhou J, Allred DC, Avis I et al Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein‐A2/B1 (hnRNP‐A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat 2001; 66: 217–24. [DOI] [PubMed] [Google Scholar]
- 27. Patry C, Bouchard L, Labrecque P et al Small interfering RNA‐mediated reduction in heterogeneous nuclear ribonucleoparticule A1/A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res 2003; 63: 7679–88. [PubMed] [Google Scholar]
- 28. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c‐Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010; 463: 364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Christofk HR, Vander Heiden MG, Harris MH et al The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452: 230–3. [DOI] [PubMed] [Google Scholar]
- 30. Bluemlein K, Gruning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011; 2: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem‐cell diseases. Nat Rev Cancer 2007; 7: 118–29. [DOI] [PubMed] [Google Scholar]
- 32. Yoshida K, Sanada M, Shiraishi Y et al Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478: 64–9. [DOI] [PubMed] [Google Scholar]
- 33. Bogner C, Leber B, Andrews DW. Apoptosis: embedded in membranes. Curr Opin Cell Biol 2010; 22: 845–51. [DOI] [PubMed] [Google Scholar]
- 34. Boise LH, Gonzalez‐Garcia M, Postema CE et al bcl‐x, a bcl‐2‐related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74: 597–608. [DOI] [PubMed] [Google Scholar]
- 35. Clarke MF, Apel IJ, Benedict MA et al A recombinant bcl‐x s adenovirus selectively induces apoptosis in cancer cells but not in normal bone marrow cells. Proc Natl Acad Sci USA 1995; 92: 11024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gottschalk AR, Boise LH, Thompson CB, Quintans J. Identification of immunosuppressant‐induced apoptosis in a murine B‐cell line and its prevention by bcl‐x but not bcl‐2. Proc Natl Acad Sci USA 1994; 91: 7350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhl JS, Krajewski S, Duran GE, Reed JC, Sikic BI. Spontaneous overexpression of the long form of the Bcl‐X protein in a highly resistant P388 leukaemia. Br J Cancer 1997; 75: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xerri L, Parc P, Brousset P et al Predominant expression of the long isoform of Bcl‐x (Bcl‐xL) in human lymphomas. Br J Haematol 1996; 92: 900–6. [DOI] [PubMed] [Google Scholar]
- 39. Ma X, Zhao Y, Li Y, Lu H, He Y. Relevance of Bcl‐x expression in different types of endometrial tissues. J Exp Clin Cancer Res 2010; 29: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Revil T, Pelletier J, Toutant J, Cloutier A, Chabot B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro‐apoptotic Bcl‐xS splice isoform. J Biol Chem 2009; 284: 21458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fortes P, Longman D, McCracken S et al Identification and characterization of RED120: a conserved PWI domain protein with links to splicing and 3′‐end formation. FEBS Lett 2007; 581: 3087–97. [DOI] [PubMed] [Google Scholar]
- 42. Puig O, Bragado‐Nilsson E, Koski T, Seraphin B. The U1 snRNP‐associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res 2007; 35: 5874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou A, Ou AC, Cho A, Benz EJ Jr, Huang SC. Novel splicing factor RBM25 modulates Bcl‐x pre‐mRNA 5′ splice site selection. Mol Cell Biol 2008; 28: 5924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor SJ, Shalloway D. An RNA‐binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature 1994; 368: 867–71. [DOI] [PubMed] [Google Scholar]
- 45. Fumagalli S, Totty NF, Hsuan JJ, Courtneidge SA. A target for Src in mitosis. Nature 1994; 368: 871–4. [DOI] [PubMed] [Google Scholar]
- 46. Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA‐binding protein Sam68 modulates the alternative splicing of Bcl‐x. J Cell Biol 2007; 176: 929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taylor SJ, Resnick RJ, Shalloway D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biol 2004; 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu K, Li L, Nisson PE, Gruber C, Jessee J, Cohen SN. Neoplastic transformation and tumorigenesis associated with sam68 protein deficiency in cultured murine fibroblasts. J Biol Chem 2000; 275: 40195–201. [DOI] [PubMed] [Google Scholar]
- 49. Bielli P, Busa R, Paronetto MP, Sette C. The RNA‐binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer 2011; 18: R91–102. [DOI] [PubMed] [Google Scholar]
- 50. Keck PJ, Hauser SD, Krivi G et al Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989; 246: 1309–12. [DOI] [PubMed] [Google Scholar]
- 51. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989; 246: 1306–9. [DOI] [PubMed] [Google Scholar]
- 52. Harper SJ, Bates DO. VEGF‐A splicing: the key to anti‐angiogenic therapeutics? Nat Rev Cancer 2008; 8: 880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bates DO, Cui TG, Doughty JM et al VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down‐regulated in renal cell carcinoma. Cancer Res 2002; 62: 4123–31. [PubMed] [Google Scholar]
- 54. Nowak DG, Woolard J, Amin EM et al Expression of pro‐ and anti‐angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci 2008; 121: 3487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Woolard J, Wang WY, Bevan HS et al VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 2004; 64: 7822–35. [DOI] [PubMed] [Google Scholar]
- 56. Pritchard‐Jones RO, Dunn DB, Qiu Y et al Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br J Cancer 2007; 97: 223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol 2001; 280: C1358–66. [DOI] [PubMed] [Google Scholar]
- 58. Ruch C, Skiniotis G, Steinmetz MO, Walz T, Ballmer‐Hofer K. Structure of a VEGF‐VEGF receptor complex determined by electron microscopy. Nat Struct Mol Biol 2007; 14: 249–50. [DOI] [PubMed] [Google Scholar]
- 59. Kawamura H, Li X, Harper SJ, Bates DO, Claesson‐Welsh L. Vascular endothelial growth factor (VEGF)‐A165b is a weak in vitro agonist for VEGF receptor‐2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 2008; 68: 4683–92. [DOI] [PubMed] [Google Scholar]
- 60. Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M. New antitumor substances, FR901463, FR901464 and FR901465 I. Taxonomy, fermentation, isolation, physico‐chemical properties and biological activities. J Antibiot (Tokyo) 1996; 49: 1196–203. [DOI] [PubMed] [Google Scholar]
- 61. Nakajima H, Hori Y, Terano H et al New antitumor substances, FR901463, FR901464 and FR901465 II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 1996; 49: 1204–11. [DOI] [PubMed] [Google Scholar]
- 62. Gozani O, Potashkin J, Reed R. A potential role for U2AF‐SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 1998; 18: 4752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti‐tumor drug spliceostatin A. Genes Dev 2011; 25: 445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Furumai R, Uchida K, Komi Y et al Spliceostatin A blocks angiogenesis by inhibiting global gene expression including VEGF. Cancer Sci 2010; 101: 2483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer‐11107 I. Taxonomy, fermentation, isolation and screening. J Antibiot 2004; 57: 173–9. [DOI] [PubMed] [Google Scholar]
- 66. Mizui Y, Sakai T, Iwata M et al Pladienolides, new substances from culture of Streptomyces platensis Mer‐11107 III. In vitro and in vivo antitumor activities. J Antibiot 2004; 57: 188–96. [DOI] [PubMed] [Google Scholar]
- 67. Kotake Y, Sagane K, Owa T et al Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 2007; 3: 570–5. [DOI] [PubMed] [Google Scholar]
- 68. Yokoi A, Kotake Y, Takahashi K et al Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J 2011; 278: 4870–80. [DOI] [PubMed] [Google Scholar]
- 69. Folco EG, Coil KE, Reed R. The anti‐tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point‐binding region. Genes Dev 2011; 25: 440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsimberidou AM, Vaklavas C, Wen S et al Phase I clinical trials in 56 patients with thyroid cancer: the M D. Anderson cancer center experience. J Clin Endocrinol Metab 2009; 94: 4423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hasegawa M, Miura T, Kuzuya K et al Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol 2011; 18: 229–33. [DOI] [PubMed] [Google Scholar]
- 72. O'Brien K, Matlin AJ, Lowell AM, Moore MJ. The biflavonoid isoginkgetin is a general inhibitor of Pre‐mRNA splicing. J Biol Chem 2008; 283: 33147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yoon SO, Shin S, Lee HJ, Chun HK, Chung AS. Isoginkgetin inhibits tumor cell invasion by regulating phosphatidylinositol 3‐kinase/Akt‐dependent matrix metalloproteinase‐9 expression. Mol Cancer Ther 2006; 5: 2666–75. [DOI] [PubMed] [Google Scholar]
- 74. Muraki M, Ohkawara B, Hosoya T et al Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem 2004; 279: 24246–54. [DOI] [PubMed] [Google Scholar]