

Abstract
Gemtuzumab ozogamicin (GO) consists of the CD33 antibody linked to calicheamicin. The binding of GO to the CD33 antigen on leukemic cells results in internalization followed by the release of calicheamicin, thereby inducing DNA strand breaks. We hypothesized that the induction of DNA strand breaks would be a surrogate marker of GO cytotoxcity. Here, two GO‐resistant variants (HL/GO‐CSA [225‐fold], HL/GO [200‐fold]) were established by serially incubating human leukemia HL‐60 cells with GO with or without a P‐glycoprotein (P‐gp) inhibitor, cyclosporine A, respectively. The CD33 positivity was reduced in both variants. The HL/GO‐CSA cells showed an increased multidrug resistance protein‐1 (MRP1) transcript, and an MRP1 inhibitor partially reversed GO resistance. The HL/GO cells had neither P‐gp nor MRP1 overexpression. Microarray analysis and Western blotting indicated elevated levels of DNA repair‐associated proteins in both variants. Two other leukemic subclones, showing either P‐gp or MRP1 overexpression, were also GO‐resistant. Using single cell gel electrophoresis analysis, it was determined that GO‐induced DNA strand breaks increased dose‐dependently in HL‐60 cells, whereas the number of breaks was reduced in the GO‐resistant cell lines. The induction of DNA strand breaks was correlated with GO sensitivity among these cell lines. The CD33 positivity and the expression levels of transporters were not proportional to drug sensitivity. Using primary leukemic cells, the induction of DNA strand breaks appeared to be associated with GO sensitivity. Thus, GO‐induced DNA strand breaks as the final output of the mechanism of action would be critical to predict GO cytotoxicity.
To improve therapeutic outcomes for patients with AML, new treatment regimes and agents are needed.1, 2, 3, 4 Gemtuzumab ozogamicin (Mylotarg; Wyeth‐Ayers Research, Pearl River, NY, USA) is a humanized mAb directed against the CD33 surface antigen that is conjugated to a derivative of the cytotoxic antibiotic calicheamicin.5 CD33 is an antigen normally expressed on early myeloid progenitor cells in normal bone marrow and on leukemic blasts in 90% of all newly diagnosed AML but not on normal stem cells.6 Because CD33 is specific to leukemic cells, GO is an attractive targeted agent that could improve the clinical outcome of AML chemotherapy without increasing toxicity. Gemtuzumab ozogamicin received marketing approval from the US Food and Drug Administration under accelerated approval regulations for the treatment of patients with CD33‐positive AML who are in the first relapse, are 60 years of age or older, and who are not considered candidates for cytotoxic chemotherapy.7 After approval, however, the Southwest Oncology Group compared GO plus standard induction therapy versus standard induction therapy alone, and found that there was no difference in disease‐free survival between the two treatment regimes.8 One major reason why the study was negative for GO's additional efficacy was that the dose of DNR was reduced in the chemotherapy + GO arm, which might mask any benefit of GO in remission induction treatment. Another problem was that there were more induction deaths in the chemotherapy + GO arm than the chemotherapy alone arm (5.4% vs. 1.4%, respectively). However, this mortality rate (5.4%) was quite similar to induction death rates in virtually all chemotherapy trials in patients of this age, approximately 5–7%. Consequently, the Food and Drug Administration recommended the withdrawal of GO from the market in the US; GO is still clinically available in some other countries including Japan. Nevertheless, a similar investigation carried out in the MRC15 trial revealed that there was a significant survival benefit for AML patients with favorable risk.9 These contradictory studies strongly suggest that there are subsets of AML that clearly benefit from the addition of GO to standard chemotherapy.10, 11 It is also suggested that GO sensitivity might vary among patients and subtypes of leukemia (i.e., acute promyelocytic leukemia) and that a predictor of drug sensitivity is needed to identify the optimal use of GO.
Mechanistically, GO binds to the CD33 antigen on the surface of leukemic cells, which results in internalization followed by the release of calicheamicin. Free calicheamicin is reduced to 1,4‐dehydrobenzene, then enters the cell nucleus, intercalates within the minor groove of the DNA helix, and consequently induces site‐specific DNA strand breaks.12, 13, 14 GO is apparently ineffective against CD33‐negative leukemia. ATP‐binding cassette transporters, such as P‐gp or MRP1, efflux GO from cells.15, 16, 17 Moreover, several DNA damage responses repair GO‐induced DNA strand breaks.12, 13, 14 In these regards, the induction of DNA strand breaks by GO is considered to be the end output of the sum of all the processes of CD33‐mediated internalization of the drug, efflux by transporters, and equilibrium between GO‐induced DNA damage and DNA repair responses. Therefore, GO‐induced DNA strand breaks would be a surrogate marker of GO cytotoxicity.
It was hypothesized that cellular sensitivity to GO would be predicted by the amount of GO‐induced DNA strand breaks in leukemic cells. To test this hypothesis, two new GO‐resistant cultured leukemic cell lines were developed. The mechanisms of resistance were investigated specifically focusing on CD33 positivity, the expression levels of transporters, DNA repair‐associated proteins, and GO‐induced DNA strand breaks. Conventional techniques as well as a comprehensive microarray analysis were used. Two additional leukemic clones showing either P‐gp or MRP1 overexpression and primary leukemic cells from patients were similarly evaluated. A correlation between GO‐induced DNA strand breaks and cellular GO sensitivity was sought.
Materials and Methods
Chemicals and reagents
Gemtuzumab ozogamicin was kindly supplied by Wyeth Japan (Tokyo, Japan) and dissolved in PBS to a stock concentration of 10 mg/mL. The P‐gp inhibitor CSA and DNR were purchased from Sigma (St. Louis, MO, USA). The MRP inhibitor MK571 was obtained from Alexis Biochemicals (Lausen, Switzerland).
Cell culture
The human leukemia cell lines HL‐60 and K562 were used. A DNR‐resistant K562 variant (K562/DNR19) and a dual ara‐C‐ and DNR‐resistant HL‐60 variant (HL/Ara‐CDNR), both of which had been established in our previous studies, were also used.18, 19 K562/DNR19 cells acquired P‐gp overexpression, whereas HL/Ara‐CDNR cells overexpressed MRP1.18, 19 All of these cell lines were cultured in RPMI‐1640 media supplemented with 10% FCS in a 5% CO2 humidified atmosphere at 37°C.
Establishment of two GO‐resistant HL‐60 variants
A GO‐resistant variant, HL/GO, was established by serial incubation of HL‐60 cells with GO followed by limiting dilution cloning. In brief, the parental HL‐60 cells were maintained with increasing concentrations of GO. The initial concentration (2 ng/mL) was one‐tenth of the concentration required to inhibit 50% growth (IC50) of HL‐60 cells. The cultures were observed daily, allowed to grow, and underwent subsequent passages with gradually increasing concentrations of GO. The passaging was repeated for 8 months, and one cell line resistant to GO (HL/GO) was cloned by the limiting dilution method. Another GO‐resistant variant (HL/GO‐CSA) was established in a similar manner by serial incubation of HL‐60 cells with increasing concentrations of GO in the presence of CSA to suppress the expression of P‐gp, followed by limiting dilution for cloning.
Proliferation assay
To evaluate the growth inhibition effects, the XTT assay was carried out according to the manufacturer's instructions (Roche, Indianapolis, IN, USA) with slight modifications.20
Quantitation of apoptotic cell death
To evaluate cytotoxicity, apoptotic cell death was determined morphologically by staining the nuclei of cells with Hoechst No. 33342 (Sigma) 24 h after treatment, as described previously.21 The nuclei, 200 per treatment condition, were then evaluated under UV illumination.
Flow cytometry
Flow cytometric analysis was carried out as described previously to detect the expression of CD33 and P‐gp using anti‐CD33 antibody and anti‐MDR1 antibody, respectively (SRL, Tokyo, Japan).
Real‐time RT‐PCR
To evaluate the expression levels of P‐gp (accession: P08183) and MRP1 (accession: AAB83983), real‐time RT‐PCR was carried out using the ABI Prism 7900 sequence detection system (Applied Biosystems, Foster City, CA, USA). The primers were prepared by Mitsubishi Chemical Medience (Tokyo, Japan), the sequences of which were not open to the public.
Gene expression profiling using DNA microarray analysis
The gene expression profiles of HL‐60 and two GO‐resistant variants (HL/GO, HL/GO‐CSA) were compared using a cDNA microarray. The total RNA was isolated from 3 × 106 cells per sample and was assessed using gel electrophoresis. The hybridization was carried out between Cy‐3‐labeled total RNA from the parental HL‐60 cells and Cy‐5‐labeled total RNA from the HL/GO cells or HL/GO‐CSA cells on microarrays of complementary DNA that contained 35 000 elements (Operon Aros, Human Genome Oligo Set, Version 4.0; Operon Biotechnologies, Tokyo, Japan). The data were retrieved as log10 (Cy5/Cy3), and final values were expressed as a fold‐change in the gene expression values between HL‐60 and GO‐resistant subclones. Values with ≥2‐fold changes were considered to be significant.22
Western blot analysis
Protein levels of XRCC5 (Ku80), RPA3, GADD45A, and PARP1 were determined by standard Western blotting. Rabbit monoclonal anti‐Ku80 (Cell Signaling Technology, Beverly, MA, USA), rabbit polyclonal anti‐RPA3 (Abgent, San Diego, CA, USA), mouse monoclonal anti‐GADD45A (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit monoclonal anti‐PARP1 (Cell Signaling Technology), and anti‐actin (Sigma) antibodies were used as primary antibodies. An anti‐rabbit IgG–HRP‐conjugated antibody (Thermo Scientific, Rockford, IL, USA) and an anti‐mouse IgG–HRP‐conjugated antibody (Zymed, San Francisco, CA, USA) were used as secondary antibodies.
Alkaline single cell gel electrophoresis (Comet) assay
Because calicheamicin induces both single‐ and double‐strand DNA breaks,12, 13 the alkaline single cell gel electrophoresis (Comet) assay was used to determine GO‐induced DNA strand breaks as described previously.23, 24 Following treatment, the mixture of the cells with agarose was fixed on a fully frosted microscope slide (Fisher Scientific, Pittsburgh, PA, USA). The slides were placed in a lysis solution (2.5 M NaCl, 10 mM Tris, 100 mM ethylenediamine tetraacetic acid, 10% DMSO, 1% Triton X‐100, pH 10.0) then soaked in electrophoretic buffer (1 mM EDTA, 300 mM NaOH, pH 13.0). Electrophoresis was carried out (15 min, 90 V, 450 mA), and the slides were then stained with ethidium bromide (20 μg/mL). The cells, 100 per treatment condition, were analyzed using a computer‐based image analysis system (Kinetic Imaging Komet system, version 4.0, Liverpool, UK). The amounts of DNA strand breaks were expressed as the “tail moment”, which combined a measurement of the length of the DNA migration and the relative DNA content therein.
The Comet assay was originally developed by Olive et al.23 and modified by Singh et al.24 Since then, the method has been widely used to determine DNA strand breaks in various fields. A conventional method to determine DNA strand breaks is an alkaline elution assay method. Compared with this old method, the Comet assay holds several advantages. First, the sample size is very small, requiring a minimum of 5000 cells per assay. Second, the Comet assay is much more sensitive and the assay procedure is simpler and takes less time than the alkakine elution assay. Third, the Comet assay can detect DNA strand breaks at the single‐cell level. Finally, the method is quantitative, as computer‐based software is available for the Comet assay that can calculate the number of DNA strand breaks. The disadvantage is that we have to set up apparatus including a fluorescence microscope, a charge‐coupled device camera, and computer‐based software.
Patient samples
Leukemic cell samples were obtained from 11 patients with AML. Prior to chemotherapy, peripheral blood was drawn into heparinized tubes, layered over Ficoll–Hypaque, and centrifuged (500g, 30 min at room temperature) to isolate the leukemic cells.21 The cells were washed twice with PBS then centrifuged (500g, 5 min at 4°C) to pellet the cells. The aliquots were resuspended in RPMI‐1640 media supplemented with 10% heat‐inactivated FCS (1 × 106 cells/mL) at 37°C in a 5% CO2 humidified atmosphere for further experiments. This study was approved by the ethics committee of The University of Fukui Hospital (Eiheiji, Japan).
Statistical analyses
All statistical analyses were carried out using Microsoft Excel 2007 software (Microsoft, Redmond, WA, USA). All graphs, linear regression lines, and curves were generated using GraphPad Prism software version 5.0 (GraphPad Software, San Diego, CA, USA). Values of P ≤ 0.05 were considered statistically significant.
Results
Establishment of two GO‐resistant HL‐60 variant cell lines
The growth inhibitory effects of GO were compared between HL‐60, HL/GO, and HL/GO‐CSA cells. The IC50 values indicated that both variants were more GO‐resistant than HL‐60 cells (Table 1). The HL/GO and HL/GO‐CSA cells were 200‐ and 225‐fold more GO‐resistant than HL‐60 cells. The variants were also more refractory to GO‐induced apoptosis than was their parental counterpart (Fig. 1A). Both variants showed cross‐resistance against DNR, a representative anthracyclin similar to calicheamicin (Table 1). Thus, the two variants showing a similar magnitude of GO resistance were successfully established from HL‐60 cells.
Table 1.
Drug sensitivity of gemtuzumab ozogamicin (GO)‐resistant HL‐60 variant human leukemia cell lines
| Drug | IC50 | ||
|---|---|---|---|
| HL‐60 | HL/GO | HL/GO‐CSA | |
| GO (μg/mL) | 0.02 | 4.00 | 4.50 |
| GO + MK571 (μg/mL) | 0.02 | 3.95 | 2.90 |
| MK571 (μM) | 58.90 | 91.00 | ND |
| DNR (μM) | 0.03 | 0.13 | 0.15 |
Cells were incubated with various concentrations of GO with or without a minimally toxic concentration of MK571 (10 μM) for 72 h. The cells were also treated with daunorubicin (DNR) in the same manner. IC50 values were then determined by using the XTT assay. CSA, cyclosporine A; ND, not determined because the IC50 value was beyond the assay range due to the high degree of resistance.
Figure 1.
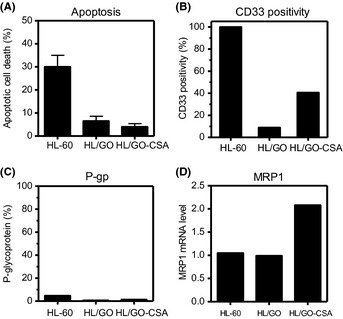
Establishment of two gemtuzumab ozogamicin (GO)‐resistant HL‐60 variants (HL/GO, HL/GO‐CSA) and their comparison with the HL‐60 human leukemia cell line. (A) Apoptotic cell death induced by GO. Cells were incubated with 1 μg/mL GO for 72 h, followed by Hoechst 33342 staining of nuclei for the evaluation of apoptotic cell death. The values are the means ± SD of triplicate determinations. (B) CD33 positivity determined by flow cytometry. (C,D) Two efflux pumps were evaluated. P‐glycoprotein (P‐gp) expression was quantitated using flow cytometry (C). Multridrug resistance protein‐1 (MRP1) was determined using real‐time RT‐PCR with the value of HL‐60 cells set as 1 (D).
Determination of CD33, P‐gp, and MRP1 expression
CD33 is required for the internalization of GO in leukemic cells, and ATP‐binding cassette transporters efflux GO from cells.15, 16, 17, 25 Flow cytometric analysis revealed that CD33 expression levels were reduced in both GO‐resistant cell lines, but the reduction was more prominent in the HL/GO cells (6% positive) than the HL/GO‐CSA cells (40% positive) (Fig. 1B). The expression of two efflux pumps, P‐gp and MRP1, was determined using flow cytometry and real‐time RT‐PCR, respectively. Neither of the GO‐resistant variants had increased P‐gp expression (Fig. 1C), but interestingly, HL/GO‐CSA cells, which had been developed with GO in the presence of the P‐gp inhibitor CSA, had an increased MRP1 transcript level (Fig. 1D). The addition of CSA might suppress the development of P‐gp and collaterally mediate the expression of MRP1,16 although the mechanism of expression was not elucidated in detail here. To confirm the role of MRP1 in the mechanism of GO resistance, cells were treated with GO in the presence of the MRP1 inhibitor MK571, and the XTT proliferation assay was carried out. The addition of a non‐toxic concentration of MK571 partially sensitized HL/GO‐CSA cells, but not HL‐60 cells or HL/GO cells, to GO (Table 1). These results suggested that CD33 positivity and MRP1 were involved in the development of GO resistance and that the mechanisms of GO resistance appeared to differ slightly between the HL/GO and HL/GO‐CSA cell lines despite the similar degree of their GO refractivity.
DNA repair‐associated factors evaluated
To further elucidate the mechanism of GO resistance, a DNA microarray was used to carry out a genome‐wide screen. The gene expression profiles were compared between each GO‐resistant variant (HL/GO, HL/GO‐CSA) and the HL‐60 cell line. Overall, thousands of upregulated or downregulated genes were detected in each GO‐resistant subclone. Because the cytotoxicity of GO depends on the induction of DNA strand breaks by calicheamicin,13 the present study focused on DNA stand break repair, and genes associated with DNA repair showing ≥2‐fold changes in both variants are listed (Table 2). Calicheamicin induces both single‐strand and double‐strand DNA breaks.13, 26 In the list of altered genes of both GO‐resistant variants (Table 2), PARP1 and GADD45A are associated with DNA excision repairs for single‐strand breaks.27, 28 XRCC5 (Ku80) and RPA3 are required for DNA double‐strand break repairs.29, 30, 31 Western blot analysis confirmed that the expression levels of PARP1, GADD45A, Ku80, and RPA3 were augmented in both the HL/GO and HL/GO‐CSA cells compared with those in the HL‐60 cells (Fig. 2). These results suggested that enhanced DNA repair functions in response to GO‐induced DNA strand breaks would contribute to the development of GO resistance in these subclones.
Table 2.
Commonly altered genes in gemtuzumab ozogamicin (GO)‐resistant HL‐60 variant human leukemia cell lines, HL/GO and HL/GO‐CSA
| Upregulated | Downregulated | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Ref. sequence | Fold change | Gene | Ref. sequence | Fold change | ||
| HL/GO | HL/GO‐CSA | HL/GO | HL/GO‐CSA | ||||
| G22P1 | NM_001469 | 4.94 | 2.51 | MAPK12 | NM_002969 | 0.35 | 0.42 |
| CCNH1 | NM_001239 | 5.85 | 3.82 | DDB2 | NM_000107 | 0.37 | 3.82 |
| RPS27A | NM_002954 | 3.26 | 2.45 | ERCC5 | NM_000123 | 0.35 | 0.47 |
| CDK7 | NM_001799 | 3.46 | 2.74 | SPI1 | NM_003120 | 0.17 | 0.26 |
| GADD45A | NM_001924 | 4.98 | 5.56 | PTPNS1 | NM_080792 | 0.34 | 0.49 |
| CETN2 | NM_004344 | 2.65 | 3.56 | FMO4 | NM_002022 | 0.34 | 0.45 |
| XRCC5 | NM_021141 | 4.86 | 4.28 | TNFRSF1A | NM_001065 | 0.32 | 0.43 |
| RPA3 | NM_002947 | 3.93 | 3.11 | BCL6 | NM_001706 | 0.31 | 0.40 |
| HSPCB | NM_007355 | 2.47 | 3.39 | CCNB2 | NM_004701 | 0.26 | 0.33 |
| PARP1 | NM_001618 | 6.68 | 2.56 | TIMP3 | NM_000362 | 0.32 | 0.43 |
| BAG3 | NM_004281 | 2.53 | 2.34 | TNFRSF14 | NM_003820 | 0.32 | 0.46 |
| CCNB1 | NM_031966 | 9.55 | 3.93 | PHB | NM_002634 | 0.36 | 0.41 |
| SOCS2 | NM_003877 | 3.60 | 3.25 | TRIB3 | NM_021158 | 0.27 | 0.37 |
| CCND3 | NM_001760 | 5.11 | 3.96 | MST1 | NM_020998 | 0.24 | 0.42 |
| PPP2CA | NM_002715 | 3.55 | 3.55 | SGK | NM_005627 | 0.37 | 0.46 |
| CCNA2 | NM_001237 | 4.33 | 2.58 | ||||
Genes that showed ≥2‐fold change values were listed.
Figure 2.
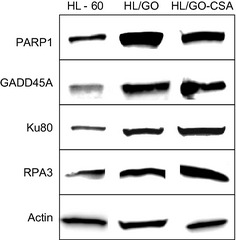
Protein expression levels of DNA repair proteins. Genes found to be altered in two gemtuzumab ozogamicin (GO)‐resistant subclones (HL/GO, HL/GO‐CSA) were evaluated for their protein expression levels by Western blotting.
GO‐induced DNA strand breaks quantitated using Comet assay
The induction of DNA strand breaks is a critical event for GO‐mediated cytotoxicity.13 The Comet assay has been successfully used to measure DNA strand breaks.21, 23, 24 After GO treatment, the HL‐60 cell tail moment values increased in a concentration‐dependent manner (Fig. 3A), indicating the production of DNA strand breaks by GO. Because DNA repair functions are complete within hours of the initiation of the DNA insult,21 the tail moments measured at 6 h would represent the final DNA damage after the capacity of DNA repair has been completed. After treatment with 10 μg/mL GO for 6 h, the tail moments of the HL/GO cells (24.2 ± 20.0, mean ± SD; P = 0.03, unpaired t‐test with two‐tailed analysis) and HL/GO‐CSA cells (24.96 ± 19.0, mean ± SD; P = 0.03, unpaired t‐test with two‐tailed analysis) were significantly smaller than that of HL‐60 cells (102.3 ± 30.7, mean ± SD) (Fig. 3B–E). These results indicated that GO‐induced DNA strand breaks were reduced in GO‐resistant variants.
Figure 3.
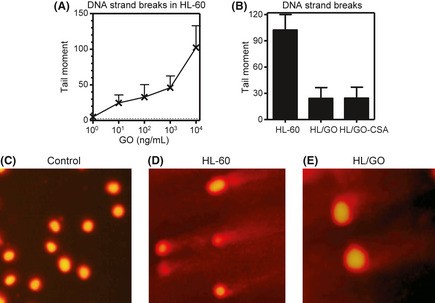
Gemtuzumab ozogamicin (GO)‐induced DNA strand breaks in human leukemia cells. (A) HL‐60 cells were incubated with various concentrations of GO for 6 h, followed by the determination of DNA strand breaks using the Comet assay. (B) Cells were incubated with 10 μg/mL GO for 6 h, followed by the determination of DNA strand breaks using the Comet assay. Typical Comet figures at 6 h (D, E) after HL‐60 cells (D) or HL/GO cells (E) had been treated with 10 μg/mL GO. (C) Control.
GO sensitivity of leukemic cells with P‐gp or MRP1 overexpression
ATP‐binding cassette transporters, especially P‐gp and MRP1, are reported to be associated with the cellular sensitivity to GO.15, 16, 17 The P‐gp‐overexpressing K562 variant K562/DNR19 (Fig. 4A) and MRP1‐overexpressing HL‐60 variant HL/Ara‐CDNR (Fig. 4B) were evaluated in the same setting. Both cell lines showed high CD33 positivity >70% (Fig. 4C), but were cross‐resistant to GO treatment (Table 3).
Figure 4.
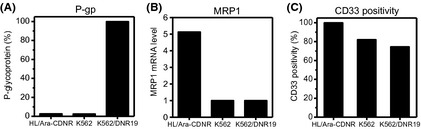
Multridrug resistance protein‐1 (MRP1)‐overexpressing HL‐60 human leukemia variant HL/Ara‐CDNR and the P‐glycoprotein (P‐gp)‐expressing K562 variant K562/DNR19. (A,B) Two efflux pumps were evaluated. P‐glycoprotein expression was quantitated using flow cytometry (A); MRP1 was determined using real‐time RT‐PCR with the value of HL‐60 cells set as 1 (B). (C) CD33 positivity determined by flow cytometry.
Table 3.
Drug sensitivity of human leukemic K562, daunorubicin (DNR)‐resistant K562 variant (K562/DNR19), and dual cytarabine and DNR‐resistant HL‐60 variant (HL/Ara‐CDNR) cells incubated with gemtuzumab ozogamicin (GO) or DNR
| Drug | IC50 | ||
|---|---|---|---|
| K562 | K562/DNR19 | HL/Ara‐CDNR | |
| GO (μg/mL) | 5.50 | ND | 3.70 |
| DNR (μM) | 0.20 | 5.60 | 0.10 |
Cells were incubated with various concentrations of GO or DNR for 72 h. IC50 values were then determined using the XTT assay. ND, not determined because the IC50 values were beyond the assay range due to the high degree of GO resistance.
Correlation between GO‐induced DNA strand breaks and GO sensitivity
The present study hypothesized that the cellular sensitivity to GO would be predicted by the amount of GO‐induced DNA strand breaks in leukemic cells. Using all the cell lines (HL‐60, HL/GO, HL/GO‐CSA, HL/Ara‐CDNR, K562), the tail moment determinants (the amount of DNA strand breaks) and the IC50 values were plotted within the same cell line. Figure 5(A) indicates a significant correlation between them (the K562/DNR19 cell line was excluded because its IC50 was beyond the detection range in Table 3). No other parameters, including CD33 positivity, P‐gp expression, and MRP1 mRNA, were proportional to GO sensitivity (Fig. 5B–D). The comparison was also made between the group of CD33‐positive/P‐gp‐negative/MRP1‐negative cell lines (HL‐60, K562) and the others (HL/GO, HL/GO‐CSA, HL/ara‐CDNR) (Fig. 5E), and there was no difference. The results suggested that the induction of DNA strand breaks would be a surrogate marker of GO‐mediated cytotoxicity.
Figure 5.
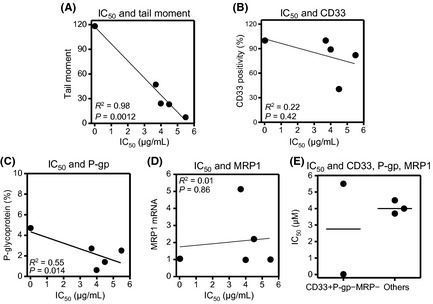
Relationship between gemtuzumab ozogamicin (GO)‐associated parameters and GO sensitivity. (A) Tail moment values (number of DNA strand breaks) and IC 50 values were plotted within the same cell line (HL‐60, HL/GO, HL/GO‐CSA, HL/Ara‐CDNR, K562). The IC 50 values were determined using the XTT assay. Tail moment values were determined by the Comet assay after cells had been treated with 10 μg/mL GO for 6 h. The values are the means of triplicate determinations. (B–D) Similarly, the correlation between the IC 50 values and each of the factors (CD33 positivity, P‐glycoprotein [P‐gp] expression, and multridrug resistance protein‐1 [MRP1] mRNA) were evaluated. (E) Comparison was made between the CD33‐positive/P‐gp‐negative/MRP1‐negative cell lines (HL‐60, K562) and the others (HL/GO, HL/GO‐CSA, HL/ara‐CDNR). Bars represent the means.
GO sensitivity in primary leukemic cells
A total of 11 patients' leukemic blast samples were similarly evaluated for CD33, the status of P‐gp and MRP, and GO‐induced DNA strand breaks and the subsequent apoptosis. The CD33 positivity varied among samples, and all of the samples were negative for P‐gp expression (Table 4). The MRP transcript levels were determined in only four samples and were all negative (Table 4). GO‐induced DNA strand breaks were also evaluated in nine samples, and the values varied widely (Fig. 6A). The extent of DNA strand breaks and the amount of apoptosis after GO treatment were not predicted by CD33 positivity (Fig. 6B,C). Importantly, a larger number of DNA strand breaks appeared to induce a greater amount of apoptosis (P = 0.07, Mann–Whitney U‐test) (Fig. 6D). These results suggested that the induction of DNA strand breaks appeared to be associated with GO sensitivity.
Table 4.
Patient characteristics
| Patient | Age, years/sex | Diagnosis | CD33, % | P‐gp, % | MRP | Apoptosis, % |
|---|---|---|---|---|---|---|
| 1 | 46/F | M3 | 97.7 | 2.3 | ND | 47 |
| 2 | 78/M | M6 | 53.9 | 2.8 | ND | ND |
| 3 | 36/M | M2 | 73.1 | 0.9 | ND | 10 |
| 4 | 88/M | M0 | 56.6 | 0.4 | – | 28 |
| 5 | 41/M | M2 | 71.8 | 0.7 | – | 58 |
| 6 | 63/M | MPD‐LT | 80.8 | 2.2 | ND | 10 |
| 7 | 71/M | MPD‐LT | 88.4 | 0.2 | ND | ND |
| 8 | 17/M | M1 | 99.4 | 0.9 | ND | ND |
| 9 | 70/M | M2 | 89.7 | 2.1 | ND | 48 |
| 10 | 71/M | M2 | 74.3 | – | – | ND |
| 11 | 75/M | M2 | 97.4 | – | – | 18 |
Flow cytometric analyses were carried out to detect the expression of CD33 and P‐glycoprotein (P‐gp) in patient samples (nos. 1–9). In nos.10 and 11, P‐gp was determined using real‐time RT‐PCR. Multidrug resistance protein (MRP) was evaluated for its transcript level using real‐time RT‐PCR in nos. 4, 5, 10, and 11. Apoptotic cell death was determined by Hoechst staining after the cells had been incubated with gemtuzumab ozogamicin (10 μg/mL) for 72 h. M0–6, French–American–British classification for acute leukemia; MPD‐LT, leukemic transformation from myeloproliferative disease; ND, not determined.
Figure 6.
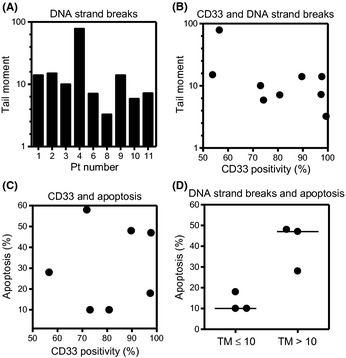
(A) Gemtuzumab ozogamicin (GO)‐induced DNA strand breaks in primary leukemic blasts. Nine samples were incubated with 10 μg/mL GO for 6 h, followed by the determination of DNA strand breaks using the Comet assay. (B,C) Relationship between CD33 positivity and GO‐induced DNA strand breaks (B) or the induction of apoptosis (C). CD33 positivity, the tail moment value, and the amount of apoptosis were plotted for the same sample. (D) GO‐induced apoptosis was compared between the patient sample group with GO‐induced tail moment (TM) ≤10 and the group with TM value >10.
Discussion
Because there are leukemia subsets that do benefit from the use of GO in the clinic, the present study was carrie out to determine cellular factors that would predict GO sensitivity. For this purpose, GO‐resistant leukemic cell lines were established (Fig. 1, Table 1) and investigated for their mechanisms of GO resistance. Previously, there was only one report that described the development of a GO‐resistant leukemic cell line and its characterization.32 However, this report revealed only a decrease in CD33 expression without any additional findings in the GO‐resistant subclone.32 Apart from this, two other studies provided insights into the mechanisms of GO resistance using cultured leukemic cell lines.33, 34 These reports showed the alteration of checkpoint kinases (Chk1/Chk2), caspase 3, or proapoptotic proteins. However, these findings were not obtained in cell lines that were established as GO‐specific resistant clones. Therefore, the present study is the first to investigate the mechanisms of GO‐specific resistance from various viewpoints.
The mechanism of resistance to a given anticancer agent is usually multifactorial. CD33 positivity and the transporters, previously well‐known factors, were evaluated in GO‐resistant cell lines (Fig. 1). The relationship between the CD33 level and GO cytotoxicity on AML blasts has been widely explored.14, 17, 25, 35, 36, 37 Usually, the levels of CD33 positivity did not closely correlate with the response to GO‐based chemotherapy.17 Here, CD33 positivity was reduced in the two GO‐resistant cell lines (Fig. 1B), but the expression level was not proportional to GO sensitivity (Fig. 5B). In the anti‐CD20 antibody rituximab, a similar antibody drug used in the treatment of malignant lymphoma, decreased CD20 positivity is one of the mechanisms of rituximab resistance. The mechanisms for the reduced CD20 antigen include genetic mutations and epigenetic changes within the CD20 coding region.38 The mechanisms of the reduction in CD33 positivity through the development of GO resistance was not elucidated in this study. However, it is speculated that the reduction in CD33 positivity might be mediated by similar mechanisms as those found in rituximab‐resistant lymphoma cells with reduced CD20 antigen. In terms of transporters, there is a general consensus that functional P‐gp‐mediated drug efflux inversely correlates with GO‐induced cytotoxicity.15, 16 The transporter MRP1 was also previously shown to attenuate GO cytotoxicity in vitro using samples from patients with AML.16 Here, P‐gp‐overexressing K562/DNR19 and MRP1‐overexpressing HL/Ara‐CDNR were highly resistant to GO (Table 3), but the expression levels of P‐gp and MRP1 were not in proportion to the cellular sensitivity to GO (Fig. 5C,D). Inhibition of MRP by the addition of MK571 was not shown mechanistically in the present study. However, the inhibitory effect of MK571 on the MRP efflux function has been widely used in published reports.39 Thus, the present study suggested that the decreased CD33 level and the presence of transporters contributed in part to the development of cellular GO resistance; nevertheless, there was no single determinant within them that would predict the sensitivity of leukemic cells to GO.
The microarray analysis identified upregulated factors that were associated with cellular responses to GO‐induced DNA strand breaks in both HL/GO cells and HL/GO‐CSA cells (Table 2). Among these, we focused on the DNA repair‐related factors, XRCC5 (Ku80), RPA, PARP1, and GADD45A, which were also upregulated at the protein level in these variants (Table 2, Fig. 2). Previous studies have shown that when calicheamicin cleaves purified DNA, it produces both double‐strand and single‐strand breaks. The double‐strand break:single‐strand break ratio in DNA was 1:1 to 1:3.13, 26 In eukaryotic cells, double‐strand breaks are repaired through two major pathways, non‐homologous end‐joining and homologous recombination.29, 30, 31 Ku80 and RPA are components of these DNA double‐strand break repairs.29, 30, 31, 40 Calicheamicin also produces DNA single‐strand breaks, which are usually repaired by excision repairs. GADD45A and PARP1 are specifically involved in DNA single‐strand break repair.27, 41, 42 Thus, these results suggested the participation of enhanced DNA repair functions in the mechanisms of GO resistance.
The HL/GO and HL/GO‐CSA cells were established by serial incubation of HL‐60 cells with GO followed by limiting dilution cloning. This followed an example of acquired resistance to GO. In the clinic, there may be not only acquired but also primary resistance to GO. To confirm GO's cytotoxicity in cells with primary GO resistance, we evaluated primary leukemic cell samples from patients. Using patients' leukemic cells, the induction of DNA strand breaks appeared to be associated with GO sensitivity, although the sample size was small (Table 4, Fig. 6). Moreover, K562/DNR19 cells, which were originally established to be DNR‐resistant,18 and HL/ara‐CDNR cells, which were originally established to be dual ara‐C‐ and DNR‐resistant,19 were both GO‐resistant with the reduction in GO‐induced DNA strand breaks (Fig. 4). This resistance was considered to be primary.
In the present study, the data suggests that CD33, P‐gp, and MRP are associated with the development of GO resistance. Nevertheless, even if given leukemic cells were highly positive for CD33 and negative for P‐gp expression and MRP expression, the drug sensitivity still varies among leukemia and is unpredictable (Fig. 5E). This is attributable to the contribution of other factors, including DNA repair. The induction of DNA strand breaks by GO is considered to be the end output of the sum of all the processes of CD33‐mediated internalization of the drug, efflux by transporters, and equilibrium between GO‐induced DNA damage and DNA repair response. This is why we have focused on the induction of DNA strand breaks. And, more to the point, we analyzed CD33 positivity, P‐gp, MRP1, DNA strand breaks, and GO sensitivity in primary cell samples from leukemic patients to confirm our hypothesis (Table 4, Fig. 6). Thus, it is suggested that the induction of DNA strand breaks is the best predictor for GO's efficacy.
For the advancement of cancer treatment, individualized chemotherapy is necessary, based on the understanding of the cellular biology of each patient's cancer cells at the molecular level. Clinical studies suggest that GO should be used in individualized regimens for specific AML subsets and not for all patients.8, 9, 10, 11 Sensitivity tests measuring GO‐induced DNA strand breaks may predict GO's clinical efficacy prior to treatment. GO‐based chemotherapy regimens can then be individualized for properly selected patients.
Disclosure Statement
The authors have no conflicts of interest.
Abbreviations
- Ara‐C
cytarabine
- CSA
cyclosporine A
- DNR
daunorubicin
- GADD45A
growth arrest and DNA damage‐45 alpha
- GO
gemtuzumab ozogamicin
- MRP
multidrug resistance protein
- PARP1
poly(ADP‐ribose) polymerase‐1
- P‐gp
P‐glycoprotein
Acknowledgments
We thank Ms. Noriko Shibata and Ms. Kanako Uzui for their technical assistance. This work was supported, in part, by a Grant‐in‐Aid from the ministry of Education, Science, and Culture of Japan (2007), a Grant from the Japanese Society of Clinical Pharmacology and Therapeutics (2007), and a Grant from Seeds for Advanced Medicine of University of Fukui (2008).
(Cancer Sci, 2012; 103: 1722–1729)
References
- 1. Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005; 106: 1154–63. [DOI] [PubMed] [Google Scholar]
- 2. Blum W, Marcucci G. New approaches in acute myeloid leukemia. Best Pract Res Clin Haematol 2008; 21: 29–41. [DOI] [PubMed] [Google Scholar]
- 3. Appelbaum FR, Gundacker H, Head DR et al Age and acute myeloid leukemia. Blood 2006; 107: 3481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fernandez HF. New trends in the standard of care for initial therapy of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2010; 56–61. [DOI] [PubMed] [Google Scholar]
- 5. Takeshita A, Ono R. Monoclonal antibody therapies for acute leukemia]. Rinsho Ketsueki 2002; 43: 353–6. [PubMed] [Google Scholar]
- 6. Bernstein ID, Singer JW, Andrews RG et al Treatment of acute myeloid leukemia cells in vitro with a monoclonal antibody recognizing a myeloid differentiation antigen allows normal progenitor cells to be expressed. J Clin Invest 1987; 79: 1153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bross PF, Beitz J, Chen G et al Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 2001; 7: 1490–6. [PubMed] [Google Scholar]
- 8. Petersdorf S, Kopecky K, Stuart RK et al Preliminary results of Southwest Oncology Group Study S0106: an international intergroup phase 3 randomized trial comparing the addition of gemtuzumab ozogamicin to standard induction therapy versus standard induction therapy followed by a second randomization to post‐consolidation gemtuzumab ozogamicin versus no additional therapy for previously untreated acute myeloid leukemia. Blood 2009; 114: 790. [Google Scholar]
- 9. Burnett AK, Hills RK, Milligan D et al Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol 2011; 29: 369–77. [DOI] [PubMed] [Google Scholar]
- 10. Clarke WT, Marks PW. Gemtuzumab ozogamicin: is there room for salvage? Blood 2010; 116: 2618–9. [DOI] [PubMed] [Google Scholar]
- 11. Ravandi F. Gemtuzumab ozogamicin: one size does not fit all–the case for personalized therapy. J Clin Oncol 2011; 29: 349–51. [DOI] [PubMed] [Google Scholar]
- 12. Ikemoto N, Kumar RA, Ling TT, Ellestad GA, Danishefsky SJ, Patel DJ. Calicheamicin‐DNA complexes: warhead alignment and saccharide recognition of the minor groove. Proc Natl Acad Sci USA 1995; 92: 10506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elmroth K, Nygren J, Mårtensson S, Ismail H, Hammarsten O. Cleavage of cellular DNA by calicheamicin γ1. DNA Repair 2003; 2: 363–74. [DOI] [PubMed] [Google Scholar]
- 14. Duong HK, Sekeres MA. Targeted treatment of acute myeloid leukemia in older adults: role of gemtuzumab ozogamicin. Clin Interv Aging 2009; 4: 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Linenberger ML, Hong T, Flowers D et al Multidrug‐resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001; 98: 988–94. [DOI] [PubMed] [Google Scholar]
- 16. Walter RB, Raden BW, Hong TC, Flowers DA, Bernstein ID, Linenberger ML. Multidrug resistance protein attenuates gemtuzumab ozogamicin–induced cytotoxicity in acute myeloid leukemia cells. Blood 2003; 102: 1466–73. [DOI] [PubMed] [Google Scholar]
- 17. Walter RB, Gooley TA, van der Velden VH et al CD33 expression and P‐glycoprotein‐mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007; 109: 4168–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Urasaki Y, Ueda T, Yoshida A et al Establishment of a daunorubicin‐resistant cell line which shows multi‐drug resistance by multifactorial mechanisms. Anticancer Res 1996; 16: 709–14. [PubMed] [Google Scholar]
- 19. Takemura H, Urasaki Y, Yoshida A, Fukushima T, Ueda T. Simultaneous treatment with 1‐beta‐d‐arabinofuranosylcytosine and daunorubicin induces cross‐resistance to both drugs due to a combination‐specific mechanism in HL60 cells. Cancer Res 2001; 61: 172–7. [PubMed] [Google Scholar]
- 20. Yamamoto S, Yamauchi T, Kawai Y et al Fludarabine‐mediated circumvention of cytarabine resistance is associated with fludarabine triphosphate accumulation in cytarabine‐resistant leukemic cells. Int J Hematol 2007; 85: 108–15. [DOI] [PubMed] [Google Scholar]
- 21. Yamauchi T, Keating MJ, Plunkett W. UCN‐01 inhibits DNA repair and induces cytotoxicity in normal lymphocytes and chronic lymphocytic leukemia lymphocytes. Mol Cancer Ther 2002; 1: 287–94. [PubMed] [Google Scholar]
- 22. Negoro E, Yamauchi T, Urasaki Y, Nishi R, Hori H, Ueda T. Characterization of cytarabine‐resistant leukemic cell lines established from five different blood cell lineages using gene expression and proteomic analyses. Int J Oncol 2011; 38: 911–9. [DOI] [PubMed] [Google Scholar]
- 23. Olive PL, Banath JP, Durand RE. Heterogeneity in radiation‐induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res 1990; 122: 86–94. [PubMed] [Google Scholar]
- 24. Singh NP, McCoy HT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175: 184–91. [DOI] [PubMed] [Google Scholar]
- 25. Jawad M, Seedhouse C, Mony U, Grundy M, Russell NH, Pallis M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia 2010; 24: 74–80. [DOI] [PubMed] [Google Scholar]
- 26. Dedon PC, Salzberg AA, Xu J. Exclusive production of bistranded DNA damage by calicheamicin. Biochemistry 1993; 32: 3617–22. [DOI] [PubMed] [Google Scholar]
- 27. Shall S, de Murcia G. Poly(ADP‐ribose) polymerase‐1: what have we learned from the deficient mouse model? Mutat Res 2000; 460: 1–15. [DOI] [PubMed] [Google Scholar]
- 28. Rosemary Siafakas A, Richardson DR. Growth arrest and DNA damage‐45 alpha (GADD45alpha). Int J Biochem Cell Biol 2009; 41: 986–9. [DOI] [PubMed] [Google Scholar]
- 29. Bolderson E, Richard DJ, Zhou B‐BS, Khanna KK. Recent Advances in Cancer Therapy Targeting Proteins Involved in DNA Double‐Strand Break Repair. Clin Cancer Res 2009; 15: 6314–20. [DOI] [PubMed] [Google Scholar]
- 30. Hartlerode AJ, Scully R. Mechanisms of double‐strand break repair in somatic mammalian cells. Biochem J 2010; 423: 157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rassool FV, Tomkinson AE. Targeting abnormal DNA double strand break repair in cancer. Cell Mol Life Sci 2010; 67: 3699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cianfriglia M, Mallano A, Ascione A, Dupuis ML. Multidrug transporter proteins and cellular factors involved in free and mAb linked calicheamicin‐A1 (gentuzumab ozogamicin, GO) resistance and in the selection of GO resistant variants of the HL60 AML cell line. Int J Oncol 2010; 36: 1513–20. [DOI] [PubMed] [Google Scholar]
- 33. Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J. Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: role of Chk1 and Chk2 phosphorylation and caspase 3. Blood 2003; 101: 4589–97. [DOI] [PubMed] [Google Scholar]
- 34. Haag P, Viktorsson K, Lindberg ML, Kanter L, Lewensohn R, Stenke L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin‐induced apoptotic cell death in AML. Exp Hematol 2009; 37: 755–66. [DOI] [PubMed] [Google Scholar]
- 35. Jilani I, Estey E, Huh Y et al Differences in CD33 intensity between various myeloid neoplasms. Am J Clin Pathol 2002; 118: 560–6. [DOI] [PubMed] [Google Scholar]
- 36. Walter RB, Raden BW, Kamikura DM, Cooper JA, Bernstein ID. Influence of CD33 expression levels and ITIM‐dependent internalization on gemtuzumab ozogamicin‐induced cytotoxicity. Blood 2005; 105: 1295–302. [DOI] [PubMed] [Google Scholar]
- 37. Morris KL, Adams JA, Liu JA. Effect of Gemtuzumab Ozogamicin on acute myeloid leukemia blast cells in vitro, as a single agent and combined with other cytotoxic cells. Br J Haematol 2006; 135: 509–12. [DOI] [PubMed] [Google Scholar]
- 38. Duman BB, Sahin B, Ergin M, Guvenc B. Loss of CD20 antigen expression after rituximab therapy of CD20 positive B cell lymphoma (diffuse large B cell extranodal marginal zone lymphoma combination): a case report and review of the literature. Med Oncol 2012; 29: 1223–6. [DOI] [PubMed] [Google Scholar]
- 39. Chen ZS, Furukawa T, Sumizawa T et al ATP‐dependent efflux of CPT‐11 and SN‐38 by the multidrug resistance protein(MRP) and its inhibition by PAK‐104P. Mol Pharmacol 1999; 55: 921–8. [PubMed] [Google Scholar]
- 40. Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non‐homologous DNA end‐joining. Nat Rev Mol Cell Biol 2003; 4: 712–20. [DOI] [PubMed] [Google Scholar]
- 41. Hollander MC, Fornace AJ Jr. Genomic instability, centrosome amplification, cell cycle checkpoints and Gadd45a. Oncogene 2002; 21: 6228–33. [DOI] [PubMed] [Google Scholar]
- 42. Audebert M, Salles B, Calsou P. Involvement of poly(ADP‐ribose) polymerase‐1 and XRCC1/DNA ligase III in an alternative route for DNA double‐strand breaks rejoining. J Biol Chem 2004; 279: 55117–26. [DOI] [PubMed] [Google Scholar]