

Abstract
This study aimed to determine the expression pattern of dickkopf‐1 (Dkk1), a potent inhibitor of Wnt signaling, in colon cancer and to assess the function and mechanism of Dkk1 in tumor progression in vitro and in vivo. We detected the protein expression of Dkk1 and some epithelial‐mesenchymal transition (EMT)‐associated markers (E‐cadherin, vimentin and β‐catenin) in 217 tissue samples of human colon cancer, upregulated Dkk1 expression in HCT116 colon cancer cells, and established a nude mouse xenograft model. Dkk1 protein overexpression was inversely related to tumor grade and the presence of metastasis and recurrence of colon cancer. Notably, the expression of Dkk1 was concomitant with reduced immunohistochemical features of EMT (e.g. increased expression of epithelial marker E‐cadherin, decreased expression of mesenchymal marker vimentin, and cytoplasmic distribution of β‐catenin). Furthermore, Dkk1 overexpression resulted in restoration of the epithelial phenotype, decreased expression of EMT transcription factors Snail and Twist, and decreased expression of markers suggestive of intestinal stem cells (e.g. cluster of differentiation 133 [CD133] and leucine‐rich‐repeat‐containing G‐protein‐coupled receptor 5 [Lgr5]). Functional analysis showed overexpression of Dkk1 reduced proliferation, migration, and invasion of colon cancer cells. Moreover, upregulation of Dkk1 led to decreased tumor‐initiating ability and suppressed colon tumor growth in nude mice. Our findings indicate that Dkk1 can suppress the progression of colon cancer, possibly through EMT inhibition, and could therefore serve as a target for tumor therapy. (Cancer Sci 2012; 103: 828–835)
The Wnt/β‐catenin pathway has a well‐established role in colorectal oncogenesis for driving proliferation and dedifferentiation in 90% of colorectal cancers.1 In the Wnt/β‐catenin signaling, Wnt binding to frizzled receptors and low‐density lipoprotein receptor‐related protein (LRP)5/6 co‐receptors leads to cytosolic stabilization and accumulation of β‐catenin. Then, β‐catenin translocates to the nucleus and forms a complex with lymphoid enhancer factor (LEF)/T cell factor proteins (TCF) family members, thereby regulating the expression of an extensive array of target genes involved in embryogenesis and tissue homeostasis.2, 3 An increasing body of evidence indicates that Wnt/β‐catenin signaling also contributes to epithelial‐mesenchymal transition (EMT),4, 5 which is a critical step in tumor progression. Activated Wnt/β‐catenin signals lead to translocation of β‐catenin into the nucleus and breakdown of cell–cell adhesion formed by β‐catenin and E‐cadherin. In particular, Wnt/β‐catenin signaling pathways have been reported to activate the transcriptional regulators of EMT,6, 7, 8, 9 such as Snail, Slug, Twist, and Zeb1.
Five classes of secreted antagonists of the Wnt pathway have been identified:10 the secreted frizzled‐related protein family, Wnt inhibitory factor 1, Xenopus cerberus, Wise, and the Dkk (Dickkopf) family. The Dkk genes function by binding to LRP5/6 and Kremen proteins, thus inducing LRP endocytosis and preventing the signal cascade. The Dkk family consists of Dkk1, Dkk2, Dkk3, Dkk4, and soggy. In particular, Dkk1 plays essential roles in vertebrate development, including head induction, bone formation, and limb patterning.11, 12, 13, 14 Also, Dkk1 has been reported to be induced by Wnt signals as a component of a negative feedback loop in normal tissues.15 In colorectal cancer, the autoregulatory mechanism of Dkk1 might be lost or abolished by epigenetic inactivation, and Dkk1 restoration in DLD‐1 colon cancer cells reduces colony formation and tumor growth in xenografts,16 suggesting that the Dkk1 gene plays a tumor suppressor function in colorectal cancer. Further investigation into the mechanisms involved in the functional roles of Dkk1 is necessary.
In this study, we evaluated the clinicopathological significance of Dkk1. We analyzed the correlation between Dkk1 expression and immunohistochemical features of EMT in tissue specimens from 217 colon cancer patients. The effects of ectopic expression of Dkk1 in colon cancer cell line HCT116 on the expression of epithelial markers and mesenchymal markers both in vivo and in vitro were studied. Changes in expression of EMT transcription factors and intestinal stem cell‐associated markers in response to Dkk1 overexpression were determined. We investigated cell proliferation, motility, and invasiveness in cell cultures with Dkk1 overexpression as well as tumor growth and tumor‐initiating ability in a colon cancer xenograft model.
Materials and Methods
Clinical samples
Tissue samples of colon cancer were harvested from 217 patients who had undergone surgery for colon cancer in Tianjin Medical University Cancer Institute and Hospital (Tianjin, China) between January 2002 and December 2004. None of the patients had received any chemotherapy or radiotherapy before their operation. Data of clinicopathological parameters were obtained from patients' clinical records and pathological reports.
Cell culture reagents and animals
Human colon cancer cell line HCT116 was obtained from the Cell Resource Center at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences/Peking Union Medical College (Beijing, China). Cells were cultured in Iscove modified Dulbecco medium with 10% FBS. The micro‐Boyden chambers were from NeuroProbe (Gaithersburg, MD, USA). Antibodies to Dkk1, β‐catenin, keratin 18, Lgr5, goat anti‐rabbit and goat anti‐mouse IgG‐FITC were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to fibronectin, Snail, Slug and Twist were from Abcam (Cambridge, UK). Antibody to CD133 was from Miltenyi Biotechnology (Auburn, CA, USA). Antibody to E‐cadherin was from BD Biosciences (San Jose, CA, USA). Antibody to vimentin was from Epitomics (Burlingame, CA, USA). Alexa Fluor 488 and 546 were from Molecular Probes (Eugene, OR, USA). BALB/C nude mice (4–5 weeks old) were obtained from Wei Tong Li Hua Experimental Animal Company (Beijing, China).
Immunohistochemical staining
The streptavidin‐biotin‐peroxidase staining method was described previously.17 Briefly, the sections were pretreated with microwaves, blocked, and incubated with a series of antibodies overnight at 4°C. Then, they were immunostained with HRP‐conjugated antibody, and the signals were revealed, with 3,3′‐diaminobenzidine buffer used as the substrate. In place of the primary antibodies for the negative control, PBS was used. The results were evaluated by assessing staining intensity according to the method described by Bittner et al.18
Plasmid transfection
Transfection with plasmid carrying Dkk1 and controlled scrambled plasmid (Genechem, Shanghai, China) was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. To establish stable HCT116 cells that overexpressed Dkk1, the G418‐resistant cells were screened.
Western blot analysis
To examine extracellular Dkk1 expression, the culture medium and the supernatant of harvested cells were collected and concentrated with a centrifugal filtering unit (Microcon YM‐30; Millipore, Billerica, MA, USA). The protein content was determined with a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA) so that a consistent amount of protein could be taken from the different samples. Protein (30–50 μg/lane) was separated by 10% SDS‐PAGE and transferred to polyvinylidene difluoride membranes. Blots were blocked and incubated with primary antibodies overnight at 4°C, incubated with secondary antibody, and detected with ECL Western blotting substrate (Millipore) according to the manufacturer's instructions.
Cell proliferation assay
Cells were separately seeded into 96‐well plates at 1 × 103/well and incubated for different periods (1, 2, 3, 4, or 5 days). After 4 h incubation at 37°C, MTT (Sigma‐Aldrich, St. Louis, MO, USA) solution was added. The purple crystals were dissolved by DMSO. The optical density was determined at 490 nm by on Spectra Max M2 (Molecular Devices, Sunnyvale, CA, USA).
Soft agar colony formation assay
To form the bottom agar, 1.5‐mL culture medium containing 0.6% agarose was added to each 35‐mm dish. Then, 1‐mL culture medium containing 0.6% agarose and 10 000 cells were mixed gently at 37°C and plated onto the bottom agar. Dishes were incubated at 37°C and 5% CO2. Six days later, colonies (>50 μm) were counted from 10 random fields in each dish.
Wound healing assay
Cells were plated in 35‐mm dishes to form a monolayer 1 day before assay. After making a straight scratch with a pipette tip, cells were incubated in a minimum medium (containing 0.5% FBS) at 37°C with 5% CO2. Cell motility was assessed by measuring the speed of wound closure at intervals.
Matrigel invasion assay
Matrigel (BD Biosciences) with a final concentration of 1.5 mg/mL was added to the upper surface of chamber filter (8‐μm pore). Next, 200‐μL cell suspension (5 × 105 cells/mL) contained in serum‐free medium was added to upper chamber, and 300‐μL culture medium supplemented with 20% FBS was added to the lower chamber. After incubation for 48 h, the passed cells were fixed and stained.
Immunofluorescence confocal microscopy
The cells were cultured on sterile glass cover slips on the day before staining. Cells were fixed with 4% paraformaldehyde, quenched with 50‐mmol/L NH4Cl, permeabilized in 0.2% Triton X‐100, and blocked in 3% BSA. The slips were incubated with the primary antibodies overnight at 4°C, labeled with the specific secondary antibodies for 1 h in the dark, mounted, and visualized with a confocal laser scanning microscopy (Leica TCS SP5, Leica Microsystems, Wetzlar, Germany).
Flow cytometry assay
With CD133 antibody, 2 × 105 cells were incubated for 30 min on ice, washed, and further incubated with Alexa Fluor 488 for 30 min on ice in the dark. Following two washings with ice‐cold PBS, cells were analyzed on a flow cytometer with CellQuest software (BD Biosciences, San Jose, CA, USA).
In vivo assay
Twenty mice were divided randomly and evenly into two groups and received either 3 × 106 control or HCT116 cells overexpressing Dkk1 (clone5) by subcutaneous injection in right groin. The tumor size was measured every 3 days for 21 days. Tumor volumes were calculated using the following formula: Volume = (Length [mm] × Width2 [mm2])/2. Tumor samples were formalin fixed and paraffin embedded.
To assess the effect Dkk1 on tumor initiation, we divided another 40 nude mice evenly into eight groups and subcutaneously injected aliquots of 104, 105, and 106 control or HCT116 cells overexpressing Dkk1 (clone5) in the right groin. The tumor incidence was monitored every day.
Statistical analysis
SPSS v.16.0 software (SPSS Inc., Chicago, IL, USA) was used for data analysis. The associations between Dkk1 and clinicopathologic parameters and the differential expression of E‐cadherin, vimentin and β‐catenin between the Dkk1‐positive and Dkk1‐negative groups were assessed with Fisher's exact test and chi‐square test. Differences or correlations between the two groups were assessed by the Mann–Whitney U‐test, Student's t‐test and Pearson's correlation test. Significance was set at P < 0.05.
Results
Association of Dkk1 expression with clinicopathological features of colon cancer cases
Among 217 samples, 131 (60.4%) showed positive Dkk1 expression, while the remaining 86 (39.6%) were negative (Fig. 1a). Tumors were categorized as positive or negative for Dkk1. The relationships between Dkk1 expression in colon cancer and any of the clinicopathologic parameters were analyzed separately (Table 1). The frequency of positive Dkk1 protein expression in Stage I, Stage II, Stage III, and Stage IV colon cancer tissue was 70.0% (7⁄10), 66.7% (92⁄138), 52.6% (30⁄57), and 16.7% (2⁄12), respectively. In 32 of 73 (43.8%) colon cancer specimens from patients with metastasis and three of 11 (27.3%) patients with recurrence, Dkk1 protein expression was detected; these rates were significantly lower than in cases without metastasis (99/144, 68.8%) and without recurrence (128/206, 62.1%). No significant correlation between Dkk1 expression and gender, age, tumor size, and histological differentiation were observed.
Figure 1.

(a) Representative Dkk1‐positive colon cancer samples with staining mainly in the cytoplasm (left) and Dkk1‐negative colon cancer samples showing almost no appreciable staining (right), ×400. (b) E‐cadherin expression was higher in Dkk1‐positive colon cancer tissue sections than in Dkk1‐negative samples. E‐cadherin is mainly localized in the membrane of Dkk1‐positive samples (red arrows) and in the cytoplasm of Dkk1‐negative samples (black arrows). Tumor cells in the Dkk1‐positive section did not express vimentin, whereas some tumor cells in the Dkk‐1‐negative section expressed vimentin. In Dkk1‐positive sections, tumor cells displayed only membranous localization of β‐catenin (red arrows), whereas tumor cells in the Dkk1‐negative section showed nuclear β‐catenin accumulation (black arrows), ×400. Dkk1, Dickkopf‐1; EMT, epithelial‐mesenchymal transition.
Table 1.
Correlation between Dickkopf‐1 and clinicopathologic characteristics of patients with colon cancer
| Variable | Total (n) | Dkk1 expression | χ2 | P‐value | |
|---|---|---|---|---|---|
| Positive (n [%]) | Negative (n [%]) | ||||
| Age | |||||
| <45 | 28 | 13 (46.4) | 15 (53.6) | 2.611 | 0.146 |
| ≥45 | 189 | 118 (62.4) | 71 (37.6) | ||
| Sex | |||||
| Men | 101 | 62 (61.4) | 39 (38.6) | 0.082 | 0.775 |
| Women | 116 | 69 (59.5) | 47 (40.5) | ||
| Tumor size | |||||
| ≥5cm | 147 | 94 (63.9) | 53 (36.1) | 2.437 | 0.138 |
| <5cm | 70 | 37 (52.9) | 33 (47.1) | ||
| Histological differentiation | |||||
| Well differentiated | 14 | 11 (78.6) | 3 (21.4) | 5.643 | 0.06 |
| Moderately differentiated | 109 | 71 (65.1) | 38 (34.9) | ||
| Poorly differentiated | 94 | 49 (52.1) | 45 (47.9) | ||
| Clinical stage | |||||
| Stage I | 10 | 7 (70.0) | 3 (30.0) | 13.681 | 0.003a |
| Stage II | 138 | 92 (66.7) | 46 (33.3) | ||
| Stage III | 57 | 30 (52.6) | 27 (47.4) | ||
| Stage IV | 12 | 2 (16.7) | 10 (83.3) | ||
| Metastasis | |||||
| Present | 73 | 32 (43.8) | 41 (56.2) | 12.568 | 0.001a |
| Absent | 144 | 99 (68.8) | 45 (31.2) | ||
| Recurrence | |||||
| Present | 11 | 3 (27.3) | 8 (72,3) | 5.305 | 0.028a |
| Absent | 206 | 128 (62.1) | 78 (37.9) | ||
P < 0.05.
Expression of Dkk1 is concomitant with decreased EMT immunohistochemical features
To assess the relationship between Dkk1 and EMT in colon cancer, we investigated the expression of several EMT‐associated markers. As shown in Table 2 and Figure 1(b), the Dkk1‐positive group showed higher E‐cadherin expression and lower vimentin and nuclear β‐catenin expression than the negative group. In the Dkk1‐positive group, β‐catenin was expressed mainly in cytoplasm, whereas it was distributed in the nucleus in the negative group. The expression of Dkk1 correlated with the expression of E‐cadherin (r = 0.291, P < 0.0001), vimentin (r = −0.313, P < 0.0001), and β‐catenin (nuclear) (r = −0.173, P < 0.01). These findings confirm that Dkk1 has an inhibitory effect on EMT.
Table 2.
Correlation between expression of Dickkopf‐1 and epithelial‐mesenchymal transition‐associated proteins
| Variable | Total (n) | DKK‐1 expression | χ2 | P‐value | |
|---|---|---|---|---|---|
| Positive (n [%]) | Negative (n [%]) | ||||
| E‐cadherin expression | |||||
| Positive | 200 | 130 (65.0) | 70 (35.0) | 22.888 | <0.001a |
| Negative | 17 | 1 (5.9) | 16 (94.1) | ||
| Vimentin expression | |||||
| Positive | 18 | 1 (5.6) | 17 (94.4) | 24.649 | <0.001a |
| Negative | 199 | 130 (65.3) | 69 (34.7) | ||
| β‐catenin nuclear expression | |||||
| Positive | 42 | 112 (64.0) | 63 (36.0) | 4.983 | 0.026b |
| Negative | 175 | 19 (45.2) | 23 (54.8) | ||
P < 0.001.
P < 0.05.
Overexpression of Dkk1 induces restoration of epithelial phenotype and decreased expression of Snail and Twist in HCT116 cells
Because the HCT116 cell line is from poorly differentiated colorectal cancer tissue and exhibits a mesenchymal phenotype, we established stable Dkk1 that overexpressed colon cancer cells to study the EMT‐reversal effect of Dkk1 on colorectal cancer cells. The chosen stable Dkk1‐overexpressing clones were clone5 and clone13. In order to rule out the clone‐to‐clone variations, we selected two clones in this study. In HCT116 cells, Dkk1 overexpression, had decreased expression of c‐myc and cyclin D1 (Fig. 2a), which are the best‐known target genes of Wnt/β‐catenin signaling,19, 20 confirming that canonical Wnt signaling and transcriptional activity of TCF/LEF were inhibited.
Figure 2.

(a) Extracellular and intracellular Dkk1 protein levels were significantly increased in clone5 and clone13, HCT116 cell pools transfected with Dkk1 plasmid. (b) Cells overexpressing Dkk1 exhibited dramatic changes in cell morphology from a scattered, spindle, fibroblastic‐like shape to a tightly packed, cuboid, epithelial‐like appearance, thus indicating EMT reversal. (c) The expression of epithelial and mesenchymal proteins was examined by immunoblotting with β‐actin as loading control. Cells overexpressing Dkk1 had a higher expression of E‐cadherin and keratin 18 but a lower expression of vimentin, fibronectin and β‐catenin. (d) Immunofluorescent staining of E‐cadherin, vimentin, and β‐catenin. Cells overexpressing Dkk1 had a higher expression of E‐cadherin and β‐catenin but alower expression of vimentin. Less β‐catenin accumulation in the nucleus was observed in cells overexpressing Dkk1 than in control cells. A green or red signal represents staining for the corresponding protein, while a blue signal represents nuclear DNA staining by DAPI. (e) Expression of EMT regulatory proteins, including Zeb1, Slug, Snail, and Twist, was examined by immunoblotting. Snail and Twist were downregulated in cells overexpressing Dkk1 compared with control cells. Clone5 and clone13, HCT116 cells overexpressing Dkk1; Dkk1, Dickkopf‐1; EMT, epithelial‐mesenchymal transition.
In HCT116 cells, Dkk1 overexpression led to morphologic changes from scattered to tightly packed colonies (Fig. 2b). Western blotting and immunofluorescence assays demonstrated cells overexpressing Dkk1 had a higher expression of E‐cadherin, keratin 18, and β‐catenin and a lower expression of vimentin and fibronectin compared with the control cells (Fig. 2c,d). Moreover, less β‐catenin accumulated in the nucleus of cells overexpressing Dkk1 than in that of control cells. These findings suggest that cells overexpressing Dkk1 are more predisposed to epithelial differentiation.
In addition to the classical EMT markers, we checked the expression of a set of EMT transcription factors, including Snail, Slug, Twist, and Zeb1. These markers could repress E‐cadherin expression via direct binding to the E‐boxes of the E‐cadherin promoter. Among them, Snail and Twist were downregulated in cells overexpressing Dkk1 compared with control cells (Fig. 2e).
Overexpression of Dkk1 impairs migration and invasion of HCT116 cells
Compared with epithelial cells, mesenchymal cells generally do not exhibit direct cell–cell contact, defined cell polarity, defined cell–ECM interactions, or cytoskeletal structures. Thus, the process of EMT can directly promote migration and invasion.
As expected, cells overexpressing Dkk1 moved slowly towards the gap compared with control cells (Fig. 3a). Significantly fewer cells overexpressing Dkk1 invaded through the Matrigel than control cells (Fig. 3b). Our findings suggest that Dkk1 upregulation leads to decreased migratory and invasive potential of HCT116 cells.
Figure 3.

Effect of Dkk1 overexpression on motility and invasion abilities of HCT116 cells. (a) Images of wound healing assay of control and HCT116 cells transfected with Dkk1 at 0 and 24 h (left), ×200. Migration distances at different times in the wound healing assay (right). Cells overexpressing Dkk1 moved slowly towards the gap compared to control cells. (b) Cells invading through the Matrigel‐coated transwell inserts were stained (left), ×200. The invading cells were counted in five predetermined fields (right),×400. Significantly fewer cells overexpressing Dkk1 invaded through the Matrigel relative to control cells. Clone5 and clone13, HCT116 cells overexpressing Dkk1; Dkk1, Dickkopf‐1; EMT, epithelial‐mesenchymal transition.
Overexpression of Dkk1 inhibits HCT116 growth in monolayer cultures and anchorage‐independent growth in soft agar
Significant differences in growth between cells overexpressing Dkk1 and control cells were observed. In the MTT cell proliferation assay, suppression of growth was observed as early as the third day in cells overexpressing Dkk1 (Fig. 4a). Anchorage‐independent growth, one of the most important malignant features of cancer cells, was also found to be significantly reduced in cells overexpressing Dkk1 (Fig. 4b).
Figure 4.

Overexpression of Dkk1 inhibits HCT116 growth in monolayer cultures and anchorage‐independent growth in soft agar. (a) MTT cell proliferation assay showed suppression of cell growth in cells overexpressing Dkk1. (b) Anchorage‐independent growth was significantly reduced in cells overexpressing Dkk1. Colonies in soft agar culture were stained (left), ×200. Histogram showing colony formation efficiency (right). Clone5 and clone13, HCT116 cells overexpressing Dkk1; Dkk1, Dickkopf‐1; EMT, epithelial‐mesenchymal transition; OD, optical density.
Dickkopf‐1 inhibits in vivo tumor growth of HCT116 cells in a xenograft mouse model, induces the expression of E‐cadherin, and decreases the expression of vimentin and β‐catenin in tumor tissues
In agreement with in vitro findings, clone5 cells overexpressing Dkk1 grew into smaller tumor masses than control cells (Fig. 5a). Of the 10 mice injected with control cells, two showed tumor invasion into skeletal muscle whereas no metastasis sites were detected in mice injected with clone5 cells. Immunohistochemical analysis showed that tumor sections from Dkk1‐transfected HCT116 cells exhibited a marked increase in E‐cadherin whereas tumor sections from control cells revealed no or weak staining. In contrast, the expression of vimentin and β‐catenin was significantly decreased in Dkk1 tumors compared with control tumors (Fig. 5b).
Figure 5.
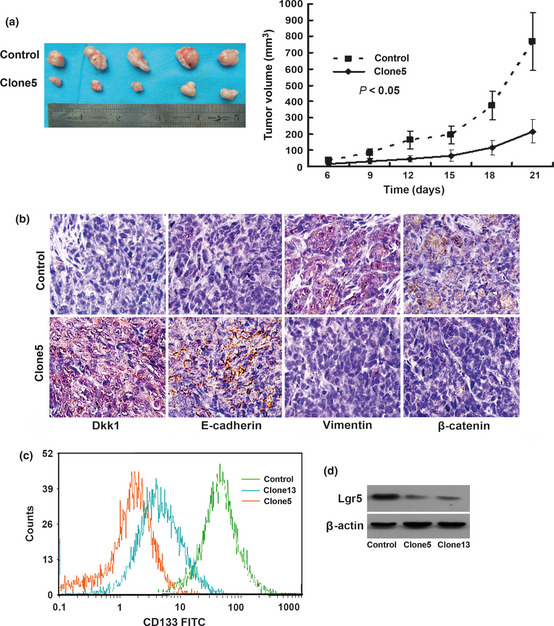
(a) Photograph of representative tumors from mice injected with control or HCT116 cells transfected with Dkk1 (left). Cells overexpressing Dkk1 produced smaller tumor masses than control cells (right). (b) Immunohistochemical staining of Dkk1, E‐cadherin, vimentin, and β‐catenin expression in harvested mouse tumor samples. Tumors overexpressing Dkk1 exhibited increased E‐cadherin expression and decreased vimentin and β‐catenin expression compared with control tumors. (c) CD133 levels measured by flow cytometry. Lgr5 expression was reduced in cells overexpressing Dkk1. (d) Expression of Lgr5 examined by immunoblotting. Lgr5 expression was reduced in cells overexpressing Dkk1. CD133, cluster of differentiation 133; Clone5 and clone13, HCT116 cells overexpressing Dkk1; Dkk1, Dickkopf‐1; EMT, epithelial‐mesenchymal transition; Lgr5, leucine‐rich‐repeat‐containing G‐protein‐coupled receptor 5.
Overexpression of Dkk1 inhibits tumor‐initiating capability and decreases expression of stem cell‐associated markers CD133 and Lgr5 in HCT116 cells
Previous reports have shown that cancer cells undergoing EMT have properties similar to stem‐like cells, such as increased self‐renewal and tumor‐initiating capabilities. Thus, these cancer cells possess greater malignant potential.21, 22
As expected, Dkk1 overexpression significantly decreased the number of tumor‐initiating HCT116 cells. At least 105 control cells were required to initiate tumor formation (Table 3). Of the five mice injected with 105 control cells, one formed a tumor. In contrast, no tumor mass arose when an equal number of clone5 cells were injected into mice. All five mice injected with 5 × 105 or 106 control cells formed tumors, whereas between two and four of the five mice injected with an equal number of clone5 cells formed tumors.
Table 3.
Tumor incidence of Dkk1/HCT116 and control/HCT116 cells injected into host mice
| Cells injected | Tumors incidence/number of injections | |||
|---|---|---|---|---|
| 104 | 105 | 5 × 105 | 106 | |
| Dkk1/HCT116 | 0/5 | 0/5 | 2/5 | 4/5 |
| Control/HCT116 | 0/5 | 1/5 | 5/5 | 5/5 |
Dkk1, Dickkopf‐1.
Surface CD133 expression was enhanced in the subpopulation of human colorectal cancer cells with tumor‐initiating properties,23 And Lgr5 has been identified as a potential intestinal stem cell marker that is expressed in a limited number of columnar cells at the crypt base.24 Surface expression of CD133 and expression of Lgr5 were measured separately with flow cytometry and Western blot (Fig. 5c,d). The CD133‐positive expression ratio of cells overexpressing Dkk1 was significantly less than that of control cells (45.6 ± 4.3% and 49.2 ± 4.0% vs 65.8 ± 3.8%, P < 0.05). Compared with control cells, cells overexpressing Dkk1 showed decreased Lgr5 expression.
Discussion
Epithelial‐mesenchymal transition is a transdifferentiation process by which epithelial cells shed their epithelial characteristics and acquire more migratory mesenchymal cell‐like properties. The resulting phenotype after EMT is suitable for tumor migration, invasion, and dissemination, thus facilitating tumor progression. Moreover, cancer cells undergoing EMT share molecular and functional characteristics with cancer stem‐like cells, such as tumor‐initiating abilities and therapeutic resistance.21, 22 Identification of EMT inhibitors shows promise for the identification of novel cancer treatment options. Although the effect of Wnt signaling in inducing EMT during physiological or pathological processes has been extensively studied,4, 25, 26, 27 the present study is the first to demonstrate the ability of Dkk1, a potent Wnt signaling inhibitor, to reverse EMT in colon cancer.
We initially investigated Dkk1 protein levels in 217 colon cancer tissue samples. We found that Dkk1 expression is inversely correlated with tumor stage and the presence of metastasis and recurrence. In Dkk1‐positive samples, we observed higher levels of E‐cadherin expression and lower levels of vimentin expression and β‐catenin nuclear distribution than in Dkk1‐negative samples. These findings suggest that the loss of Dkk1 in colon cancer may contribute to its progression and that Dkk1 is possibly associated with a reversal of the EMT process.
The transition between epithelial and mesenchymal cell types is characterized by cellular morphology, functional and behavioral phenotype, and the expression of differentiation markers. After isolation of stable clones, cells overexpressing Dkk1 were morphologically more epithelial‐like and less mesenchymal‐like than control cells. In accordance with the morphological changes, vimentin, β‐catenin, and fibronectin were downregulated and E‐cadherin and keratin 18 were upregulated in colon cancer cells transfected with Dkk1. We further demonstrated that Dkk1 could downregulate the expression of Snail and Twist, two transcription factors considered to be key regulators of EMT. Moreover, we investigated alterations in cell behavior and found that Dkk1 can significantly impair the proliferation, invasion, and migration abilities of colon cancer cells. We also found that Dkk1 has significant inhibitory effects on tumor growth in nude mice. Previous reports have indicated that cancer cells undergoing EMT share the properties of stem cell‐like cells and, thus, have more malignant properties.19, 20 In the present study, we found that cancer stem cell‐like traits (e.g. tumor‐initiating capabilities, expression of intestinal stem cell‐associated proteins CD133 and Lgr5) were significantly decreased in cells overexpressing Dkk1. This study's findings demonstrate that ectopic expression of Dkk1 exerts EMT reversing effects, thereby suppressing the progression of colon cancer.
β‐catenin is the central molecule of Wnt signaling. Various downstream effectors of β‐catenin/TCF involve versatile functional modules of Wnt signaling responsible for cell proliferation, dedifferentiation,3, 28 inhibition of apoptosis, and tumor progression.29 Nuclear β‐catenin has been shown to induce EMT and is used as a mesenchymal marker.30, 31 Accounting for low levels of Wnt signaling activity, Dkk1 overexpression decreases the expression and intracellular distribution of β‐catenin as well as the expression of c‐myc and cyclin D1. Furthermore, Snail, Twist, CD133, and Lgr5 have been reported to be Wnt/β‐catenin signaling‐responsive proteins.7, 9, 32, 33 Thus, decreased Wnt/β‐catenin may account for tumor suppression, cell differentiation, and other phenotypic changes. The EMT reversal effect caused by Dkk1 in colon cancer may be, at least in part, caused by the removal of the inhibitory effect of Dkk1 on the Wnt/β‐catenin pathway.
There is evidence that Wnt acting independently of β‐catenin is also involved in the dynamic transition between epithelial and mesenchymal cell types.34, 35, 36 Further studies are required to determine whether Dkk1 is involved in the regulation of EMT via the non‐classical Wnt pathway and whether it has additional tumor suppressive effects not associated with the Wnt pathway.
In conclusion, the findings of this study confirm that Dkk1 has inhibitory effects on colon cancer progression and suggest that Dkk1 exhibits antitumor activity associated with EMT reversal. These findings may be important for future studies on the mechanism of colon cancer progression and for effective targeting of EMT via Dkk1, which may offer new hope for anti‐cancer therapy.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This study was supported by grants from the Key Project of Nature Science Foundation of China (No. 30830049), the Cooperation of China‐Sweden (No. 09ZCZDSF04400) and the Tianjin Natural Science Foundation (No. 08JCZDJC23500 and No. 09JCYBJC12100).
References
- 1. Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 2006; 5: 997–1014. [DOI] [PubMed] [Google Scholar]
- 2. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev 1997; 11: 3286–305. [DOI] [PubMed] [Google Scholar]
- 3. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–50. [DOI] [PubMed] [Google Scholar]
- 4. Gupta S, Iljin K, Sara H et al FZD4 as a mediator of ERG oncogene‐induced WNT signaling and epithelial‐to‐mesenchymal transition in human prostate cancer cells. Cancer Res 2010; 70: 6735–45. [DOI] [PubMed] [Google Scholar]
- 5. Bates RC, Mercurio AM. The epithelial‐mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther 2005; 4: 365–70. [DOI] [PubMed] [Google Scholar]
- 6. Ohira T, Gemmill RM, Ferguson K et al WNT7a induces E‐cadherin in lung cancer cells. Proc Natl Acad Sci U S A 2003; 100: 10429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yook JI, Li XY, Ota I et al A Wnt‐Axin2‐GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol 2006; 8: 1398–406. [DOI] [PubMed] [Google Scholar]
- 8. Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E‐cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer 2009; 9: 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howe LR, Watanabe O, Leonard J, Brown AM. Twist is up‐regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res 2003; 63: 1906–13. [PubMed] [Google Scholar]
- 10. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci 2003; 116: 2627–34. [DOI] [PubMed] [Google Scholar]
- 11. Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf‐1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998; 391: 357–62. [DOI] [PubMed] [Google Scholar]
- 12. Morvan F, Boulukos K, Clement‐Lacroix P et al Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 2006; 21: 934–45. [DOI] [PubMed] [Google Scholar]
- 13. Mukhopadhyay M, Shtrom S, Rodriguez‐Esteban C et al Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cell 2001; 1: 423–34. [DOI] [PubMed] [Google Scholar]
- 14. Kuhnert F, Davis CR, Wang HT et al Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf‐1. Proc Natl Acad Sci USA 2004; 101: 266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez‐Sancho JM, Aguilera O, Garcia JM et al The Wnt antagonist DICKKOPF‐1 gene is a downstream target of beta‐catenin/TCF and is downregulated in human colon cancer. Oncogene 2005; 24: 1098–103. [DOI] [PubMed] [Google Scholar]
- 16. Aguilera O, Fraga MF, Ballestar E et al Epigenetic inactivation of the Wnt antagonist DICKKOPF‐1 (DKK‐1) gene in human colorectal cancer. Oncogene 2006; 25: 4116–21. [DOI] [PubMed] [Google Scholar]
- 17. Zhang S, Qi L, Li M et al Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J Exp Clin Cancer Res 2008; 27: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bittner M, Meltzer P, Chen Y et al Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000; 406: 536–40. [DOI] [PubMed] [Google Scholar]
- 19. He TC, Sparks AB, Rago C et al Identification of c‐MYC as a target of the APC pathway. Science 1998; 281: 1509–12. [DOI] [PubMed] [Google Scholar]
- 20. Tetsu O, McCormick F. Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–6. [DOI] [PubMed] [Google Scholar]
- 21. Santisteban M, Reiman JM, Asiedu MK et al Immune‐induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res 2009; 69: 2887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mani SA, Guo W, Liao MJ et al The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang DD, Kim YJ, Lee CN et al Expansion of CD133(+) colon cancer cultures retaining stem cell properties to enable cancer stem cell target discovery. Br J Cancer 2010; 102: 1265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barker N, Ridgway RA, van Es JH et al Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 2009; 457: 608–11. [DOI] [PubMed] [Google Scholar]
- 25. Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol 2010; 338: 127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29: 4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shin SY, Rath O, Zebisch A, Choo SM, Kolch W, Cho KH. Functional roles of multiple feedback loops in extracellular signal‐regulated kinase and Wnt signaling pathways that regulate epithelial‐mesenchymal transition. Cancer Res 2010; 70: 6715–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van de Wetering M, Sancho E, Verweij C et al The beta‐catenin/TCF‐4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111: 241–50. [DOI] [PubMed] [Google Scholar]
- 29. Stommel JM, Wahl GM. A new twist in the feedback loop: stress‐activated MDM2 destabilization is required for p53 activation. Cell Cycle 2005; 4: 411–7. [DOI] [PubMed] [Google Scholar]
- 30. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial‐mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–42. [DOI] [PubMed] [Google Scholar]
- 31. Gavert N, Ben‐Ze'ev A. Epithelial‐mesenchymal transition and the invasive potential of tumors. Trends Mol Med 2008; 14: 199–209. [DOI] [PubMed] [Google Scholar]
- 32. Lewis A, Segditsas S, Deheragoda M et al Severe polyposis in Apc(1322T) mice is associated with submaximal Wnt signalling and increased expression of the stem cell marker Lgr5. Gut 2010; 59: 1680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bisson I, Prowse DM. WNT signaling regulates self‐renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res 2009; 19: 683–97. [DOI] [PubMed] [Google Scholar]
- 34. Garriock RJ, Krieg PA. Wnt11‐R signaling regulates a calcium sensitive EMT event essential for dorsal fin development of Xenopus. Dev Biol 2007; 304: 127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dissanayake SK, Wade M, Johnson CE et al The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 2007; 282: 17259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuzugullu H, Benhaj K, Ozturk N et al Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer 2009; 8: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]