

Abstract
Amphiregulin (AR) is derived from a membrane‐anchored form (proAR) by ectodomain shedding, and is a ligand that activates epidermal growth factor receptor (EGFR). We have recently shown that proAR translocates from the plasma membrane to the nucleus after truncation of 11 amino acids at the C‐terminus, which is independent of the conventional EGFR signaling pathway. Although proAR immunoreactivity has reportedly been detected in the nucleus of cancer cells, its biological meaning has never been investigated. This study was performed to investigate the roles of proAR nuclear translocation in human gastric cancer. We constructed proAR truncated 11 amino acids at the C‐terminus (proARΔC11) that spontaneously translocates to the nucleus, and established proARΔC11‐expression regulatable gastric cancer cells (MKN45, MKN28) using the tet‐off system. Using these cells, we found that proAR nuclear translocation significantly induced chemoresistance in vitro and in vivo. Analyzing the relationship between immunoreactive localization of proAR and the clinical outcome for 46 advanced gastric cancer cases treated with chemotherapy, median survival time was 311 days in 16 patients with AR‐positive staining in the nucleus and 387 days in 30 patients with AR‐negative staining (P < 0.05). The present study demonstrates that proAR nuclear translocation increases resistance to anti‐cancer drugs, which might be associated with poor prognosis in human gastric cancer. (Cancer Sci 2012; 103: 708–715)
All of the epidermal growth factor (EGF) receptor (EGFR) ligands are cleaved from membrane‐anchored precursors (pro‐forms) at the plasma membrane in a process termed “ectodomain shedding.” The ligand binding to EGFR induces activation of intracellular signaling cascades implicated in the regulation of a wide variety of cellular processes, including growth, differentiation, apoptosis, adhesion and migration.1 Deregulated expression and activation of EGFR is now widely accepted to play a role in cancer progression. As a result, EGFR has become a therapeutic target and many EGFR inhibitors are in clinical use.
Epidermal growth factor receptor is highly expressed in 33–74% of gastric cancers,2 and elevated levels have been reported as an independent indicator of poor prognosis.3, 4, 5 Furthermore, increased expression of EGFR ligands, such as transforming growth factor (TGF)‐α, heparin‐binding EGF‐like growth factor (HB‐EGF) and amphiregulin (AR), is associated with a poor clinical prognosis in many cancers, including gastric cancer.6, 7 In addition, the EGFR and EGFR ligand gene families have been associated with growth regulation and gastric wall invasion in vitro.8
Despite aberrant enhancement of both EGFR and EGFR ligand expression in gastric cancers, EGFR‐targeted therapeutics does not clinically show sufficient benefit against gastric cancer.2 These results suggest that other signal pathways might be present in the EGFR–ligand signal cascade besides the conventional signal that activates EGFR by binding with ligands. We have previously demonstrated a new function of proHB‐EGF that is not associated with the conventional EGFR signaling pathway. In this mechanism, the carboxyl‐terminal fragment (HB‐EGF‐CTF) generated by ectodomain shedding of proHB‐EGF translocates from the plasma membrane into the inner nuclear membrane and regulates transcriptional repressors.9, 10, 11 Moreover, we have reported that HB‐EGF‐CTF might offer a new molecular target for treating gastric cancer.12 However, mechanisms besides the EGFR‐binding cascade are little understood in terms of other EGFR ligands.
Amphiregulin is also a member of the EGF family, and its increased expression has been reported in many epithelial cancers, including gastric cancer.13, 14, 15, 16, 17, 18 AR is considered to be a new serological marker for breast cancer and colorectal cancer.19, 20, 21 In non‐small cell lung cancer, AR is associated with poor prognosis.22 Therefore, AR has been recognized as one of the most important factors in the EGF family for cancer therapy. Many studies have detected AR immunoreactivity in human gastric cancer tissues, in the cytoplasm and nucleus of tumor cells.14, 23 However, the biological function of AR in the cell nucleus has long been unclear.20 We recently reported that a significant amount of the precursor of AR (proAR) translocates from the plasma membrane to the nuclear membrane and that truncation of the proAR C‐terminal tail is required for the nuclear translocation. ProAR that translocates to the nucleus suppresses global transcription (Fig. 1A).24 However, the mechanisms through which proAR nuclear translocation affects human cancer remain unclear. We presumed that proAR nuclear translocation, along with proHB‐EGF, might have important effects on progression and chemotherapy for gastric cancer. To investigate the specific role of proAR localized to the nucleus, we require a good system to induce spontaneous translocation of proAR to the nucleus, because we have to exclude side effects of shedding stimulants that has multiple effects on cells. Therefore, we constructed a mutant of proAR, proARΔC11, which lacks C‐terminal 11 amino acids and spontaneously translocates to the nucleus without the production of released AR, and established human gastric cancer cells with inducible proARΔC11 expression, using a tetracycline (tet)‐regulating system, the tet‐off system.
Figure 1.
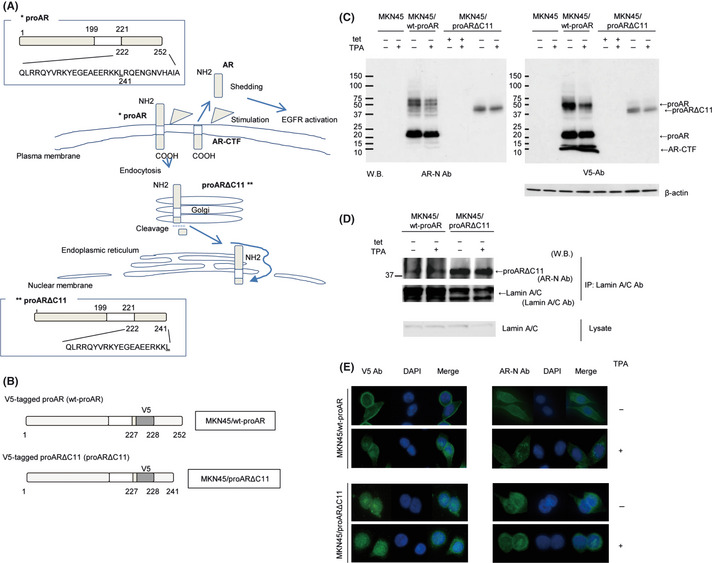
(A) Amphiregulin (AR) is expressed on the plasma membrane as proAR, consisting of a predicted 252 amino acids. ProAR primarily localizes at the plasma membrane. Stimulation induces partial proAR‐ectodomain shedding, resulting in production of soluble AR and the carboxyl‐terminal fragment of proAR (AR‐CTF). In addition, stimulation causes endocytosis of the residual proAR. During translocation to the nucleus, proAR is truncated to the 11 amino acids at the C‐terminus (proARΔC11), and proARΔC11 is targeted to the endoplasmic reticulum (ER). ER‐localized proARΔC11 subsequently diffuses or is actively transported to the nucleus. (B) Schematic presentation of V5‐tag inserted proAR and its deletion mutants. V5‐tagged proAR protein was termed wt‐proAR, and V5‐tagged proARΔC11 protein was proARΔC11. MKN45 lines transfected with the tet‐off system for consistent and conditional induction of wt‐proAR and proARΔC11 were named “MKN45/wt‐proAR” and “MKN45/proARΔC11,” respectively. (C) Western blot analysis of proAR and AR‐CTF expression in MKN45, MKN45/wt‐proAR and MKN45/proARΔC11. Each lane contained 100 μg of protein. The concentration of 12‐0‐tertadecanoylphorbor‐13‐acetate (TPA) was 200 nmol/L to induce ectodomain shedding. We used anti‐V5 antibody (V5 Ab) to recognize the cytoplasmic region of wt‐proAR and proARΔC11, and anti‐AR antibody (AR‐N Ab) to recognize the proAR ectodomain. Loading control comprised β‐actin. (D) Immunoprecipitation and western blot analysis of MKN45/wt‐proAR and MKN45/proARΔC11. The concentration of TPA was 200 nmol/L. Cell lysates were immunoprecipitated with anti‐lamin A/C antibody (Lamin A/C Ab), and precipitated proteins were analyzed by western blotting using AR‐N Ab and Lamin A/C Ab. The cell lysates were analyzed using Lamin A/C Ab. (E) Intracellular localization of proAR after TPA‐inducible processing in MKN45/wt‐proAR and MKN45/proARΔC11 under immunofluorescence microscopy. The concentration of TPA was 200 nmol/L. Nucleus stained blue with DAPI, AR stained green with AR‐N Ab and AR‐CTF stained green with V5 Ab.
We investigate the biological behavior of proARΔC11 nuclear translocation using this system and present the novel finding that proAR nuclear translocation increases chemoresistance and represents a new target for gastric cancer therapy.
Materials and Methods
Materials
Cisplatin (CDDP) was purchased from Nippon Kayaku (Tokyo, Japan), paclitaxel (PTX) from Bristol‐Myers Squibb (New York, NY, USA) and 5‐fluorouracil (5‐FU) from Kyowa Hakko Kirin (Tokyo, Japan). Anti‐AR antibody (AR‐N Ab) (R&D Systems, Minneapolis, MN, USA) was used to recognize the proAR ectodomain. Anti‐V5 antibody (V5 Ab) is a mouse monoclonal IgG antibody, purchased from Invitrogen (Carlsbad, CA, USA).
Cell culture
MKN45 gastric cancer cells (Japan Health Science Research Resources Bank, Tokyo, Japan) were maintained in RPMI1640 medium (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FBS. Cells were cultured at 37°C in 5% CO2 humidified air. MKN28 gastric cancer cells (Japan Health Science Research Resources Bank) were maintained under the same conditions as MKN45.
Transfection (tet‐off system)
We have previously described cDNA constructs pME18s‐wt‐AR and pME18s‐ARΔC11, which contain an inserted sequence encoding the V5 epitope tag in AR cDNA (Fig. 1B).24 AR cDNA derived from these plasmids were subcloned into pTRE2hyg (Clontech Laboratories, Mountain View, CA, USA), and the cDNA constructs were verified by DNA sequencing using a CEQ 8000 DNA Analysis System (Beckman Coulter, Brea, CA, USA). The tet‐off system25 was used for consistent and conditional induction of wt‐proAR and proARΔC11, essentially as described previously.26 Briefly, MKN45 cells were first co‐transfected with two vectors to establish a tet‐off parental MKN45 cell line. These vectors were a pCAG 20‐1 plasmid carrying a tet response element plus three tandem repeats of the VP‐16 minimal transcriptional activation domain (tTA) gene driven by a CAG promoter, and pUHD 10‐3 PURO with a puromycin resistance gene driven by a tet‐responsive promoter. After puromycin selection, tet‐off parental MKN45 cell lines were established. Next, pTRE2hyg plasmids carrying wt‐proAR or proARΔC11 cDNA, driven by a tet‐responsive promoter as described above, were transfected into the established tet‐off parental cells. After hygromycin selection, drug‐resistant clones were examined for the induction/expression of AR proteins by western blot analysis. Established gastric cancer cell lines were termed “MKN45/wt‐proAR” and “MKN45/proARΔC11,” respectively. These cells were maintained in RPMI1640 medium supplemented with 10% FBS, 200 μg/mL hygromycin B (Invitrogen) and 1 μg/mL tet (Sigma‐Aldrich). Tet withdrawal induced each form of proAR expression. “MKN28/proARΔC11” was established, as well as MKN45/proARΔC11.
Immunofluorescence microscopy
Samples were incubated with AR‐N Ab or V5 Ab as primary antibodies after fixation with ethanol and acetone. After incubation with the appropriate secondary antibodies, all sections were counterstained using DAPI (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA). Images were obtained using an Eclipse 80i fluorescence microscope (Nikon, Tokyo, Japan).
Each cell line was cultured in medium with or without tet, then switched to serum‐free medium for 24 h and incubated for another 60 min in the absence (controls) or presence of 200 nmol/L 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA) (Cell Signaling Technology, Boston, MA, USA) for 60 min. Cells were stimulated by TPA, which induces ectodomain shedding of proAR to produce soluble AR.
Western blotting
Aliquots of sample were fractionated on 4–20% sodium dodecyl sulfate‐polyacrylamide gel electorophoresis and then electroblotted to nitrocellulose membranes. The membranes were blocked with 5% skim milk in PBS (−). Then, the membranes were incubated with secondary antibodies after incubation with primary antibodies. The membranes were treated with enhanced chemiluminescence detection reagents (Amersham Biosciences, Buckinghamshire, UK) and exposed to scientific imaging films (Eastman Kodak, New York, NY, USA), and proteins were visualized as bands. Monoclonal β‐actin antibody (Abcam, Cambridge, MA, USA) was used to probe an internal control.
Immunoprecipitation and nuclear extraction
Total cell lysates were incubated with anti‐lamin A/C antibody (Lamin A/C Ab) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h at 4°C. Protein‐G‐Sepharose beads (Amersham Biosciences) were then added to the mixture. After incubation for 1 h, the suspension was centrifuged and the collected protein‐G‐Sepharose beads were washed three times with lysis buffer. The bound proteins were analyzed by western blotting. Nuclear fractions were prepared using ProteoExtract Subcellular Proteome Extraction Kit (Merck, Darmstadt, Germany) according to the instructions from the manufacturer. Anti‐Histon H1 antibody (Histon H1 Ab) (Santa Cruz Biotechnology) was used to confirm samples as appropriate nuclear fractions.
Cell proliferation assay
Each cell line was seeded at 1.0 × 104 cells on 6‐cm diameter dishes in RPMI1640 containing 10% FBS with or without 1 μg/mL tet. Cells were counted on days 4, 7 and 10 using a Bürker–Türk counting chamber (Minato Medical, Tokyo, Japan).
Growth inhibition assay
Each cell line was seeded with or without tet in 96‐well plates (1.0 × 104 cells/well), and cultured for 24 h. Anti‐cancer drugs were then added to triplicate wells (200 μL/well) with CDDP at concentrations of 0–20 μmol/L, PTX at 0–25 μmol/L, or 5‐FU at 0–500 μmol/L in MKN45/wt‐proAR and MKN45/proARΔC11. In addition, CDDP at 0–30 μmol/L, PTX at 0–50 μmol/L, or 5‐FU at 0–5000 μmol/L were added in MKN28/proARΔC11, respectively. After incubation at 37°C in 5% CO2 for 48 h, the number of living cells was measured using an 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt (MTS) assay (Promega, Madison, WI, USA) according to the instructions from the manufacturer.
Xenograft into mice
Four‐week‐old specific‐pathogen‐free conditioned female BALB/c nude mice were purchased from Charles River Japan (Yokohama, Japan). At 6–7 weeks old, each mouse was injected subcutaneously into the left flank with 3.0 × 106 MKN45/proARΔC11 cells suspended in 0.2 mL PBS (−). MKN45/proARΔC11 cells in this study were regulated through the drinking water to mice in the form of doxycycline (dox) (Clontech Laboratories), a derivative of tetracycline. Drinking water in the non‐proARΔC11‐expressing group was supplemented with 1 mg/mL of dox.
MKN45/wt‐proAR cells were transplanted, as well as MKN45/proARΔC11 cells.
Treatment in vivo
The abovementioned nude mice were divided into four groups (four mice/group): chemotherapy group with dox (dox [+]); chemotherapy group without dox (dox [−]); control group dox (+); and control group dox (−). After implantation, mice in chemotherapy groups were treated by i.p. administration of each anti‐cancer drug. The total amount of injected liquid was set to 1.0 mL to optimize the spread of the administered drugs throughout the entire peritoneal cavity. The dose of CDDP in each mouse was 12 mg/kg once on day 7, PTX was fixed at 20 mg/kg once on days 7, 14 and 21, and 5‐FU was 60 mg/kg once on days 7, 11 and 15. Tumor size was measured twice a week by the same observer using calipers, and volume was calculated as length × width2 × 0.5. Mice were weighed on the day of tumor measurement and maximum body weight loss was used as a measure of treatment toxicity. All mice were sacrificed on day 28 after implantation.
Immunohistochemistry
Immunohistochemical staining was performed using V5 Ab (dilution 1:500) for xenograft tumors and AR‐N Ab (dilution 1:400) for clinical biopsy samples. Consecutive sections (4‐μm thick) were deparaffinized and dehydrated. After inhibition of endogenous peroxidase activity by immersion in 3% H2O2/methanol solution, antigen retrieval was achieved by heating the samples in 10 mmol/L citrate buffer (pH 6.0) in a microwave oven for 10 min in at 98°C. Sections were then incubated with primary antibodies. After thorough washing in PBS (−), samples were incubated with biotinylated secondary antibodies and then with avidin–biotin horseradish peroxidase complexes (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA, USA). Finally, immune complexes were visualized by incubation with 0.01% H2O2 and 0.05% 3,3′‐diaminobenzidine tetrachloride. Nuclear counterstaining was accomplished using Mayer's hematoxylin.
Clinical patients and assessment
We used the computerized database of the institution to identify 46 patients with unresectable advanced gastric cancer treated using chemotherapy between April 2003 and December 2007 at Nagoya City University Hospital. Informed consent was provided by all patients. All 46 patients were diagnosed with gastric cancer based on biopsy results and biopsy specimens were immunostained using AR‐N Ab.
All immunostained specimens were assessed by two investigators who were blinded to all clinical information. When more than 10% of all cancer cells in each section were stained, immunostaining was defined as positive.
Response to treatment was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST) (version 1.0),27 using computed tomography. Overall survival time was calculated from the initial date of chemotherapy until death from any cause or last confirmation of survival.
Chemotherapy schedule
The treatment schedule with oral tegafur, 5‐chloro‐2,4‐dihydropyrimidine and potassium oxonate (S‐1) involved oral administration at a dose that did not exceed 40 mg/m2 based on the body surface area (BSA) of the patient: BSA < 1.25 m2, 40 mg; 1.25 m2 ≤ BSA < 1.5 m2, 50 mg; and BSA ≥ 1.5 m2, 60 mg. This dose was administered twice daily for 28 consecutive days, followed by 2 weeks of rest. In patients treated with S‐1 plus CDDP, the same dose of S‐1 was given orally, twice daily for the first 3 consecutive weeks of a 5‐week cycle, and 60 mg/m2 CDDP was administered intravenously on day 8 of each cycle. In patients treated with S‐1 plus PTX, the same dose of S‐1 was administered for the first 2 consecutive weeks and 50 mg/m2 PTX was given intravenously on days 1 and 8, and repeated every 3 weeks.
Statistical analysis
Values are expressed as mean ± SD. Data were analyzed using Welch's t‐test and the χ2 test, as appropriate. Values of P < 0.05 were considered statistically significant. Survival curves were calculated using the Kaplan–Meier method and compared with the log‐rank test.
Results
Wt‐proAR and proARΔC11 overexpression in MKN45 tet‐off system
We first evaluated gene expression of endogenous AR in several human gastric cancer cell lines by real‐time PCR. Among tested cell lines, MKN45 expressed AR mRNA at the lowest levels (data not shown). We evaluated protein levels of proAR and the carboxyl‐terminal fragment of proAR (AR‐CTF) in MKN45, MKN45/wt‐proAR and MKN45/proARΔC11 by western blotting (Fig. 1C). MKN45 and MKN45/proARΔC11 with medium containing tet showed the undetectable level of proAR. MKN45/wt‐proAR expressed a sufficient level of proAR and AR‐CTF on tet withdrawal. MKN45/proARΔC11 also expressed a sufficient level of proARΔC11 on tet withdrawal, but not AR‐CTF. The same pattern of expression was observed even with TPA in MKN45/proARΔC11, indicating that proARΔC11 did not undergo shedding. We thus confirmed that each form of proAR could be induced by tet withdrawal in both MKN45/wt‐proAR and MKN45/proARΔC11. These results demonstrate that wt‐proAR is processed by ectodomain shedding, but that proARΔC is not.
ProARΔC11 spontaneously translocates to the nucleus without TPA treatment.24 Immunoprecipitation with Lamin A/C Ab was used to confirm the physical interaction between proAR and nuclear lamins, which form the nuclear lamina on the interior of the nuclear membrane, as described previously.24 In MKN45/wt‐proAR, a certain protein level of precipitated proAR was observed, and TPA treatment enhanced precipitated proAR. In MKN45/proARΔC11, a sufficient level of precipitated proARΔC11 was observed, and the same pattern was observed even with TPA. In addition, immunoprecipitated proAR and proARΔC11 proteins were the same size (Fig. 1D). We also confirmed the nuclear localization of proAR with nuclear extraction. In MKN45/wt‐proAR, TPA treatment enhanced proAR proteins in nuclear fractions. In MKN45/proARΔC11, proARΔC11 proteins were observed regardless of presence or absence of TPA in nuclear fractions (Fig. S1).
Immunofluorescence microscopy was used to confirm the localization of proAR. In MKN45/wt‐proAR, immunostaining signal of AR‐N and V5 was shifted from the cytoplasm to the perinucleus on addition of TPA, which activates the processing of wt‐proAR. However, in MKN45/proARΔC11, both AR‐N and V5 immunostaining were observed at the perinucleus regardless of presence or absence of TPA (Fig. 1E). These results demonstrate that wt‐proAR translocates from plasma membrane to the perinucleus in response to ectodomain shedding, but proARΔC11 spontaneously translocates to the perinucleus without shedding stimuli. We thus successfully established a gastric cancer cell line (MKN45/proARΔC11) that can spontaneously translocate proARΔC11 to the nucleus. We also confirmed that established MKN28/proARΔC11 can work as well as MKN45/proARΔC11 (Fig. S2).
ProARΔC11 suppresses cell growth
No significant differences were noted for cell growth between parental MKN45 with and without tet (Fig. 2A). In contrast, MKN45/proARΔC11 grew significantly slower in the absence of tet (tet [−]) than in the presence of tet (tet [+]), while MKN45/wt‐proAR did the opposite (Fig. 2B,C). These results demonstrate that spontaneous nuclear translocation of proARΔC11 slows gastric cancer cell proliferation in vitro.
Figure 2.
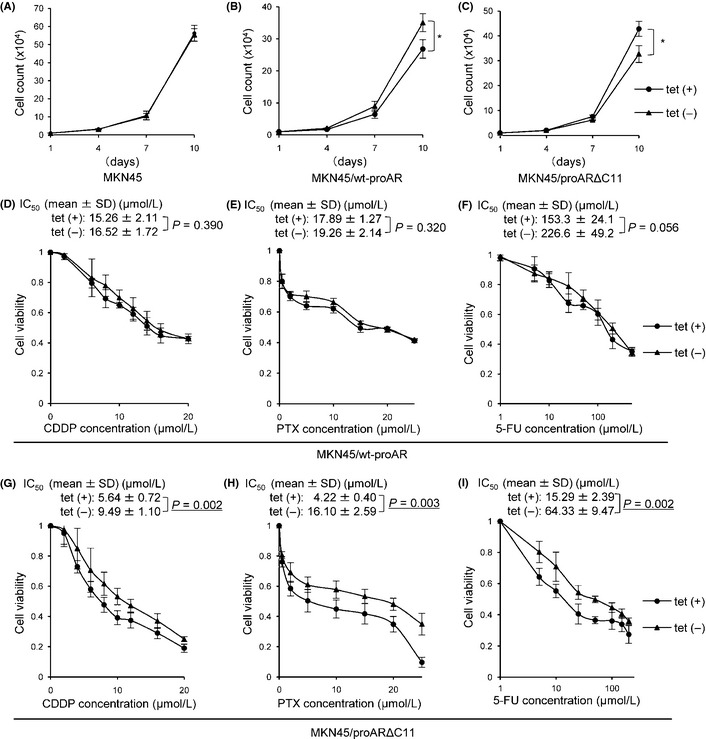
Cell proliferation assay and MTS assay. Cell proliferation assay in MKN45 (A), MKN45/wt‐proAR (B) and MKN45/proARΔC11 (C). MTS assay showed growth‐inhibitory effects on MKN45/wt‐proAR tet (+)/tet (−) (D–F) and MKN45/proARΔC11 tet (+)/tet (−) (G–I) induced by any anti‐cancer drugs (D and G, CDDP; E and H, PTX; F and I, 5‐fluorouracil [5‐FU]) for 48 h. Filled circle, each cell with tetracycline (tet [+]); filled triangle, each cell without tetracycline (tet [−]). Data are shown as means of four independent experiments. Bars, SD. *P < 0.01: cell proliferation of MKN45/wt‐proAR tet (+) vs cell proliferation of MKN45/wt‐proAR tet (−), and MKN45/proARΔC11 tet (+) vs MKN45/proARΔC11 tet (−). IC 50, 50% inhibitory concentration.
ProARΔC11 induces chemoresistance
MKN45/wt‐proAR tended to show chemoresistance by tet withdrawal, but no significant alteration of chemoresistance to all three drugs in MKN45/ wt‐proAR was observed with or without tet (Fig. 2D–F). However, compared to MKN45/proARΔC11 tet (+), MKN45/proARΔC11 tet (−) showed significant chemoresistance against all three drugs, and 50% inhibitory concentration (IC50) for CDDP, PTX and 5‐FU was significantly higher in MKN45/proARΔC11 tet (−) than in MKN45/proARΔC11 tet (+) (Fig. 2G–I). We also confirmed that tet withdrawal induced significant chemoresistance against all three drugs in MKN28/proARΔC11, as well as MKN45/proARΔC11 (Fig. S3). These results suggest that proARΔC11 nuclear translocation induces chemoresistance to anti‐cancer drugs in vitro.
Xenograft
No chemotherapies resulted in death or significant loss of body weight, demonstrating that these regimens had low‐grade toxicity in nude mice (data not shown). In all tumors, expression of proARΔC11 was immunohistologically confirmed to be properly controlled by dox (Fig. 3A; a–d). We also confirmed proper expression of wt‐proAR (data not shown). Immunostaining with large magnification revealed that wt‐proAR and proARΔC11 mainly localized at the plasma membrane and nuclear membrane, respectively (Fig. 3A; b,e). Tumor growth of MKN45/proARΔC11 dox (−) was slower than dox (+). Although tumor growth of MKN45/proARΔC11 dox (+) was obviously suppressed by all of three drugs, MKN45/proARΔC11 dox (−) was barely suppressed with the administration of any anti‐cancer drug (Fig. 3B–D). These results demonstrate that proARΔC11 nuclear translocation reduces tumor growth and induces chemoresistance to anti‐cancer drugs in vivo, as well as in vitro.
Figure 3.
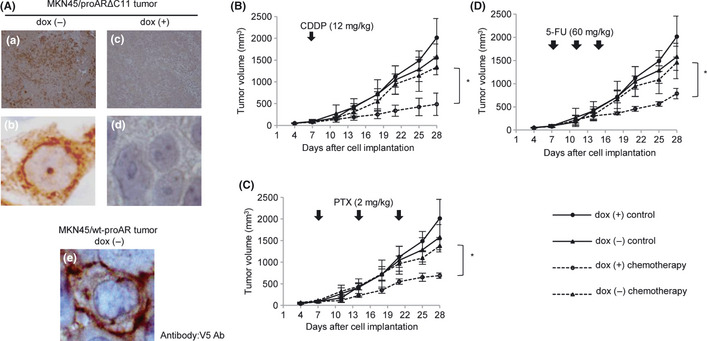
(A) Expression of proARΔC11 and wt‐proAR in xenograft tumor. We used V5 Ab to recognize transfected proARΔC11 and wt‐proAR. Immunohistochemical staining of proARΔC11 xenograft tumor without doxycycline (dox [−]) (a, ×100; b, ×1000). Immunohistochemical staining of proARΔC11 xenograft tumor with doxycycline (dox [+]) (c, ×100; d, ×1000). Immunohistochemical staining of wt‐proAR xenograft tumor dox (−) with large magnification (e, ×1000). (B–D) Subcutaneous MKN45/proARΔC11 tumor growth curves for nude mice after treatment with any anti‐cancer drugs (B, Cisplatin [CDDP]; C, paclitaxel [PTX]; D, 5‐fluorouracil [5‐FU]). ●, no chemotherapy dox (+); ▲, no chemotherapy dox (−); ○, each anti‐cancer drug dox (+); △, each anti‐cancer drug dox (−). Data are shown as means of four independent experiments. Bars, SD. (B) Intraperitoneal administration of 12 mg/kg of CDDP once on day 7. (C) Intraperitoneal administration of 2 mg/kg of PTX once weekly for 3 weeks. (D) Intraperitoneal administration of 60 mg/kg of 5‐FU once on days 7, 11 and 15. *, P < 0.05: treatment by each anti‐cancer drug (CDDP, PTX, 5‐FU) in dox (+) vs treatment by each anti‐cancer drug in dox (−).
In contrast, tumor growth of MKN45/wt‐proAR dox (−) was much faster than dox (+), as well as in culture. Although tumor growth of MKN45/wt‐proAR dox (+) was obviously suppressed by CDDP, MKN45/wt‐proAR dox (−) was barely suppressed, even with administration of CDDP (Fig. S4). These results demonstrate that upregulation of wt‐proAR expression enhanced tumor growth and induced chemoresistance to CDDP in vivo, which differed in part from in vitro.
Patient characteristics
Characteristics of patients in this study are shown in Table 1. AR was detected in both the nucleus and cytoplasm in all cases showing AR‐positive staining (AR [+]) (Fig. 4A; a–d). Among the 46 patients, 16 (34.8%) showed AR (+), and 30 (65.2%) showed AR‐negative staining (AR [−]). The clinical stage for all 16 patients in the AR (+) group and 27 of 30 patients in the AR (−) group was metastasis, and three patients in the AR (−) group showed local advance. All patients in both groups received an S‐1‐based regimen as the first‐line chemotherapy. Patients in the AR (+) group were administered S‐1 alone in nine cases, S‐1 plus CDDP in two cases and S‐1 plus PTX in five cases, compared to S‐1 alone in 11 cases, S‐1 plus CDDP in eight cases and S‐1 plus PTX in 11 cases for the AR (−) group. No significant differences were noted between AR (+) and AR (−) groups in terms of gender ratio, median age, performance status, extent of disease, histological type, number of metastatic organs or first‐line chemotherapeutic regimen used.
Table 1.
Patient characteristics
| AR (+) | AR (−) | P‐value | |
|---|---|---|---|
| (n = 16) | (n = 30) | ||
| Gender | |||
| Male/female | 13/3 | 20/10 | 0.295 |
| Age | |||
| Median | 67 | 65.5 | 0.133 |
| Range | 56–85 | 36–80 | |
| PS | |||
| 0 | 9 | 17 | |
| 1 | 5 | 11 | 0.780 |
| 2 | 2 | 2 | |
| Extent of disease | |||
| Local advance | 0 | 3 | 0.191 |
| Metastasis | 16 | 27 | |
| The number of metastatic organ | |||
| 0 | 0 | 3 | |
| 1 | 4 | 15 | 0.119 |
| 2 | 9 | 8 | |
| ≥3 | 3 | 4 | |
| Histological type | |||
| Intestinal type | 2 | 6 | 0.523 |
| Diffuse type | 14 | 24 | |
| First line chemotherapy | |||
| S1 | 9 | 11 | |
| S1+CDDP | 2 | 8 | 0.373 |
| S1+PTX | 5 | 11 | |
CDDP, cisplatin; PTX, paclitaxel; PS, performance status; S1, oral tegafur, 5‐chloro‐2,4‐dihydropyrimidine and potassium oxonate.
Figure 4.
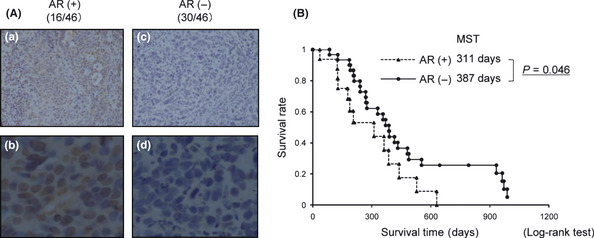
(A) Amphiregulin (AR) expression in pretreatment endoscopic biopsy samples of gastric cancer. Immunohistochemical staining of biopsy samples from human gastric cancer. The antibody used for immunostaining was AR‐N Ab. (a,b) Immunohistochemical staining in the AR‐positive (AR [+]) group. (c,d) Immunohistochemical staining in the AR‐negative (AR [−]) group. Original magnification: a and c, ×100; b and d, ×400. (B) Overall survival. Filled triangle, AR (+) group; filled circle, AR (−) group; MST, median survival time.
Survival and response to chemotherapy
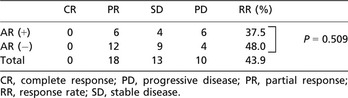
Overall survival times in the AR (+) and AR (−) groups are shown in Figure 4(B). Median survival time was 311 days in the AR (+) group, which was significantly shorter than the 387 days in the AR (−) group (P = 0.046). Target lesions evaluated according to RECIST criteria were present for all 16 patients (100%) in the AR (+) group and for 25 patients (83%) in the AR (−) group. Response rate was 37.5% (6/16) in the AR (+) group and 48.0% (12/25) in the AR (−) group. No significant differences in response rate were apparent between AR (+) and AR (−) groups (P = 0.509) (Table 2).
Table 2.
Response rate
Discussion
Epidermal growth factor receptor and EGFR ligands have been well studied as molecular targets for cancer therapy.28, 29, 30 AR has been characterized as a growth factor involved in cell proliferation and differentiation,1 AR is recognized as an important factor affecting cancer therapy.14, 15, 16, 17, 18, 19, 20, 21, 22 We recently reported that a significant amount of proAR is internalized without ectodomain shedding in response to shedding stimuli and translocates to the nucleus.24 This signal transduction induces heterochromatin formation and suppression of global transcription. However, the relationship between proAR nuclear localization and cancer has not yet been reported. Therefore, the present work focuses on proAR nuclear translocation as a new molecular target for gastric cancer. In completing this study, we successfully established gastric cancer cell lines with tet‐regulated inducible proAR nuclear translocation, MKN45/proARΔC11. We were thus able to selectively analyze the basic effects of proAR nuclear translocation, both in vitro and in vivo.
Our study revealed that spontaneous nuclear translocation of proARΔC11 significantly slowed proliferation and induced chemoresistance to three anti‐cancer drugs, CDDP, PTX and 5‐FU, in vitro and in vivo. Overexpression of wt‐proAR enhanced cell growth, which is explained by the production of released AR and EGFR activation because constitutive shedding of proAR in MKN45/wt‐proAR was observed (Fig. 1C). However, chemoresistance of MKN45/wt‐proAR to anti‐cancer drugs did not show significant correlation, which might be explained by the different extent of nuclear translocation of proAR.
Furthermore, we immunohistochemically investigated AR expression on specimens of human gastric cancers. Our data indicate that the AR (+) group experienced significantly poorer prognosis. Although significant differences were not noted, probably due to the small sample size, response rates tended to be lower in the AR (+) group than in the AR (−) group. These results supported the present data shown in vitro and in vivo. Although many previous reports have shown AR immunoreactivity in cancers, including gastric cancer, to be detected in both the cytoplasm and the nuclei of tumor cells, the significance of AR localization in the nucleus has not been clarified.1, 31, 32, 33 In the present study, AR was expressed in both the nucleus and the cytoplasm in all AR (+) cases. AR immunoreactivity in the nucleus must show translocated proAR. These results suggest that proAR nuclear localization might play a crucial role in the determination of some tumor properties.
We have previously reported that HB‐EGF‐CTF translocates from the plasma membrane to the nucleus and regulates the cell cycle by abrogation of transcriptional repressors.9, 10 Moreover, we have shown that it is important for gastric cancer therapy to inhibit not only the conventional EGFR signal pathway, but also HB‐EGF‐CTF nuclear translocation.12 The present study shows that proAR nuclear translocation might offer a new molecular target for gastric cancer progression, along with HB‐EGF. The present study also indicates that nuclear translocation as well as overexpression of proAR could be critical for molecular diagnosis of gastric cancer. We found some squamous cell carcinoma cell lines presenting spontaneous nuclear translocation of endogenous proAR (Shigeki Higashiyama, unpublished observation). Although we have not yet elucidated the existence of mutant gene expressing proARΔC11 in living cells, C‐terminal modification of proAR, such as proteolytic cleavage, and its mediating factors might be good molecular targets for diagnosis and therapy.
Cancer cells can acquire resistance to chemotherapy by a range of mechanisms. In general, chemoresistance is attributed to mutation or overexpression of the drug target, inactivation of the drug, or elimination of the drug from the cell. However, these considerations cannot confer full resistance to multiple anti‐cancer drugs with different mechanisms of action. We have not yet elucidated how proAR nuclear translocation induces chemoresistance. Further investigation remains necessary, as the detailed mechanisms are not yet fully understood, and clarifying these mechanisms would certainly result in the development of new cancer therapies.
In conclusion, this study is the first report to show basic and clinical behavior of proAR nuclear translocation. Our results indicate that proAR nuclear translocation induces chemoresistance to anti‐cancer drugs and might be associated with poor prognosis in human gastric cancer. ProAR nuclear translocation might offer a prognostic marker and a new molecular target for gastric cancer therapy.
Disclosure Statement
All authors have no conflict of interest to declare.
Supporting information
Fig. S1. Western blot analysis with nuclear fractions.
Fig. S2. Western blot analysis and immunofluorescence study of proARΔC11 in MKN28/proARΔC11.
Fig. S3. MTS assay in MKN28/proARΔC11.
Fig. S4. Subcutaneous MKN45/wt‐proAR tumor growth curves after treatment with Cisplatin (CDDP).
Acknowledgments
This study was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 22790665). We thank Takumi Era (Kumamoto University, Kumamoto, Japan) for providing the tet‐off system, and Yukimi Hashizume for technical assistance.
References
- 1. Shoyab M, Plowman GD, McDonald VL, Bradley JG, Todaro GJ. Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science 1989; 243: 1074–6. [DOI] [PubMed] [Google Scholar]
- 2. Rojo F, Tabernero J, Albanell J et al Pharmacodynamic studies of gefitinib in tumor biopsy specimens from patients with advanced gastric carcinoma. J Clin Oncol 2006; 24: 4309–16. [DOI] [PubMed] [Google Scholar]
- 3. Kopp R, Ruge M, Rothbauer E et al Impact of epidermal growth factor (EGF) radioreceptor analysis on long‐term survival of gastric cancer patients. Anticancer Res 2002; 22: 1161–7. [PubMed] [Google Scholar]
- 4. Garcia I, Vizoso F, Martin A et al Clinical significance of the epidermal growth factor receptor and HER2 receptor in resectable gastric cancer. Ann Surg Oncol 2003; 10: 234–41. [DOI] [PubMed] [Google Scholar]
- 5. Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37(Suppl. 4): S9–15. [DOI] [PubMed] [Google Scholar]
- 6. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor‐related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19: 183–232. [DOI] [PubMed] [Google Scholar]
- 7. Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Target‐based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer 2003; 10: 1–21. [DOI] [PubMed] [Google Scholar]
- 8. Piontek M, Hengels KJ, Porschen R, Strohmeyer G. Antiproliferative effect of tyrosine kinase inhibitors in epidermal growth factor‐stimulated growth of human gastric cancer cells. Anticancer Res 1993; 13: 2119–23. [PubMed] [Google Scholar]
- 9. Nanba D, Mammoto A, Hashimoto K, Higashiyama S. Proteolytic release of the carboxy‐terminal fragment of proHB‐EGF causes nuclear export of PLZF. J Cell Biol 2003; 163: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kinugasa Y, Hieda M, Hori M, Higashiyama S. The carboxyl‐terminal fragment of pro‐HB‐EGF reverses Bcl6‐mediated gene repression. J Biol Chem 2007; 282: 14797–806. [DOI] [PubMed] [Google Scholar]
- 11. Hieda M, Isokane M, Koizumi M et al Membrane‐anchored growth factor, HB‐EGF, on the cell surface targeted to the inner nuclear membrane. J Cell Biol 2008; 180: 763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shimura T, Kataoka H, Ogasawara N et al Suppression of proHB‐EGF carboxy‐terminal fragment nuclear translocation: a new molecular target therapy for gastric cancer. Clin Cancer Res 2008; 14: 3956–65. [DOI] [PubMed] [Google Scholar]
- 13. Cook PW, Pittelkow MR, Keeble WW, Graves‐Deal R, Coffey RJ Jr, Shipley GD. Amphiregulin messenger RNA is elevated in psoriatic epidermis and gastrointestinal carcinomas. Cancer Res 1992; 52: 3224–7. [PubMed] [Google Scholar]
- 14. Akagi M, Yokozaki H, Kitadai Y et al Expression of amphiregulin in human gastric cancer cell lines. Cancer 1995; 75: 1460–6. [PubMed] [Google Scholar]
- 15. Johnson GR, Saeki T, Gordon AW, Shoyab M, Salomon DS, Stromberg K. Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J Cell Biol 1992; 118: 741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Damstrup L, Kuwada SK, Dempsey PJ et al Amphiregulin acts as an autocrine growth factor in two human polarizing colon cancer lines that exhibit domain selective EGF receptor mitogenesis. Br J Cancer 1999; 80: 1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Funatomi H, Itakura J, Ishiwata T et al Amphiregulin antisense oligonucleotide inhibits the growth of T3M4 human pancreatic cancer cells and sensitizes the cells to EGF receptor‐targeted therapy. Int J Cancer 1997; 72: 512–7. [DOI] [PubMed] [Google Scholar]
- 18. Castillo J, Erroba E, Perugorria MJ et al Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res 2006; 66: 6129–38. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Masri S, Phung S, Chen S. The role of amphiregulin in exemestane‐resistant breast cancer cells: evidence of an autocrine loop. Cancer Res 2008; 68: 2259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Willmarth NE, Ethier SP. Amphiregulin as a novel target for breast cancer therapy. J Mammary Gland Biol Neoplasia 2008; 13: 171–9. [DOI] [PubMed] [Google Scholar]
- 21. Lievre A, Laurent‐Puig P. Predictive factors of response to anti‐EGFR treatments in colorectal cancer. Bull Cancer 2008; 95: 133–40. [DOI] [PubMed] [Google Scholar]
- 22. Ishikawa N, Daigo Y, Takano A et al Increases of amphiregulin and transforming growth factor‐alpha in serum as predictors of poor response to gefitinib among patients with advanced non‐small cell lung cancers. Cancer Res 2005; 65: 9176–84. [DOI] [PubMed] [Google Scholar]
- 23. Kitadai Y, Yasui W, Yokozaki H et al Expression of amphiregulin, a novel gene of the epidermal growth factor family, in human gastric carcinomas. Jpn J Cancer Res 1993; 84: 879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Isokane M, Hieda M, Hirakawa S et al Plasma‐membrane‐anchored growth factor pro‐amphiregulin binds A‐type lamin and regulates global transcription. J Cell Sci 2008; 121: 3608–18. [DOI] [PubMed] [Google Scholar]
- 25. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline‐responsive promoters. Proc Natl Acad Sci USA 1992; 89: 5547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Era T, Wong S, Witte ON. Analysis of Bcr‐Abl function using an in vitro embryonic stem cell differentiation system. Methods Mol Biol 2002; 185: 83–95. [DOI] [PubMed] [Google Scholar]
- 27. Therasse P, Arbuck SG, Eisenhauer EA et al New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National cancer institute of the United States, National cancer institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 28. Xu H, Yu Y, Marciniak D et al Epidermal growth factor receptor (EGFR)‐related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol Cancer Ther 2005; 4: 435–42. [DOI] [PubMed] [Google Scholar]
- 29. Perera RM, Narita Y, Furnari FB et al Treatment of human tumor xenografts with monoclonal antibody 806 in combination with a prototypical epidermal growth factor receptor‐specific antibody generates enhanced antitumor activity. Clin Cancer Res 2005; 11: 6390–9. [DOI] [PubMed] [Google Scholar]
- 30. Rocha‐Lima CM, Soares HP, Raez LE, Singal R. EGFR targeting of solid tumors. Cancer Control 2007; 14: 295–304. [DOI] [PubMed] [Google Scholar]
- 31. Johnson GR, Saeki T, Auersperg N et al Response to and expression of amphiregulin by ovarian carcinoma and normal ovarian surface epithelial cells: nuclear localization of endogenous amphiregulin. Biochem Biophys Res Commun 1991; 180: 481–8. [DOI] [PubMed] [Google Scholar]
- 32. Modrell B, McDonald VL, Shoyab M. The interaction of amphiregulin with nuclei and putative nuclear localization sequence binding proteins. Growth Factor 1992; 7: 305–14. [DOI] [PubMed] [Google Scholar]
- 33. Nylander N, Smith LT, Underwood RA, Piepkorn M. Topography of amphiregulin expression in cultured human keratinocytes: colocalization with the epidermal growth factor receptor and CD44. In Vitro Cell Dev Biol Anim 1998; 34: 182–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Western blot analysis with nuclear fractions.
Fig. S2. Western blot analysis and immunofluorescence study of proARΔC11 in MKN28/proARΔC11.
Fig. S3. MTS assay in MKN28/proARΔC11.
Fig. S4. Subcutaneous MKN45/wt‐proAR tumor growth curves after treatment with Cisplatin (CDDP).