

Abstract
High dysadherin expression has been recognized as a biological predictor of metastasis and poor prognosis for many different cancer types; however, the molecular mechanisms of how dysadherin affects cancer progression are still poorly understood. In this study, we examined whether AKT signaling could link dysadherin expression with downstream events that promote the metastatic potential of human breast cancer cells. Immunohistochemical analysis of breast cancer tissues showed that dysadherin expression was highly associated with elevated expression of phospho‐AKT. The introduction of dysadherin cDNA into BT‐474, MCF‐7 and T‐47D breast cancer cell lines enhanced their levels of AKT phosphorylation, while knockdown of dysadherin in MDA‐MB‐231 and Hs578T breast cancer cell lines suppressed AKT phosphorylation. Treatment with the AKT inhibitor triciribine suppressed dysadherin‐mediated pro‐metastatic effects, including epithelial–mesenchymal transition, cell motility and drug resistance. These findings suggest that dysadherin might contribute to breast cancer progression through AKT activation. (Cancer Sci 2012; 103: 1280–1289)
Breast cancer is one of the most commonly detected malignancies in women worldwide.1 The latest advances in cancer diagnosis and therapy have greatly improved survival of breast cancer patients. Metastasis is a destructive consequence of cancer progression and is the most common cause for treatment failure and death in breast cancer.2 There is much to discover about the biological and molecular mechanisms that control the metastatic process.
Dysadherin is a cancer cell membrane‐associated glycoprotein that has been recognized as a prognostic indicator of metastasis and/or poor survival in many different cancer types.3, 4, 5 The presence of dysadherin expression contributes causally to cancer metastasis in a number of ways, including downregulation of E‐cadherin‐mediated cell adhesion, upregulation of tumor‐promoting chemokine production and enhancement of cancer stem‐cell properties.5, 6, 7 Based on these findings, dysadherin might represent a molecular target for the treatment of advanced cancer. The detailed molecular mechanisms that link dysadherin expression to downstream events that promote cancer metastasis have not been fully clarified. The elucidation of dysadherin signaling mechanisms in specific aspects of metastasis might lead to the development of effective therapies for treating advanced cancer.
Nam et al.5 show, in a breast cancer model system, that NF‐κB transcriptional activity is enhanced by dysadherin expression. Specifically, NF‐κB regulates the expression of genes involved in many cellular processes that play a key role in the development and progression of cancer.8 NF‐κB is intimately intertwined with cancer growth and metastasis;9 the NF‐κB transcription factor has been identified as a target of the AKT signaling pathway.10 AKT is a serine/threonine kinase that plays a critical role in cell survival by negatively interacting with proteins that promote apoptosis and by interacting with proteins involved in cell proliferation, cell growth, cell motility and invasion.11 Increased AKT protein expression and activity have been detected in various types of cancers, including those of the stomach, prostate, ovary, breast and brain.12, 13 The detection of activated AKT in breast cancer patients is associated with poor prognosis, with a higher probability of relapse accompanied by distant metastasis.14 Clinical data suggest that AKT is involved in a broad spectrum of chemoresistance, as well as radioresistance, in breast cancer; therefore, AKT is considered to be an important target for advanced breast cancer therapies.15, 16
Collectively, these studies prompted us to ask whether there is a possible link between dysadherin expression and AKT signaling in cancer progression. In the present study, we introduce dysadherin to breast cancer cell lines to clarify the involvement of AKT signaling in mediating the pro‐metastatic effect of dysadherin. The purpose of this work is to gain a better understanding of the molecular mechanisms of dysadherin for development of improved anti‐cancer strategies.
Materials and Methods
Cell culture and reagents
The human breast cancer cell lines BT‐474, MCF‐7, T‐47D, MDA‐MB‐231 and Hs578T were cultured in DMEM (Welgene, Daegu, Korea) containing 10% FBS and 1% penicillin/streptomycin (Invitrogen, Grand Island, NY, USA), as previously described.5, 17 Paclitaxel and AKT inhibitor triciribine were purchased from Sigma‐Aldrich (St. Louis, MO, USA) and Calbiochem (La Jolla, CA, USA), respectively.
Quantitative RT‐PCR (qRT‐PCR)
Real‐time qRT‐PCR was carried out using the ABI 7300 Real‐Time PCR system with the SYBR green dye (Applied Biosystems, Foster City, CA, USA). First‐strand cDNA was prepared from total RNA using a RevertAid first‐strand synthesis kit (Fermentas, Hanover, MD, USA). The qRT‐PCR was performed in triplicate. Human dysadherin mRNA levels were normalized with human cyclophilin A (PPIA) mRNA. The primer sets used in this study are as follows: Dysadherin, 5′‐TCCCACTGATGACACCACGA‐3′ (forward) and 5′‐AAAACCAGATGGCTTGAGGGT‐3′ (reverse); PPIA, 5′‐TGCCATCGCCAAGGAGTAG‐3′ (forward) and 5′‐TGCACAGACGGTCACTCAAA‐3′ (reverse).
Immunoblot analysis and immunofluorescent staining
Immunoblot analysis and immunofluorescent staining were performed, as previously described.18 Antibodies to the following proteins were used: Dysadherin (NCC‐M53; provided by Dr Setsuo Hirohashi, National Cancer Center, Tokyo, Japan),19 PI3K‐p85, pPI3K‐p85, AKT, phospho‐AKT (pAKT), GSK‐3β and pGSK‐3β (Cell Signaling Technology, Beverly, MA, USA), β‐actin (Sigma‐Aldrich), E‐cadherin, Fibronectin, N‐cadherin and Vimentin (BD Biosciences, San Jose, CA, USA). Human breast cancer tissue arrays (AccuMax Array, cat. no. A312) were purchased from Isu Abxis (Seoul, Korea). Rhodamine‐phalloidin (Cytoskeleton, Denver, CO, USA) was used to visualize F‐actin. Dysadherin and pAKT expression were measured as the intensity of fluorescence divided by the area of the nucleus, as determined by DAPI staining, in three randomly selected fields for each specimen using the Image‐Pro Plus program (Media Cybernetics, Silver Spring, MD, USA). Clinico‐pathological characteristics of breast tissue samples that were used in this analysis are described in Supporting information Table S1.
Generation of dysadheirn‐knockdown cells and dysadherin‐overexpressing stable cells
Cells were transduced with lentiviral‐based plasmids containing shRNA targeting dysadherin using dysadherin‐coding siRNA oligos5 or transfected with dysadherin expression vector (pcDNA‐L3HSV; provided by Dr Setsuo Hirohashi, National Cancer Center, Tokyo, Japan).3 The knockdown or overexpression of the Dysadherin gene was confirmed by qRT‐PCR and immunoblot analysis. Stably transfected cells were isolated and expanded under puromycin (knockdowns) or G418 selection (overexpressors).
Cell proliferation and apoptosis analysis
Cell proliferation was assessed using 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetra‐zolium bromide (MTT; Sigma‐Aldrich) assay and cell cycle assay, as previously described.7 Apoptosis was detected by the FACS assay for annexin V (BD Biosciences) and the immunoblot analysis for cleaved poly‐ADP ribose polymerase (PARP; Cell Signaling Technology).
Cell motility and wound‐healing migration analysis
Cell motility was assessed using the HitKit HSC reagent kit (Cellomics, Pittsburgh, PA, USA), as previously described.20 Briefly, fluorescent microspheres were covered on 96‐well plates that had been coated with type I collagen (BD Bioscience). After incubation for 16–18 h at 37°C, the cells were fixed in formaldehyde and were stained with rhodamine‐conjugated phalloidin. Track and cell areas were analyzed using the Cellomics Array Scan HCS reader (Cellomics). Cell motility was calculated by dividing the track area by the cell area, which was performed for each visual field from three independent experiments. A wound‐healing migration assay was performed, as previously described.21 Briefly, cells were seeded on 6‐well plates. When cells were confluent, wounds were made on the cell monolayer using a micropipette tip. Cells were subsequently washed with PBS, replenished with DMEM with 1% FBS, and photographed using an inverted microscope (Nikon, Tokyo, Japan). Dishes were placed in the incubator for 48–92 h and the matched wound regions were photographed.
Statistical analysis
All experiments were conducted in triplicate (n = 3) or greater, and the results are presented as the mean ± SD. Statistical analyses of these data were conducted via unpaired parametric Student's t‐test or nonparametric Mann–Whitney U‐test.
Results
Modification of the Dysadherin gene in breast cancer cell lines
Before modification of Dysadherin in breast cancer cells, we confirmed the expression profile of dysadherin in breast cancer cells. Specifically, qRT‐PCR and immunoblot analysis showed that BT‐474, MCF‐7 and T‐47D breast cancer cells possessed little or no dysadherin expression. Conversely, metastatic MDA‐MB‐231 and Hs578T breast cancer cells had high dysadherin expression (Fig. 1A,B), which is consistent with previous findings.5 In the present study, we introduced dysadherin cDNA (Dys) into BT‐474, MCF‐7 and T‐47D cells. Through qRT‐PCR and immunoblot analysis, we observed that expression of dysadherin was efficiently increased in breast cancer cells transfected with Dys compared with the parental cells (none, non‐transfected cells) and cells transfected with empty vector (mock). In addition, we stably knocked down dysadherin expression in MDA‐MB‐231 and Hs578T cells using lentiviral shRNA. The qRT‐PCR and immunoblot analyses showed that expression of dysadherin was efficiently suppressed in breast cancer cells transfected with dysadherin shRNA (shDys), whereas the parental cells (none, non‐transfected cells) and cells transfected with control shRNA (shCon) preserved dysadherin expression (Fig. 1C,D).
Figure 1.
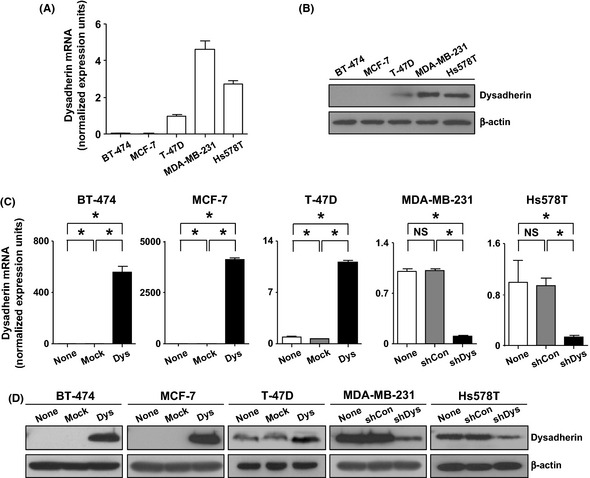
Modification of Dysadherin in breast cancer cell lines. (A,B) qRT‐PCR and immunoblot analysis for dysadherin in breast cancer cell lines. (C,D) qRT‐PCR and immunoblot analysis for dysadherin in control (none, non‐transfected cells), mock (empty vector), Dys (Dysadherin cDNA), shCon (control shRNA) or shDys‐ (Dysadherin shRNA) transfected cells. Dysadherin mRNA were normalized with peptidylprolyl isomerase A mRNA in each sample. β‐actin was used as a loading control in the immunoblot analysis. Data are the mean ± SD (n = 3). *P < 0.01. NS, not significant.
Role of dysadherin in breast cancer progression
To investigate the functional role of dysadherin in breast cancer progression, we determined whether dysadherin expression regulates cell proliferation, invasion potential and apoptosis inhibition. First, we examined the cell proliferation rates of both the dysadherin overexpressing and knockdown cells by MTT assay and cell cycle analysis. We found that overexpression or knockdown of dysadherin itself did not influence cell proliferation (Fig. 2A,B).
Figure 2.
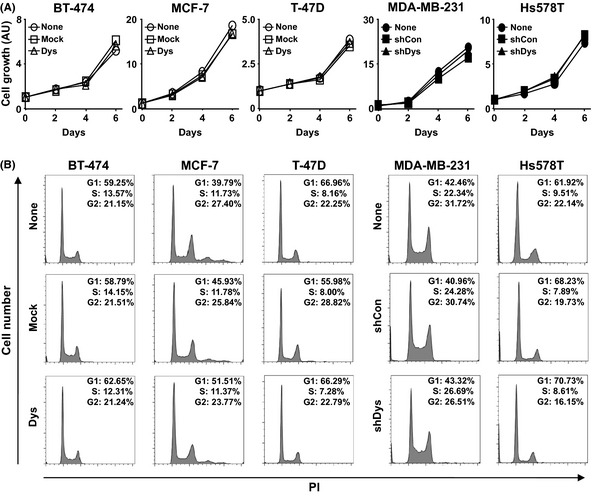
Effect of dysadherin on breast cancer cell proliferation. (A) MTT assay of breast cancer cells after modification of the Dysadherin gene. Data are the mean ± SD (n = 3). AU, arbitary units. (B) Cell cycle analysis of breast cancer cells after modification of the Dysadherin gene. PI, propidium.
Second, we investigated whether modification of Dysadherin was able to regulate the invasiveness of breast cancer cells in vitro. Cell motility assay showed that dysadherin overexpression significantly increased the motility of the cancer cells, and the knockdown of dysadherin expression significantly reduced the motility of the cancer cells compared with control counterparts (Fig. 3A). The wound‐healing assay also showed that dysadherin‐expressing MCF‐7 cells possessed a more enhanced closure, and dysadherin‐knockdown MDA‐MB‐231 cells showed delayed closure compared with control counterparts (Fig. 3B). These findings are supported by previous observations that the knockdown of dysadherin significantly reduces the invasion of breast cancer cell lines through Matrigel (BD Biosciences Discovery Labware).5 The actin cytoskeleton is thought to be an important mediator of cell migration in the process of cancer cell invasion.22 Observation by confocal microscopy demonstrated that dysadherin overexpression increased the cortical actin layer at the edges of MCF‐7 cells, and dysadherin knockdown decreased actin at the edges of MDA‐MB‐231 cells compared with their control counterparts (Fig. 3C). The epithelial–mesenchymal transition (EMT) is considered a critical biological process in epithelial tumor invasion.23 Therefore, we examined whether modification of Dysadherin was able to influence EMT. An immunofluorescent study demonstrated that dysadherin expressing MCF‐7 cells showed a decrease of E‐cadherin and an increase of fibronectin but no change in N‐cadherin and vimentin expression compared with control counterparts. Dysadherin‐knockdown MDA‐MB‐231 cells showed a decrease in fibronectin expression but no change in E‐cadherin, N‐cadherin and vimentin compared to control counterparts (Fig. 3D).
Figure 3.
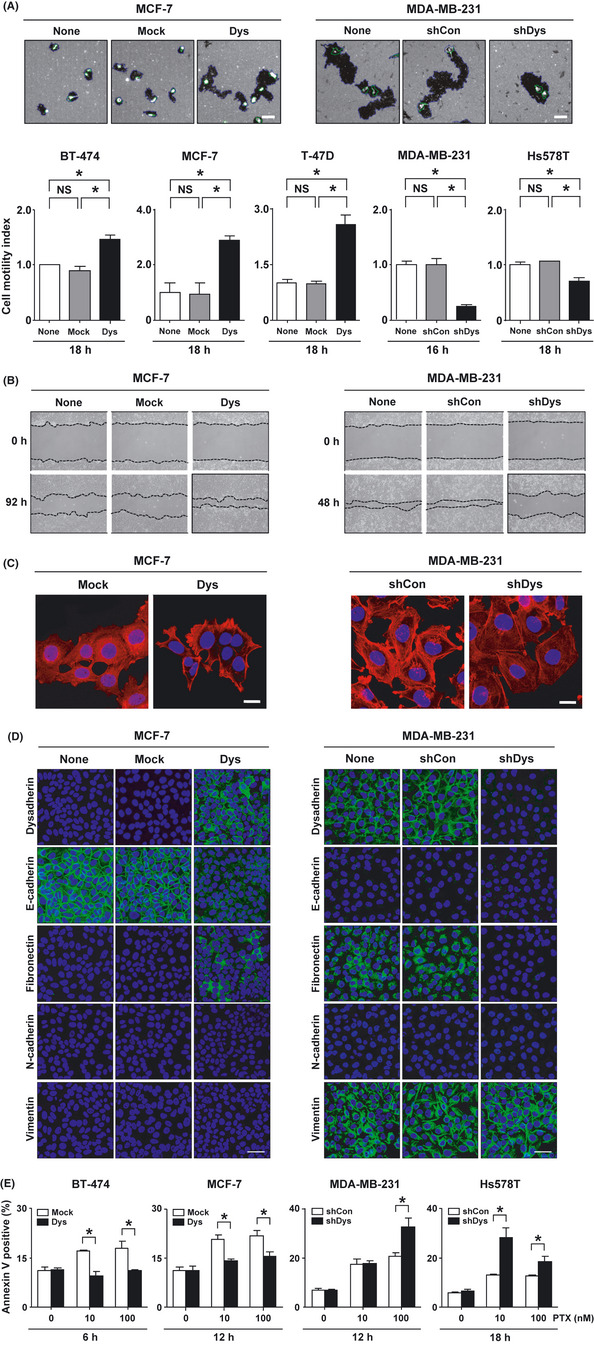
Effect of dysadherin on cell motility, cell migration, actin organization, epithelial–mesenchymal transition (EMT) and anti‐apoptotic capacity in breast cancer cell lines. (A) Cell motility assay of breast cancer cells after modification of the Dysadherin gene. Blue line represents the track area of a cell. Green line represents the cell area. Data are the mean ± SD (n = 3). Scale bar, 100 µm. (B) Cell migration assay of breast cancer cells. Confluent cell cultures were scraped (“wounded”) with a micropipette tip (top). Cells were photographed at 48–92 h after wounding by phase contrast microscopy (bottom). (C) Immunofluorescent analysis of F‐actin in breast cancer cells. All nuclei were counterstained with DAPI (blue). Scale bar, 100 µm. (D) Immunofluorescent analysis of EMT‐related protein expression in breast cancer cells. All nuclei were counterstained with DAPI (blue). Scale bar, 50 µm. (E) Analysis of apoptosis in breast cancer cells after treatment with 10 and 100 nM paclitaxel (PTX) for 6–18 h. Apoptosis was measured using the annexin V assay. Data are the mean ± SD (n = 3). * P < 0.01. NS, not significant.
Third, we determined whether modification of Dysadherin was able to regulate anti‐apoptotic capacity. Paclitaxel is used widely in the treatment of breast cancer;24 therefore, we determined the rate of paclitaxel‐induced apoptosis in the dysadherin‐expressing or dysadherin‐knockdown breast cancer cells after the cells were cultured with paclitaxel for 6–18 h. Apoptosis analysis showed that the dysadherin overexpression significantly reduced the rate of paclitaxel‐induced apoptosis, and dysadherin knockdown significantly enhanced the rate of paclitaxel‐induced apoptosis compared to the control cells (Fig. 3E).
Identification of AKT signaling as a downstream target of dysadherin
Our previous findings suggest that the dysadherin expression is associated with the enhanced activity of the NF‐κB pathway in a breast cancer model system.5 The activation of NF‐κB has been recognized as a key target of AKT signal pathway.10 To investigate whether dysadherin can propagate the AKT signal within the cancer cells, we first analyzed the correlation between dysadherin and pAKT in breast cancer samples. Immunofluorescent staining showed that both dysadherin and pAKT expression were highly elevated in cancerous tissues compared with non‐cancerous tissues from the same breast cancer patient. In cancerous tissues, the pAKT expression increased when dysadherin expression was strongly present (Fig. 4A). Analysis of fluorescent intensity also showed that increased expression of dysadherin was significantly correlated with pAKT expression in clinical breast cancer specimens (Supporting information Fig. S1, Supporting information Table S1).
Figure 4.
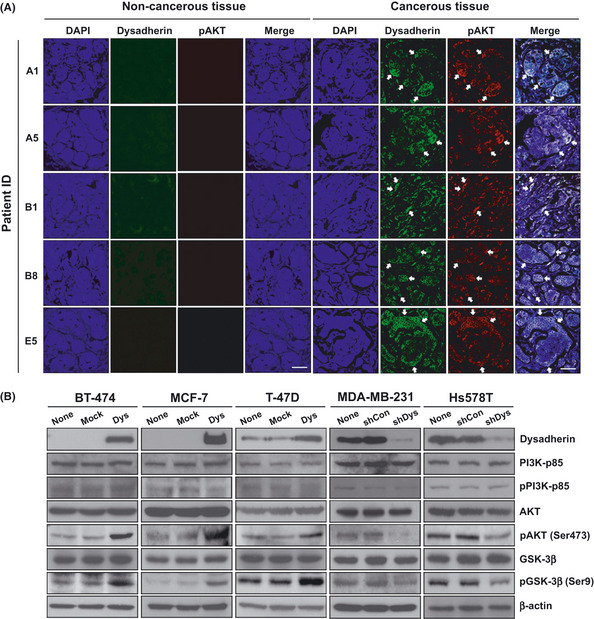
Correlation between dysadherin and phospho‐AKT (pAKT) expression in breast cancer tissues and cell lines. (A) Immunofluorescent staining of dysadherin (green) and pAKT (red) in breast cancer tissues compared with matched normal breast tissues. Arrows indicate the colocalization of dysadherin and pAKT in breast cancerous tissue. All nuclei were counterstained with DAPI (blue). Scale bar, 100 μm. (B) Immunoblot analysis of dysadherin and AKT downstream signaling in breast cancer cell lines after modification of the Dysadherin gene. β‐actin was used as a loading control.
AKT is recruited to the plasma membrane by increased phosphoinositide 3‐kinase (PI3K) signaling. Subsequently, AKT is phosphorylated and transferred to several subcellular locations, where it can phosphorylate downstream targets in various pathways. PI3K is a heterodimer composed of a p85 regulatory and a p110 catalytic subunit of which there are several isoforms. The p85 subunit is known to control the sequential activation of PI3K.25 Glycogen synthase kinase‐3β (GSK‐3β) is known as a downstream molecule associated with AKT activation.11 To determine whether dysadherin expression influences AKT signaling in breast cancer cells, we examined PI3K‐p85, AKT and GSK‐3β expression in dysadherin‐expressing or dysadherin‐knockdown cells. Modification of Dysadherin in breast cancer cells did not affect the expression of PI3K‐p85 and pPI3K‐p85, or total expression of AKT. Dysadherin overexpression in breast cancer cells increased pAKT and activation of the downstream molecule, pGSK‐3β. In contrast, dysadherin knockdown in breast cancer cells suppressed pAKT and pGSK‐3β compared with the control cells (Fig. 4B). These results demonstrate that dysadherin might enhance AKT activation in breast cancer cell lines.
Treatment with an AKT inhibitor can suppress dysadherin‐mediated pro‐metastatic effects
The above observation prompted us to ask whether AKT signaling is related to dysadherin‐mediated pro‐metastatic effects, including invasive activity and resistance to apoptosis. To investigate this possibility, dysadherin‐expressing or dysadherin‐knockdown breast cancer cells were cultured with the AKT inhibitor triciribine, and cell motility, actin organization, EMT and anti‐apoptosis were assessed. Before these assays were conducted, we determined the cytotoxic concentration of triciribine in MCF‐7 and MDA‐MB‐231 cells. MTT analysis showed that triciribine did not induce growth inhibition of human breast cancer cells at concentrations of up to 100 nM (Supporting information Fig. S2); therefore, concentrations of <100 nM were used to study of the effect of triciribine on the dysadherin‐mediated pro‐metastatic effects. In addition, to rule out the possibility that triciribine treatment might have any unexpected effect on dysadherin expression, we analyzed the change in dysadherin protein expression in MCF‐7 cells after transfection with mock or Dys following 48 h treatment with 100 nM triciribine. Triciribine treatment did not affect the dysadherin protein expression (Supporting information Fig. S3).
First, cell motility assay showed that triciribine treatment reduced the ability of dysadherin to promote cell motility in breast cancer cells (Fig. 5A). Second, F‐actin staining showed that triciribine treatment decreased the ability of dysadherin to enhance the cortical actin layer at the cell membrane of MCF‐7 cells (Fig. 5B). These results suggest that the ability of dysadherin to promote tumor cell invasion in vitro is dependent on AKT activation. Third, to test whether triciribine treatment influences dysadherin‐induced EMT, dysadherin expressing MCF‐7 cells were cultured with tricirbine. Immunofluorescent staining showed that dysadherin‐expressing cells had a decrease of E‐cadherin and an increase of fibronectin but no change in N‐cadherin and vimentin. Tricirbine treatment reversed dysadherin‐induced EMT (Fig. 5C). qRT‐PCR analysis showed that tricirbine treatment did not affect E‐cadherin mRNA level in MCF‐7 cells after transfection with mock or Dys (Supporting information Fig. S4). Therefore, these results support the possibility that dysadherin‐mediated AKT activation downregulates expression of E‐cadherin through a post‐transcriptional mechanism and enhances the EMT phenotype.
Figure 5.
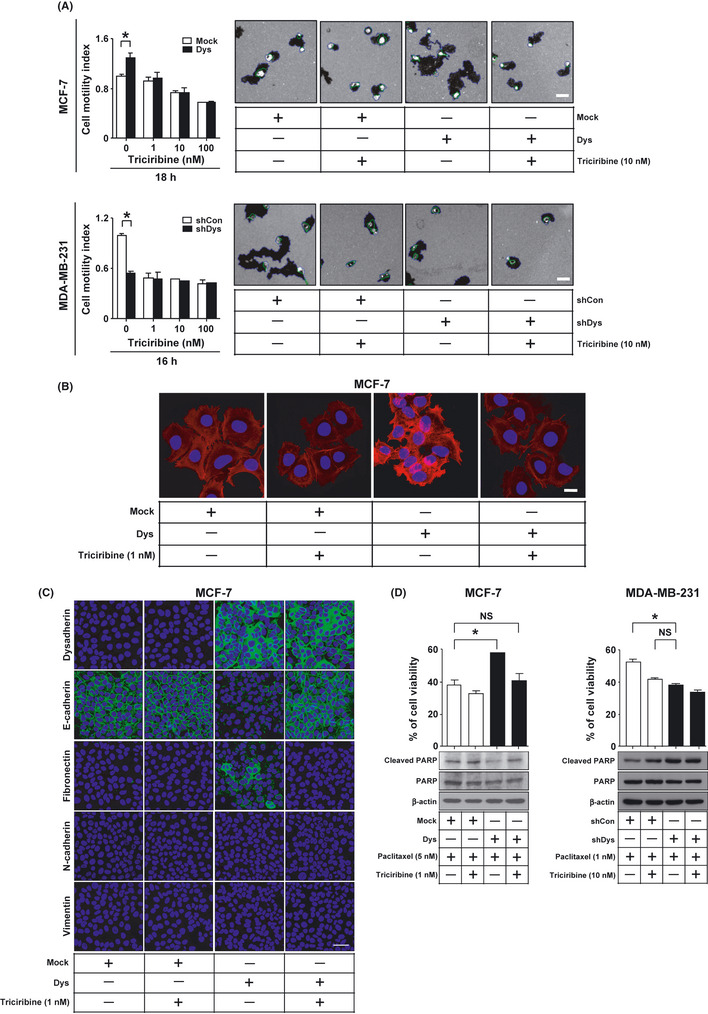
Effect of AKT inhibitor on dysadherin‐mediated metastatic potential in breast cancer cell lines. (A) Motility assay of breast cancer cells after modification of the Dysadherin gene following 16–18 h treatment with 1, 10 and 100 nM triciribine. Data are the mean ± SD (n = 3). Scale bar, 100 µm. (B) Immunofluorescent analysis of F‐actin in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 24 h treatment with 1 nM triciribine. All nuclei were counterstained with DAPI (blue). Scale bar, 100 µm. (C) Immunofluorescent analysis of epithelial–mesenchymal transition‐related proteins in MCF‐7 cells after transfection with mock or Dys following 72 h treatment with 1 nM triciribine. All nuclei were counterstained with DAPI (blue). Scale bar, 50 µm. (D) Immunoblot analysis of cleaved poly‐ADP ribose polymerase (PARP) and MTT assay in breast cancer cells after modification of the Dysadherin gene following 72 h treatment with paclitaxel and triciribine. Data are the mean ± SD (n = 3). *P < 0.01. NS, not significant. b‐actin was used as a loading control.
To test whether triciribine treatment influences dysadherin‐induced apoptotic resistance to paclitaxel, dysadherin‐expressing or dysadherin‐knockdown cells were cultured with triciribine and paclitaxel for 72 h. MTT and immunoblot analysis showed that the presence of dysadherin protected breast cancer cells from apoptosis induced by paclitaxel treatment compared with control cells. Triciribine treatment reversed dysadherin‐mediated protection from apoptosis by paclitaxel treatment (Fig. 5D). These data suggest that AKT activity might mediate the anti‐apoptotic effects of dysadhrein.
Discussion
Dysadherin is recognized as an important player in cancer metastasis;6 however, the signaling pathways downstream of dysadherin in the metastatic process remain largely uncharacterized. To the best of our knowledge, our study is the first experimental report showing that AKT activation plays an important role in mediating the pro‐metastatic effects of dysadherin in breast cancer.
The in vitro phenotype of cell motility reflects the metastatic potential of the cancer cell in vivo. High dysadherin expression in human pancreatic and breast cancer cell lines facilitated cell motility by the recruitment of filamentous actin at the cell membrane.4, 5 Despite these observations, the signal transduction downstream of dysadherin expression that promotes cell motility is still poorly understood. Our work is the first evidence that AKT activation may mediate the ability of dysadherin to promote cell motility in breast cancer.
Dysadherin overexpression promoted breast cancer invasiveness by a mechanism that involves the upregulation of chemokine (C–C motif) ligand 2 (CCL2).5 Therefore, we investigated the interaction between CCL2 production and dysadherin‐mediated AKT activation in human breast cancer cell lines. In preliminary studies, qRT‐PCR analysis showed that triciribine treatment suppressed the CCL2 mRNA expression in MDA‐MB‐231 and Hs578T breast cancer cell lines (Supporting information Fig. S5). The addition of recombinant CCL2 protein did not reverse the effect of dysadherin knockdown to attenuate ATK phosphorylation in MDA‐MB‐231 cells (Supporting information Fig. S6). Therefore, these results support the possibility that the production of tumor‐promoting chemokine, CCL2, might be a downstream target of dysadherin‐mediated AKT activation in human breast cancer cells.
Epithelial–mesenchymal transition is implicated in the conversion of early‐stage tumors into invasive malignancies.23 A hallmark of EMT is the loss of E‐cadherin expression; the loss of E‐cadherin increases tumor‐cell invasiveness in vitro and contributes to the transition of adenoma to carcinoma in animal models.26 A body of clinical data suggests that dysadherin expression has significant negative correlation with E‐cadherin expression in many different cancer types.6 Experimental data also suggest that the transfection of Dys into PLC/PRF/5 liver cancer cells causes the change in E‐cadherin protein expression and function without affecting its expression on the mRNA level;3 however, the regulatory mechanism governing this process still remains unclear. This study provides the first evidence that AKT activation may be a signal by which dysadherin can alter E‐cadherin function.
Dysadherin overexpression had no effect on cell proliferation, but supported cell survival. These results suggest that dysadherin might not affect RAS‐MAPK signaling. Therefore, we investigated whether the dysadherin expression affects the level of pMEK expression, a downstream molecule of RAS‐MAPK signaling. Immunoblot analysis showed that overexpression or knockdown of the Dysadherin gene did not cause the change in pMEK protein expression in breast cancer cells (Supporting information Fig. S7). Therefore, these results support the possibility that dysadherin contributes to cell survival through a mechanism that does not involve the activation of RAS signaling.
Development of chemoresistance is a serious problem during the treatment of cancer. Drug resistance in cancer is recognized as the doorway for cancer recurrence and metastasis. Elevated expression of dysadherin is associated with sensitivity to combined chemotherapy with irinotecan and 5‐fluorouracil in the treatment of colorectal cancer.27 The transfection of Dys into PLC/PRF/5 liver cancer cells enhances resistance to doxorubicin‐induced apoptosis.7 The present work shows the involvement of dysadherin in paclitaxel chemoresistance of human breast cancer cells. This regulatory mechanism indicates that AKT activation might mediate the ability of dysadherin to promote chemoresistance in breast cancer cells. NF‐κB transcriptional activity is reported to inhibit apoptosis and to induce drug resistance in cancers.28 We have previously shown that expression of dysadherin is associated with enhanced activity of the NF‐κB pathway. In preliminary promoter‐reporter studies, inhibition of AKT activity suppressed the ability of dysadherin to promote the activation of the NF‐κB pathway (Supporting information Fig. S8). To further explore whether inhibition of NF‐κB signaling affects dysadherin‐mediated AKT activation, we examined the change in AKT phosphorylation in T‐47D cells after transfection with mock or Dys following 24 h treatment with 1 μM 6‐amino‐4‐(4‐phenoxyphenylethylamino)quinazoline (NF‐κB inhibitor). Immunoblot analysis showed that inhibition of NF‐κB activity slightly decreased the level of pAKT expression in mock‐expressing T‐47D cells, but did not affect the level of pAKT expression in dysadherin‐overexpressing T‐47D cells (Supporting information Fig. S9). Therefore, these results support that NF‐κB activation might be a downstream signaling of dysadherin‐mediated AKT activation. Deregulation of the AKT pathway is associated with resistance to doxorubicin and 4‐hydroxyl‐tamoxifen, which is a chemotherapeutic drug and estrogen receptor (ER) antagonist used in breast cancer therapy.29 These data suggest that dysadherin status may be worth evaluating as a useful predictor of drug response for breast cancer.
Some breast cancer cell lines including MCF‐7 and MDA‐MB‐231 harbor activating mutations of PIK3CA gene resulted in AKT activation.30 Therefore, first, we determined whether modification of the Dysadherin gene affects PIK3CA expression in breast cancer cells. Immunoblot analysis showed that overexpression or knockdown of the Dysadherin gene did not affect the expression of PIK3CA at protein level in breast cancer cells (Supporting information Fig. S10). Second, we examined the change in AKT phosphorylation in MCF‐7 cells after transfection with mock or Dys following 1 h treatment with 10 μM LY294002 (PIK3CA inhibitor). Immunoblot analysis showed that LY294002 treatment largely decreased the level of pAKT expression in dysadherin‐negative MCF‐7 cells, but slightly decreased the level of pAKT expression in dysadherin‐overexpressing MCF‐7 cells (Supporting information Fig. S11). These results suggest that PIK3CA activation might not be required for dysadherin‐mediated AKT activation.
Collectively, these mechanistic links between dysadherin and AKT in cancer progression suggest new approaches to the treatment of advanced breast cancer. Our present work does not fully elucidate whether AKT activation is a primary or secondary effect of dysadherin expression; therefore, further research is necessary to determine the molecular mechanisms through which dysadherin modulates AKT activation. One possible mechanism by which dysadherin modulates AKT activation is via its unusually long extracellular domain, which may facilitate interactions with other membrane proteins or with extracellular matrix components and may affect signaling dynamics. This hypothesis is supported by several reports. The expression of Episialin (MUC1), a cell membrane glycoprotein, downregulated E‐cadherin‐mediated cell–cell adhesion by means of its highly glycosylated extracellular domain.31 Treatment with benzyl‐α‐GalNAc, a modulator of O‐glycosylation, upregulated E‐cadherin expression in dysadherin‐transfected PLC/PRF/5 liver cancer cells but did not affect E‐cadherin expression in mock expressing cells.32 Given these observations, additional systematic analysis of the dysadherin protein interactome would be helpful for insight into possible mechanisms through which dysadherin modulates AKT activation. The other possible mechanism is that effects on the NaK‐ATPase may mediate the pro‐metastatic effect of dysadherin. Dysadherin is a member of the FXYD family (FXYD5), a family of proteins expressed in a tissue‐specific manner that regulate the function of NaK‐ATPase.3 NaK‐ATPase has been shown to act as a signal transducer in addition to being an ion pump.33, 34 Barwe and colleagues report that NaK‐ATPase is associated with the regulatory subunit of PI3‐kinase that modulates phosphorylation of AKT. The inhibition of NaK‐ATPase in highly motile Moloney sarcoma virus‐transformed Madian–Darby canine kidney cells suppresses cell motility.35 These findings support the possibility that dysadherin might contribute to AKT activation through mechanisms involving changes in NaK‐ATPase; therefore, these molecular details are under active investigation.
Estrogen‐receptor‐negative breast cancer is generally more aggressive and has a poorer prognosis than ER‐positive breast cancer. The experimental restoration of ER‐α to the ER‐negative cell line MDA‐MB‐231 resulted in a reduction of dysadherin expression.5 The ER‐dependent signaling pathway has been shown to be involved in the activation of AKT and the downstream molecules.36 Therefore, further investigating a possible link between ER‐status and dysadherin‐mediated AKT activation will surely lead to a better understanding of the dysadherin biology in breast cancer progression.
In summary, we have shown that dysadherin might play an important role in breast cancer by promoting invasion and metastasis through a mechanism that involoves enhanced AKT activation. Dysadherin could potentially be exploited as a new molecular target for the visualization, prevention or treatment of advanced cancer.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. The correlation between dysadherin and phospho‐AKT (pAKT) expression in breast cancer tissues. Fig. S2. Cell viability analysis in breast cancer cell lines. Fig. S3. Immunoblot analysis of dysadherin in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 48 h treatment with 100 nM triciribine. Fig. S4. qRT‐PCR analysis of E‐cadherin in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 48 h treatment with 1 nM triciribine. Fig. S5. qRT‐PCR analysis of CCL2 in MDA‐MB‐231 and Hs578T cells after 24 h treatment with 100 nM triciribine. Fig. S6. Immunoblot analysis of AKT activity level in MDA‐MB‐231 cells after transfection with shCon (control shRNA) or shDys (Dysadherin shRNA) following 24 h treatment with recombinant CCL2 protein (R&D Systems, Minneapolis, MN, USA; 100 ng/mL). Fig. S7. Immunoblot analysis of the level of MEK and pMEK after modification of the Dysadherin gene in MCF‐7 and MDA‐MB‐231 breast cancer cell lines. Fig. S8. The effect of Akt inhibition on dysadherin‐mediated NF‐κB activation. Fig. S9. Immunoblot analysis of AKT activity level in T‐47D cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 24 h treatment with 1 μM 6‐amino‐4‐(4‐phenoxyphenylethylamino)quinazoline (Calbiochem; NF‐κB inhibitor). Fig. S10. Immunoblot analysis of PIK3CA after modification of the Dysadherin gene in breast cancer cell lines. Fig. S11. Immunoblot analysis of AKT activity level in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 1 h treatment with 10 μM LY294002 (Calbiochem; PIK3CA inhibitor).
Table S1. Clinicopathological characteristics of breast cancer tissue samples by immunofluorescent analysis.
Acknowledgments
We thank Dr Setsuo Hirohashi for providing antibody NCC‐M53 and vector pcDNA‐L3HSV. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2010‐0016028).
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Moody SE, Perez D, Pan TC et al The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 2005; 8: 197–209. [DOI] [PubMed] [Google Scholar]
- 3. Ino Y, Gotoh M, Sakamoto M, Tsukagoshi K, Hirohashi S. Dysadherin, a cancer‐associated cell membrane glycoprotein, down‐regulates E‐cadherin and promotes metastasis. Proc Natl Acad Sci USA 2002; 99: 365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimamura T, Yasuda J, Ino Y et al Dysadherin expression facilitates cell motility and metastatic potential of human pancreatic cancer cells. Cancer Res 2004; 64: 6989–95. [DOI] [PubMed] [Google Scholar]
- 5. Nam JS, Kang MJ, Suchar AM et al Chemokine (C‐C motif) ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res 2006; 66: 7176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nam JS, Hirohashi S, Wakefield LM. Dysadherin: a new player in cancer progression. Cancer Lett 2007; 255: 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Park JR, Kim RJ, Lee YK et al Dysadherin can enhance tumorigenesis by conferring properties of stem‐like cells to hepatocellular carcinoma cells. J Hepatol 2011; 54: 122–31. [DOI] [PubMed] [Google Scholar]
- 8. Dolcet X, Llobet D, Pallares J, Matias‐Guiu X. NF‐kB in development and progression of human cancer. Virchows Arch 2005; 446: 475–82. [DOI] [PubMed] [Google Scholar]
- 9. Prasad S, Ravindran J, Aggarwal BB. NF‐kappaB and cancer: how intimate is this relationship. Mol Cell Biochem 2010; 336: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LoPiccolo J, Granville CA, Gills JJ, Dennis PA. Targeting Akt in cancer therapy. Anticancer Drugs 2007; 18: 861–74. [DOI] [PubMed] [Google Scholar]
- 11. Liu W, Bagaitkar J, Watabe K. Roles of AKT signal in breast cancer. Front Biosci 2007; 12: 4011–9. [DOI] [PubMed] [Google Scholar]
- 12. Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A 1987; 84: 5034–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun M, Wang G, Paciga JE et al AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol 2001; 159: 431–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perez‐Tenorio G, Stal O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 2002; 86: 540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle 2008; 7: 2991–6. [DOI] [PubMed] [Google Scholar]
- 16. Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal 2009; 21: 470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nam JS, Suchar AM, Kang MJ et al Bone sialoprotein mediates the tumor cell‐targeted prometastatic activity of transforming growth factor beta in a mouse model of breast cancer. Cancer Res 2006; 66: 6327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim RJ, Kim SR, Roh KJ et al Ras activation contributes to the maintenance and expansion of Sca‐1pos cells in a mouse model of breast cancer. Cancer Lett 2010; 287: 172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shimamura T, Sakamoto M, Ino Y et al Dysadherin overexpression in pancreatic ductal adenocarcinoma reflects tumor aggressiveness: relationship to e‐cadherin expression. J Clin Oncol 2003; 21: 659–67. [DOI] [PubMed] [Google Scholar]
- 20. Nam JS, Ino Y, Kanai Y, Sakamoto M, Hirohashi S. 5‐aza‐2′‐deoxycytidine restores the E‐cadherin system in E‐cadherin‐silenced cancer cells and reduces cancer metastasis. Clin Exp Metastasis 2004; 21: 49–56. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Nagase H, Watanabe T et al Inhibition of MMP‐9 transcription and suppression of tumor metastasis by pyrrole‐imidazole polyamide. Cancer Sci 2010; 101: 759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci 2005; 96: 379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang Y, Massague J. Epithelial‐mesenchymal transitions: twist in development and metastasis. Cell 2004; 118: 277–9. [DOI] [PubMed] [Google Scholar]
- 24. Nakayama S, Torikoshi Y, Takahashi T et al Prediction of paclitaxel sensitivity by CDK1 and CDK2 activity in human breast cancer cells. Breast Cancer Res 2009; 11: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jimenez C, Hernandez C, Pimentel B, Carrera AC. The p85 regulatory subunit controls sequential activation of phosphoinositide 3‐kinase by Tyr kinases and Ras. J Biol Chem 2002; 277: 41556–62. [DOI] [PubMed] [Google Scholar]
- 26. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 27. Graudens E, Boulanger V, Mollard C et al Deciphering cellular states of innate tumor drug responses. Genome Biol 2006; 7: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bentires‐Alj M, Barbu V, Fillet M et al NF‐kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003; 22: 90–7. [DOI] [PubMed] [Google Scholar]
- 29. McCubrey JA, Steelman LS, Abrams SL et al Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 2006; 46: 249–79. [DOI] [PubMed] [Google Scholar]
- 30. Eckert LB, Repasky GA, Ulku AS et al Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res 2004; 64: 4585–92. [DOI] [PubMed] [Google Scholar]
- 31. Makiguchi Y, Hinoda Y, Imai K. Effect of MUC1 mucin, an anti‐adhesion molecule, on tumor cell growth. Jpn J Cancer Res 1996; 87: 505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsuiji H, Takasaki S, Sakamoto M, Irimura T, Hirohashi S. Aberrant O‐glycosylation inhibits stable expression of dysadherin, a carcinoma‐associated antigen, and facilitates cell‐cell adhesion. Glycobiology 2003; 13: 521–7. [DOI] [PubMed] [Google Scholar]
- 33. Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. Multiple signal transduction pathways link Na+/K + ‐ATPase to growth‐related genes in cardiac myocytes. The roles of Ras and mitogen‐activated protein kinases. J Biol Chem 1998; 273: 15249–56. [DOI] [PubMed] [Google Scholar]
- 34. Mohammadi K, Kometiani P, Xie Z, Askari A. Role of protein kinase C in the signal pathways that link Na+/K + ‐ATPase to ERK1/2. J Biol Chem 2001; 276: 42050–6. [DOI] [PubMed] [Google Scholar]
- 35. Barwe SP, Anilkumar G, Moon SY et al Novel role for Na,K‐ATPase in phosphatidylinositol 3‐kinase signaling and suppression of cell motility. Mol Biol Cell 2005; 16: 1082–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsai EM, Wang SC, Lee JN, Hung MC. Akt activation by estrogen in estrogen receptor‐negative breast cancer cells. Cancer Res 2001; 61: 8390–2. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The correlation between dysadherin and phospho‐AKT (pAKT) expression in breast cancer tissues. Fig. S2. Cell viability analysis in breast cancer cell lines. Fig. S3. Immunoblot analysis of dysadherin in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 48 h treatment with 100 nM triciribine. Fig. S4. qRT‐PCR analysis of E‐cadherin in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 48 h treatment with 1 nM triciribine. Fig. S5. qRT‐PCR analysis of CCL2 in MDA‐MB‐231 and Hs578T cells after 24 h treatment with 100 nM triciribine. Fig. S6. Immunoblot analysis of AKT activity level in MDA‐MB‐231 cells after transfection with shCon (control shRNA) or shDys (Dysadherin shRNA) following 24 h treatment with recombinant CCL2 protein (R&D Systems, Minneapolis, MN, USA; 100 ng/mL). Fig. S7. Immunoblot analysis of the level of MEK and pMEK after modification of the Dysadherin gene in MCF‐7 and MDA‐MB‐231 breast cancer cell lines. Fig. S8. The effect of Akt inhibition on dysadherin‐mediated NF‐κB activation. Fig. S9. Immunoblot analysis of AKT activity level in T‐47D cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 24 h treatment with 1 μM 6‐amino‐4‐(4‐phenoxyphenylethylamino)quinazoline (Calbiochem; NF‐κB inhibitor). Fig. S10. Immunoblot analysis of PIK3CA after modification of the Dysadherin gene in breast cancer cell lines. Fig. S11. Immunoblot analysis of AKT activity level in MCF‐7 cells after transfection with mock (empty vector) or Dys (Dysadherin cDNA) following 1 h treatment with 10 μM LY294002 (Calbiochem; PIK3CA inhibitor).
Table S1. Clinicopathological characteristics of breast cancer tissue samples by immunofluorescent analysis.