

Abstract
The chemokine CC motif receptor 5 (CCR5) and its ligands have been reported to be associated with cancer progression and metastasis. Although recent researches have demonstrated a fundamental role of hypoxia in cancer, the effect of hypoxia on the expression and function of CCR5 and its ligands in cancer cells is unknown. Here, we investigated the status of CCR5 and its ligands in cancer cells under hypoxic conditions. Quantitative polymerase chain reaction, western blotting and immunofluorescence staining showed that hypoxia induced a strong increase of CCR5 expression. Dual luciferase assay and mRNA stability analysis indicated that hypoxia‐induced CCR5 mRNA expression relied on both transcriptional and posttranscriptional mechanisms. We detected the expression of CCR5 ligands and found that chemokine CC motif ligand 5 (CCL5) was induced under hypoxia. Recombinant human CCL5 stimulated cell migration rather than cell proliferation under hypoxia, and neutralization of CCL5 inhibited hypoxia‐induced migration of cancer cells. Similarly, overexpression of CCR5 increased cell migration, and knockdown of CCR5 attenuated hypoxia‐mediated cell migration. We further showed that hypoxia‐inducible factor‐1α (HIF‐1α) was involved in CCR5 and CCL5 regulation under hypoxia. HIF‐1α mRNA levels were highly correlated with CCR5 mRNA and CCL5 mRNA levels in clinical samples. CCR5 and CCL5 were highly expressed in breast cancer lymph nodes metastases. Taken together, our data suggest that CCR5‐CCL5 interaction promotes cancer cell migration under hypoxia. (Cancer Sci 2012; 103: 904–912)
Chemokines are initially described as regulators of leukocyte trafficking for their ability to stimulate migration of leukocytes during inflammatory processes. They bind to their cognate receptors, most of which belong to the G protein‐coupled receptor family.1 The CC chemokine receptor 5 (CCR5) is a member of CC chemokine receptor family. CCR5 is identified as the receptor for CCL3 (macrophage inflammatory protein 1α (MIP 1α), CCL4 (MIP 1β) and CCL5 (RANTES).2 CCR5 and its ligands stimulate the migration of memory/effector Th1 cells, macrophages, natural killer (NK) cells, and immature dendritic cells to damaged or infected sites.3 Recent evidence has suggested the involvement of CCR5 and/or its ligands in tumorigenesis. A variety of human cancer cells, including breast cancer cells,4 lung cancer cells,5 Hodgkin's lymphoma cells6 and prostate cancer cells7 are found to express CCR5, or release its ligands. It has been demonstrated that cancer cells stimulate the secretion of CCL5 from mesenchymal stem cells, which then acts in a paracrine fashion to enhance cancer metastasis.4 CCL5‐CCR5 interaction is reported to provide cancer cells with a proliferative advantage.8 However, conflicting reports also demonstrate that by boosting T‐cell responses to tumors, CCR5 has a specific, ligand‐dependent role in optimizing antitumor activity.9, 10 CCR5 reduces chemical‐induced fibrosarcoma incidence and growth, and CCL5 blockade improves the efficacy of cancer immunochemotherapy.11 CCR5 and/or its ligands may play a dual role by contributing to cancer progression while at the same time attracting antitumor T cells.
Tumors emerge from the somatic evolution of a genetically diversified cancer‐cell population under selective pressures of local environment. Hypoxia is a major selective factor that promotes the growth of tumors with a diminished susceptibility to radiation and chemotherapy. Hypoxia has been reported to associate with cancer progression, cancer metastasis, and thus poor prognosis. Hypoxia greatly influences cellular phenotypes by altering the expression of genes that promote angiogenesis, cell survival, and metastasis.12 Presently, the status of CCR5 and its ligands in cancer cells under hypoxia is unknown. We therefore investigated the effect of hypoxia on the expression of CCR5 and its ligands in MDA‐MB‐231 and MDA‐MB‐435 cells, and explored the role of CCR5 and CCL5 in cancer cells under hypoxia. We postulated a mechanism involving hypoxia‐inducible factor‐1α (HIF‐1α) in regulating CCR5 and CCL5 expression.
Materials and Methods
Tissue samples
Tissue samples were obtained from patients who underwent surgery at the Department of Surgery, The Fourth People's Hospital, Xuzhou, China. They were obtained from patients with breast cancer. The age range was 28–67 years. In total, 14 primary breast tumors and 14 matched metastatic carcinomas in lymph nodes were studied. All the samples were classified according to World Health Organization (WHO) criteria. None of the patients had received pre‐operative chemotherapy or radiotherapy. Pathologists confirmed the histopathological diagnosis for each specimen. The sampling and usage of all cancer tissues in this study were approved by the ethics committee of China Pharmaceutical University, and informed consent for participation in this study was obtained from each patient.
Cell culture and regents
MDA‐MB‐231 cells were cultured as monolayers in Roswell Park Memorial Institute (RPMI)‐1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 μg/mL streptomycin. MDA‐MB‐435 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma) with 10% FCS. To create hypoxic conditions, 70–80% confluent cells were placed in a tissue culture incubator in an atmosphere of 1% O2 and 5% CO2 at 37°C. Recombinant human CCL5 was purchased from R&D Systems (Minneapolis, MN, USA). YC‐1, the molecular inhibitor of HIF‐1α, was obtained from Sigma.
Immunohistochemical analysis
Immunohistochemical studies were performed in a standard protocol (for details, see Data S1).
Quantitative and semi‐quantitative RT‐PCR
Total RNA was isolated from cultured cell lines or frozen samples using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Quantitative polymerase chain reaction (PCR) was carried out on an iCycler Real‐time PCR Detection System (Bio‐Rad, Hercules, CA, USA) (for details, see Data S1).
Western blotting
Total cellular extracts were prepared using standard procedures. Western blotting was performed using specific antibodies against CCR5 (Abcam, Cambridge, UK) and β‐actin (Sigma) (for details, see Data S1).
Immunofluorescence
Cells were seeded on coverslips and incubated in hypoxic conditions for 12 h. Cells were then cultured with anti‐CCR5 primary antibody and labeled with fluorescein isothiocyanate (FITC)‐conjugated secondary antibody (for details, see Data S1).
Migration assay
Cell migration assays were conducted in a double chamber transwell (8‐mm pore size; Corning Costar, Acton, MA, USA)13 (for details, see Data S1).
Proliferation assay
Cell proliferation assay was performed by using standard methods explained briefly in Data S1.
Enzyme‐linked immunosorbent assay
Cells were incubated under hypoxic conditions with serum deprived medium for 12 h and the supernatants were harvested. Each supernatant was centrifuged at 2000g and stored at −70°C until analysis. Enzyme‐linked immunosorbent assay (ELISA) was performed according to the manufacturer's instructions for human CCL5 (R&D Systems).
Luciferase reporter gene assay
The 1040bp CCR5 promoter14 was cloned into the pGL‐3 basic vector (Promega, Madison, WI, USA), between the KpnI and NheI site, immediately 5′ upstream of the luciferase gene. Transfections were done using Lipofectamine 2000 transfection reagents (Invitrogen) according to manufacturer instructions. Briefly, MDA‐MB‐231 cells were seeded in 24‐multi‐well plates and were cotransfected with reporter plasmids (pGL3‐CCR5) and renilla luciferase reporter vector pRL‐TK. After 6 h, the transfection mixture was replaced with fresh medium and cells were allowed to recover for 18 h. Cells were then treated for different times under hypoxia or normoxia, and luciferase activities were determined and data were normalized with respect to renilla luciferase activity.
Plasmids construction and cell transfection
Plasmids construction and cell transfection were performed as previously described.15 Briefly, full‐length cDNA for CCR5 and HIF‐1α were amplified and cloned into pGEM‐T vectors (Promega). Cloned fragments were recovered and ligated into pcDNA3.1+ (Invitrogen). Transfections were done using Lipofectamine 2000 according to manufacturer instructions. Cells transfected with empty vectors were used as controls. To select stable transfectants, 48 h after transfection, cells were passaged and grown in medium containing G418. A limited dilution was used in a 96‐well plate for repeated colony selection. After 3 weeks of culture, visible colonies were picked up, expanded and maintained. Western blot was done to confirm the overexpression of target genes.
Short hairpin RNA and RNA interference
The human CCR5 and HIF‐1α gene sequence were analyzed for a potential siRNA target with web‐based siRNA target finder and design tools. pSilencer2.1 (Ambion, Austin, TX, USA) was used according to the manufacturer's protocol for construction of human CCR5 and HIF‐1α siRNA vectors. Cells were transfected with pSilencer2.1‐CCR5 (shCCR5) or pSilencer2.1‐HIF1α (shHIF1α) and selected as hygromycin‐resistant pools. The shRNA sequences were as follows (targeting sequence): shCCR5‐1, 5′‐GAGCATGACTGACATCTAC‐3′ (target position: 543);16 shCCR5‐2, 5′‐CTCTGCTTCGGTGTCGAAA‐3′ (target position: 1016); shHIF1α‐1, 5′‐CCATATAGAGATACTCAAA‐3′ (target position: 2376);15 shHIF1α‐2, 5′‐CTAACTGGACACAGTGTGTTT‐3′ (target position: 783). CCR5 and HIF‐1α knockdown were confirmed by quantitative PCR and western blot.
Statistical analyses
Results were mean ± standard error of the mean (SEM) of three independent experiments. Unpaired Student's t‐test and Spearman correlation coefficient analysis were used. P‐values were two‐sided: 0.05 was considered statistically significant.
Results
Hypoxia increases CCR5 and CCL5 expression in cancer cells
Hypoxia is a consequence of a structurally and functionally disturbed tumor microenvironment and has been demonstrated to regulate the expression of some chemokine receptors (e.g. CXCR4). To determine whether hypoxia mediated CCR5 expression in cancer cells, we investigated the level of CCR5 expression in response to hypoxia. As shown in Figure 1(a), MDA‐MB‐231 and MDA‐MB‐435 cells, cultured in hypoxia for 1–12 h, showed increased CCR5 mRNA expression, as compared with normoxic controls. Cells also showed up‐regulation of CCR5 protein under hypoxic conditions (Fig. 1b). These results were confirmed by immunofluorescence staining (Fig. 1c), wherein hypoxia induced a strong increase of CCR5 expression in MDA‐MB‐231 cells. Clearly, these results showed that the expression of CCR5 was selectively controlled by hypoxia.
Figure 1.
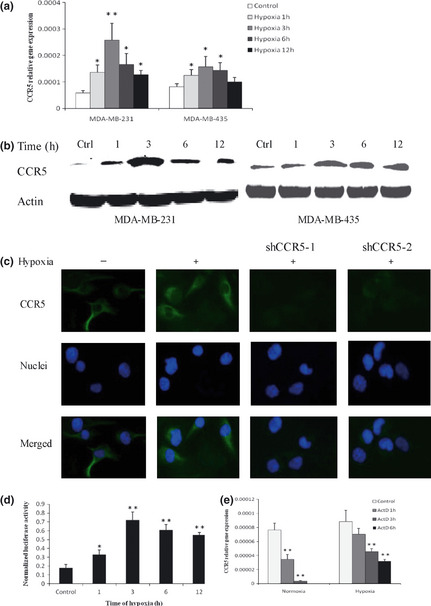
Effect of hypoxia on CCR5 expression. (a) Relative gene expression of CCR5. Cells were cultured under hypoxic conditions for 1–12 h, or under normoxia. Total RNA was tested for CCR5 mRNA by quantitative polymerase chain reaction (qPCR). (b) Western blotting for CCR5 protein under hypoxic or normoxic conditions. Results presented are representative of at least three independent experiments. (c) MDA‐MB‐231 cells were cultured under hypoxia for 12 h, and immunofluorescence staining of CCR5 assayed. Cells in which CCR5 expression was inhibited by shRNA knockdown were used as controls. Nuclei were visualized using Hochest 33342. All experiments were repeated three times. (d) MDA‐MB‐231 cells were co‐transfected with pGL3‐CCR5 and pRL‐TK reporter vectors and relative luciferase activity was measured after exposure to hypoxia for different times. (e) Stabilization of CCR5 mRNA by hypoxia. MDA‐MB‐231 cells were cultured for 6 h under normoxia or hypoxia, and in the presence or absence of 1 μg/mL actinomycin D. Total RNA was extracted and analyzed by quantitative PCR for CCR5 mRNA levels. *P < 0.05; **P < 0.01.
To assess the mechanisms of hypoxia action, dual luciferase reporter gene assay was performed to estimate the activity of CCR5 promoter under hypoxia. Cells were transiently cotransfected with pGL3‐CCR5 and pRL‐TK and exposed to hypoxia for 1–12 h. We found that hypoxia significantly increased CCR5 promoter activity (Fig. 1d). To further investigate posttranscriptional mechanisms of hypoxia, CCR5 mRNA stability in hypoxic conditions was measured. Cells were cultured either in normoxia or hypoxia in the presence or absence of 1 μg/mL actinomycin D. Total RNA was extracted at different times as indicated. As shown in Figure 1(e), the decay of CCR5 mRNA was greatly reduced when cells were cultured in hypoxic conditions, suggesting that hypoxia‐induced CCR5 mRNA expression relied on both transcriptional and posttranscriptional mechanisms.
CCR5 acts through three ligands: CCL3 (MIP 1α), CCL4 (MIP 1β) and CCL5 (RANTES). We investigated CCL3, CCL4 and CCL5 in cancer cells by semi‐quantitative reverse transcription‐polymerase chain reaction (semi‐qRT‐PCR). Substantial amounts of CCL4 and CCL5 mRNA were found; however, CCL3 was not expressed in both cell lines (Fig. 2a). We next investigated the effect of hypoxia on CCL4 and CCL5 expression by cancer cells. As shown in Figure 2(b), CCL5 mRNA showed a strong increase under hypoxia for a period of 12 h. Hypoxia did not induce any significant changes on CCL4 mRNA. These results showed that the ligand for CCR5 under hypoxia in cancer cells should be CCL5. As CCL5 is known to act on target cells via three high affinity receptors, termed CCR1, CCR3 and CCR5, we proceeded to investigate whether CCR1 and CCR3 also interact with CCL5 under hypoxia. We showed that MDA‐MB‐231 and MDA‐MB‐435 cells expressed the highest levels of CCR5 mRNA, compared with CCR1 and CCR3. CCR1 and CCR3 mRNA levels were not much affected by hypoxia (Fig. 2c). Moreover, no significant differences in the expression of CCR1 and CCR3 between breast cancer tumors and tumor‐adjacent tissues were found (Fig. 2d). Thus, the main receptor for CCL5 under hypoxia was determined to be CCR5. We further measured CCL5 production by ELISA. Notably, the level of CCL5 secreted by MDA‐MB‐231 cells cultured under hypoxia for 12 h accumulated to a level approximately 1.9‐fold higher than that produced under normoxia, and CCL5 produced by MDA‐MB‐435 obtained a approximately 1.6‐fold higher than normoxic controls (Fig. 2e). These data showed that CCL5 was induced under hypoxia by cancer cells, and indicated that CCR5‐CCL5 interaction might play a role in tumor hypoxic environment.
Figure 2.
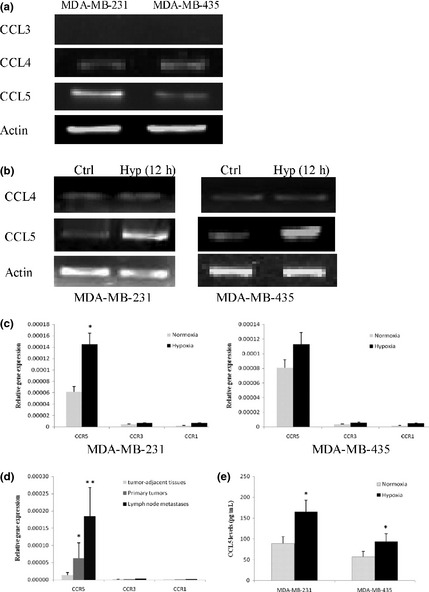
CCL5 secretion under hypoxia. (a) Gene expression of CCL3, CCL4 and CCL5 in cancer cells. Total RNA was subjected to reverse transcription‐polymerase chain reaction (RT‐PCR) using specific primers. (b) Gene expression of CCL4 and CCL5 in MDA‐MB‐231 and MDA‐MB‐435 cells. Cells were cultured under hypoxic conditions for 12 h, or under normoxia. Total RNA was tested for CCL4 and CCL5 mRNA levels by conventional PCR. The data in the figure were representatives of at least three independent experiments. (c) Effects of hypoxia on CCR1, CCR3 and CCR5 expression by MDA‐MB‐231 and MDA‐MB‐435 cells. Cells were incubated for 12 h in normoxia or hypoxia, and CCR1, CCR3 and CCR5 gene expression were determined by quantitative PCR. (d) Relative gene expression of CCR1, CCR3 and CCR5 were analyzed by quantitative PCR in clinical samples. (e) CCL5 production in MDA‐MB‐231 cells was measured by enzyme linked immunosorbent assay (ELISA) after exposure to hypoxia for 12 h. *P < 0.05.
CCR5‐CCL5 interaction is essential in hypoxia‐induced cell migration
To investigate possible contributions of elevated CCL5 secretion under hypoxia, recombinant human CCL5 was administered to MDA‐MB‐231 and MDA‐MB‐435 cells to determine if increased CCL5 production could confer any proliferative advantages to cancer cells. The stable transfectants of MDA‐MB‐231 and MDA‐MB‐435 cells overexpressing CCR5 were used (Fig. S1). As shown in Figure 3(a), we observed significant increases in cell number in transfected MDA‐MB‐231 and MDA‐MB‐435 cells grown in the presence of 1 or 5 ng/mL CCL5, which was not observed in mock cells. Overexpression of CCR5 alone also did not promote cancer cell proliferation. The data suggested that both increased production of CCL5 and high levels of CCR5 were necessary to promote cancer cell proliferation. Although CCL5 treatment in combination with ectopic CCR5 expression greatly enhanced cell survival under hypoxia, they did not protect MDA‐MB‐231 and MDA‐MB‐435 cells from hypoxia‐induced cell death (Fig. 3b).
Figure 3.
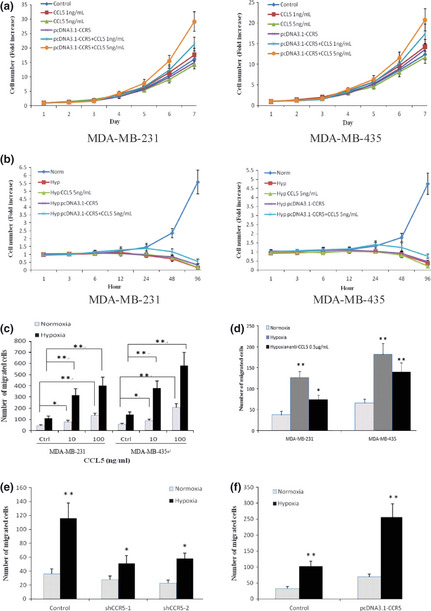
CCR5 and CCL5 in hypoxia‐induced cell migration. (a) MDA‐MB‐231 and MDA‐MB‐435 cells were stably transfected with pcDNA3.1‐CCR5, and then cultured in the presence of CCL5 or not for 7 days (2% fetal calf serum [FCS]‐containing medium). Growth curves were shown. Untransfected cells were used as controls. (b) Transfected cells were cultured under hypoxia and CCL5 was added (2% FCS‐containing medium). Growth curves were shown. Untransfected cells in normoxia were used as controls. (c) Recombinant human CCL5 induced cell migration under normoxic or hypoxic conditions. Different concentrations of CCL5 were added to the lower chamber and serum‐free base medium was used as control. Average numbers of cells migrating in five randomly selected fields were counted 12 h after seeding. (d) Neutralization of CCL5 inhibited hypoxia‐induced cell migration. Cells were pretreated with 0.5 μg/mL anti‐CCL5 antibody for 1 h, and subjected to migration assay. Cells without pretreatment were used as controls. The chemoattractant was 2% fetal calf serum (FCS). (e) Blocking CCR5 expression with shRNA knockdown in MDA‐MB‐231 cells decreased the ability of hypoxia to induce cell migration. Forty‐eight hours after shRNA transfection, cells were subjected to migration assay under normoxia or hypoxia. The chemoattractant was 2% FCS. (f) The migration of MDA‐MB‐231 cells increased after they were transfected with pcDNA3.1‐CCR5. The chemoattractant was 2% FCS. *P < 0.05; **P < 0.01.
Cell migration assay was assessed to investigate the role of CCL5 in cancer metastasis under hypoxia. We found that CCL5 stimulated the migratory responses of MDA‐MB‐231 and MDA‐MB‐435 cells both in normoxic or hypoxic conditions, and responses under low oxygen conditions were higher (Fig. 3c). Notably, exposure of cancer cells to hypoxia stimulated cell migration even without the addition of CCL5. To evaluate possible contributions of increased CCL5 secretion to hypoxia‐induced migration, cell migration was assayed in the presence of anti‐human CCL5 neutralizing antibody. As shown in Figure 3(d), neutralization of CCL5 attenuated hypoxia‐induced migration of cancer cells. Inhibition of CCL5 resulted in approximately 50% reduction of hypoxia‐mediated cell migration in MDA‐MB‐231 cells and approximately 30% in MDA‐MB‐435 cells, indicating that the actions of CCL5 were responsible for much, although not all, of the observed hypoxia‐induced cancer cell migration.
We showed that CCR5 expression was increased by cancer cells under hypoxia, supporting the notion that CCL5 might act primarily in an autocrine fashion in hypoxic conditions. To probe whether the observed hypoxia‐induced cell migration required CCR5–CCL5 interactions, we inhibited CCR5 expression in MDA‐MB‐231 cells through shRNA knockdown (Fig. S2). Inhibition of CCR5 expression greatly attenuated the ability of hypoxia to enhance the metastasis of MDA‐MB‐231 cells (Fig. 3e). In contrast, overexpression of CCR5 stimulated cancer cell migration. Hypoxia was more effective at eliciting transfected cell migration (Fig. 3f). Taken together, these results indicated a clear effect of CCR5‐CCL5 interaction on cancer cell migration in hypoxic conditions.
Effect of HIF‐1α on CCR5 and CCL5
The cellular response to hypoxia involves the stabilization of a HIF‐1 transcriptional complex that activates genes that promote angiogenesis, cell survival and metastasis. To investigate the role of HIF‐1α in the hypoxic induction of CCR5, we inhibited HIF‐1α expression in MDAMB‐231 cells by more than 80% through shRNA knockdown (Fig. S3). MDA‐MB‐231 cells transiently transfected with HIF‐1α shRNA constructs were cultured under normoxic or hypoxic conditions for 12 h, and total RNA was tested for CCR5 mRNA levels by real‐time PCR. As shown in Figure 4(a), knockdown of HIF‐1α abrogated the upregulation of CCR5 mRNA under hypoxia. Furthermore, YC‐1, a small molecule inhibitor of HIF‐1,17, 18 also prevented the hypoxic induction of CCR5 mRNA. Western blot analysis showed that CCR5 protein accumulation caused by hypoxia was blocked either by HIF‐1α shRNA knockdown or by the small molecule inhibitor YC‐1 (Fig. 4b). We next overexpressed HIF‐1α in MDA‐MB‐231 cells and analyzed its effects on CCR5 expression (Fig. S4). The overexpressed HIF‐1α induced significant accumulation of CCR5 mRNA and protein levels both in normoxic and hypoxic conditions, as shown in Figure 4(c,d). Indeed, overexpression of HIF‐1α induced approximately fivefold higher levels of CCR5 mRNA when cultured under hypoxic conditions relative to normoxic conditions. These results showed that CCR5 expression was regulated by HIF‐1α.
Figure 4.
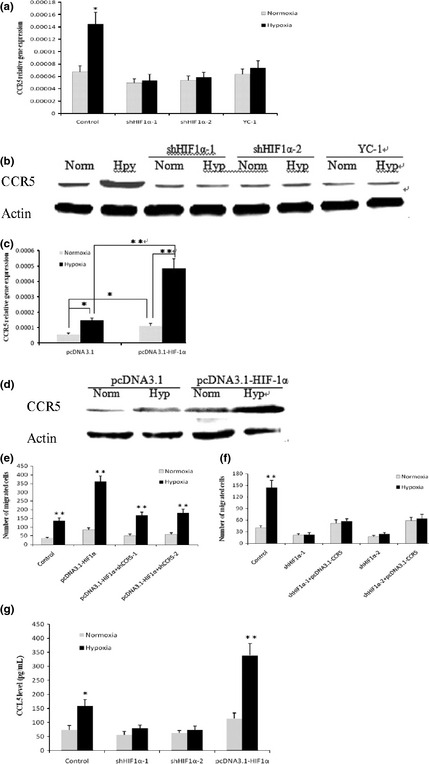
Hypoxia‐inducible factor‐1α (HIF‐1α)‐dependent upregulation of CCR5 and CCL5 by hypoxia. (a) MDA‐MB‐231 cells were transfected with shRNA vectors targeting HIF‐1α (shHIF1α‐1 and shHIF1α‐2), or pretreated with 50 μM YC‐1 for 30 min, and incubated under normoxia or hypoxia for 12 h. CCR5 mRNA was measured by quantitative polymerase chain reaction (PCR) and (b) CCR5 protein was tested by western blot. Data shown are representative of three independent experiments. (c) MDA‐MB‐231 cells were transfected with pcDNA3.1‐HIF‐1α, and cultured in normoxia or hypoxia for 24 h. CCR5 mRNA levels were tested by quantitative PCR and (d) CCR5 protein was measured by western blot. Cells transfected with empty vectors were used as controls. (e) CCR5 in HIF‐1α‐mediated cancer cell migration. MDA‐MB‐231 cells were stably transfected with pcDNA3.1‐HIF1α, and then transiently transfected with shCCR5 vectors. Cells were subjected to migration assay under normoxia or hypoxia. (f) MDA‐MB‐231 cells were stably transfected with shHIF1α‐1 or shHIF1α‐2, and then transiently transfected with pcDNA3.1‐CCR5. Cell migratory ability was measured in normoxic or hypoxic conditions. The chemoattractant was 2% fetal calf serum (FCS). (g) MDA‐MB‐231 cells showed elevated CCL5 production after they were transiently transfected with pcDNA3.1‐HIF1α; HIF‐1α knockdown abolished hypoxia‐induced CCL5 secretion. *P < 0.05; **P < 0.01.
We proceeded to explore the possible involvement of CCR5 in HIF‐1α‐mediated cancer cell migration. HIF‐1α has been demonstrated to promote cancer metastasis by enhancing the expression of key factors implicated in cancer cell migration and invasion. To probe whether the HIF‐1α‐mediated cell migration required CCR5, we stably transfected MDA‐MB‐231 cells with pcDNA3.1‐HIF1α, and then inhibited CCR5 expression through shRNA knockdown in MDA‐MB‐231/HIF1α cells. As shown in Figure 4(e), overexpression of HIF‐1α increased MDA‐MB‐231 cell migration, and silencing of CCR5 greatly attenuated the HIF‐1α‐stimulated cell mobility both under normoxic and hypoxic conditions. Next, we inhibited the expression of HIF‐1α in MDA‐MB‐231 cells, and then tested whether overexpression of CCR5 restores cell migratory ability. We found that knockdown of HIF‐1α abrogated hypoxia‐induced cell migration (Fig. 4f). Notably, silencing of HIF‐1α reduced the motility of MDA‐MB‐231 cells by approximately 50% in normoxic conditions, and overexpression of CCR5 fully restored cell migratory ability. Finally, we investigated the effect of HIF‐1α on the production of CCL5. Inhibition of HIF‐1α expression in MDA‐MB‐231 cells, achieved using shRNA knockdown, abrogated the effect of hypoxia to enhance the production of CCL5. In contrast, overexpression of HIF‐1α increased CCL5 secretion both in normoxic or hypoxic conditions, as measured by ELISA assay (Fig. 4g). Taken together, these data indicated that HIF‐1α contributed to CCR5 and CCL5 regulation under hypoxia.
Expression of CCR5 and CCL5 in primary breast cancer and lymph node metastases
We investigated the status of CCR5 and CCL5 in 14 breast cancer patients who developed lymph node metastases. Quantitative PCR analyses of tumor‐adjacent tissues, primary tumors and lymph nodes metastases revealed significant increases of CCR5 and CCL5 expression in breast cancer, suggesting the increase of CCR5 and CCL5 expression during malignant progression of cancer (Fig. 5a). Our data also showed that HIF‐1α mRNA levels were highly correlated with both CCR5 mRNA (R = 0.7835, P = 0.001091) and CCL5 mRNA levels (R = 0.6985, P = 0.003561) in clinical samples (14 primary tumors and 14 lymph node metastases), as shown in Figure 5(b). The immunohistochemical analysis showed that CCR5 was expressed in nine of 14 tumor sections analyzed, and overexpressed in 11 of 14 lymph nodes metastases examined. CCL5 expression was significantly elevated in 13 of 14 lymph node metastases, whereas it present at low levels (n = 11) in matched primary tumors (Fig. 5c). CCR5 and CCL5 primarily located in the cytoplasm. We have previously shown that HIF‐1α is highly expressed in breast cancer metastasis;15 these findings demonstrated that CCR5 and CCL5 were also present in breast cancer metastasis, and suggested that CCR5‐CCL5 interaction might play a role in cancer metastasis in an autocrine fashion.
Figure 5.
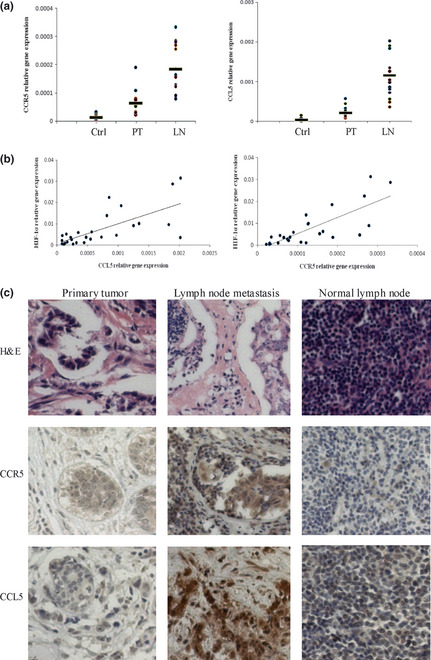
Overexpression of CCR5 and CCL5 in samples obtained from breast cancer patients (n = 14). (a) Expression of CCR5 and CCL5 were analyzed by quantitative polymerase chain reaction (PCR) in tumor‐adjacent tissues (Ctrl), primary tumors (PT) and lymph nodes metastases (LN). (b) HIF‐1α, CCR5 and CCL5 mRNA were measured by quantitative PCR. The correlations between HIF‐1α and CCR5, HIF‐1α and CCL5 were shown. (c) Expression of CCR5 and CCL5 in primary tumors and lymph node metastases. Hematoxylin and eosin (H&E) stain confirmed the pathologic characters of clinical samples.
Discussion
It is well established that hypoxia is a powerful driving force in tumor progression. Hypoxia is known to induce genes involved in the regulation of cell proliferation, extracellular matrix production, cell adhesion, and other hallmarks of tumorigenesis.19, 20, 21 Here, we reported that hypoxia increased CCR5 and CCL5 expression, which contributed substantially to cancer cell migration in an autocrine fashion. We also showed that HIF‐1α was involved in the regulation of CCR5 and CCL5 in hypoxic conditions.
It is intriguing that hypoxia inhibited cancer cell growth while at the same time increasing the expression of CCR5 and CCL5, which exerted pro‐survival functions. This was likely caused by the moderate mediation of CCR5 and CCL5 during hypoxia. CCL5‐induced cancer cell proliferation was dependent on high levels of CCR5 protein. Incubation of MDA‐MB‐231/CCR5 and MDA‐MB‐435/CCR5 cells in the presence of CCL5 resulted in an increase of cell growth, but untransfected cells were not affected. It is also reported that MCF‐7 cells that overexpressed CCR5, but not control cells, exhibited a significant increase in cell number after grown in the presence of CCL5.8 Although higher levels than normoxic control were still present at 12 h, CCR5 protein levels decreased after exposure to hypoxia for 6–12 h. Furthermore, hypoxia‐induced CCL5 secretion was much lower than the CCL5 concentrations used to stimulate cancer cell growth.
Interestingly, although the effect of hypoxia on CCR5 expression in cancer cells is unknown, hypoxia has been shown to induce or repress CCR5 expression in other target cells. The hypoxic inhibition of a cluster of inflammatory genes, including CCR5, is found in human peripheral blood monocytes22 and mouse macrophages.23 However, other studies demonstrate that hypoxia strongly upregulates the expression of CCR5 in human dendritic cells24 and human hepatic stellate cells.25 The discrepancy of these reports may indicate that the effect of hypoxia varies depending on the specific cell functional status rather than on the gene under consideration. Mononuclear phagocyte migration to hypoxic pathological sites is a highly regulated process. Hypoxia may provide a stop signal to their concentration within hypoxic tissues by decreasing the expression of specific chemokine receptors.22 In contrast, hypoxia strongly enhances the innate immune functions of dendritic cells by inhibiting their maturation, increasing both their production of inflammatory cytokines and their chemokine receptors expression.24 Our data suggested that, in response to hypoxia, cancer cells highly expressed CCR5 and CCL5. Our in vivo data also revealed abundant expression of CCR5 and CCL5 in breast cancer lymph nodes metastases, whereas moderate expression of CCR5 was detected in primary tumors. The increase of CCL5 expression in lymph node metastases was more significant because only weak expression of CCL5 was found in primary tumors. Functional CCR5‐CCL5 interaction in cancer cells might have an important role in facilitating invasion and migration of cancer cells to target organs.
Under hypoxia, the most global regulator identified is the transcriptional activator HIF‐1. It comprises HIF‐1β, a constitutively expressed subunit, and an oxygen‐sensitive inducible subunit, HIF‐1α. Hypoxia increases HIF‐1α protein stabilization, and allows recruitment of transcriptional cofactors and activates HIF transcriptional complexes. Once stabilized and activated, HIF binds to the consensus hypoxia‐responsive element (HRE), which is present in the oxygen‐regulated elements of many known HIF‐1 target genes. Our in vitro and in vivo results clearly suggested that HIF‐1α was involved in the regulation of CCR5 and CCL5 under hypoxic conditions. Whether there are HRE sequences in CCR5 and CCL5 regulatory regions is presently being explored further in our laboratory. We searched the available gene sequence for the core HRE element (5′‐RCGTG‐3′) representing putative HIF‐1 binding sites. A consensus HRE sequence ACGTG was found in the 3′ untranslated region (UTR) of human CCL5 gene. Two possible HIF‐1 binding sites, one located in CCR5 promoter and another in an intron within 5′ UTR of CCR5, were also detected. Whether HIF‐1 transcriptional complexes bind to these sites, thereby inducing gene expression in hypoxic conditions, remains to be investigated.
Although the CCR5‐CCL5 chemokine loop clearly contributed to cancer cell migration under hypoxia, the role of CCL4 is less obvious, despite our demonstration that MDA‐MB‐231 and MDA‐MB‐435 cells expressed both CCL4 and CCL5 and other studies showing that CCR5 binds both CCL4 and CCL5.
It has been demonstrated that chemo‐attracted leukocytes may damage tumors by increasing the immunogenicity through the action of antigen‐presenting cells or by stimulating the adaptive immune response through T‐helper or cytotoxic T cells and NK cells. Activated CCR5+ host T cells with anti‐tumor activity are reported to infiltrate into and accumulate at tumor locations, thereby promoting immune‐mediated tumor destruction and bringing about therapeutic benefits.9, 10 The possible tumor‐suppressive activity seems, however, greatly influenced by the activation state of the host's immune system.26, 27 If the recruited lymphocytes are activated, the resulting immune response will reduce or even prevent tumorigenesis and metastasis. However, tumor cells under immune surveillance can undergo a process referred to as immune editing, and become resistant to immune system. In the tumor microenvironment, tumor‐associated macrophages are skewed to immature or immunosuppressive phenotype characterized by decreased resistance against intracellular parasites and tumors.26, 28 Meanwhile, tumor‐associated macrophages can increase cancer growth by producing factors that promote tissue remodeling and angiogenesis.29
In conclusion, our results underscored the critical importance of the CCR5–CCL5 autocrine interaction in hypoxia‐induce cancer cell migration. CCR5 and CCL5 represent significant therapeutic targets that should be considered in the future treatment of cancer metastasis.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Data S1. Materials and Methods.
Fig. S1. Western blotting for CCR5 protein in MDA‐MB‐231 and MDA‐MB‐435 cells transfected with pcDNA3.1‐CCR5.
Fig. S2. Knockdown of CCR5 by short hairpin RNA specifically designed against human CCR5.
Fig. S3. Knockdown of hypoxia‐inducible factor‐1α (HIF‐1α) by short hairpin RNA specifically designed against human HIF‐1α.
Fig. S4. Western blotting for hypoxia‐inducible factor‐1α (HIF‐1α) protein in MDA‐MB‐231 cells transfected with pcDNA3.1‐ HIF‐1α.
Acknowledgments
This work was financially supported by the National Science and Technology Major Projects for “New Drug Innovation and Development” (no. 2009ZX09302‐002).
References
- 1. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol 2000; 18: 217–42. [DOI] [PubMed] [Google Scholar]
- 2. Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Molecular cloning and functional expression of a new human CC‐chemokine receptor gene. Biochemistry 1996; 35: 3362–7. [DOI] [PubMed] [Google Scholar]
- 3. Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol 2006; 7: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karnoub AE, Dash AB, Vo AP et al Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007; 449: 557–63. [DOI] [PubMed] [Google Scholar]
- 5. Borczuk AC, Papanikolaou N, Toonkel RL et al Lung adenocarcinoma invasion in TGFβRII‐deficient cells is mediated by CCL5/RANTES. Oncogene 2008; 27: 557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aldinucci D, Lorenzon D, Cattaruzza L et al Expression of CCR5 receptors on Reed‐Sternberg cells and Hodgkin lymphoma cell lines: involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int J Cancer 2008; 122: 769–76. [DOI] [PubMed] [Google Scholar]
- 7. Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate 2006; 66: 124–34. [DOI] [PubMed] [Google Scholar]
- 8. Murooka TT, Rahbar R, Fish EN. CCL5 promotes proliferation of MCF‐7 cells through mTOR‐dependent mRNA translation. Biochem Biophys Res Commun 2009; 387: 381–6. [DOI] [PubMed] [Google Scholar]
- 9. González‐Martín A, Gómez L, Lustgarten J, Mira E, Mañes S. Maximal T cell‐mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res 2011; 71: 5455–66. [DOI] [PubMed] [Google Scholar]
- 10. Nesbeth Y, Scarlett U, Cubillos‐Ruiz J et al CCL5‐mediated endogenous antitumor immunity elicited by adoptively transferred lymphocytes and dendritic cell depletion. Cancer Res 2009; 69: 6331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conforti R, Ma Y, Morel Y et al Opposing effects of toll‐like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res 2010; 70: 490–500. [DOI] [PubMed] [Google Scholar]
- 12. Semenza GL. HIF‐1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002; 8: S62–7. [DOI] [PubMed] [Google Scholar]
- 13. Liang Z, Wu T, Lou H et al Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res 2004; 64: 4302–8. [DOI] [PubMed] [Google Scholar]
- 14. Moriuchi H, Moriuchi M, Fauci AS. Cloning and analysis of the promoter region of CCR5, a coreceptor for HIV‐1 entry. J Immunol 1997; 159: 5441–9. [PubMed] [Google Scholar]
- 15. Lin S, Sun L, Hu J et al Chemokine C‐X‐C motif receptor 6 contributes to cell migration during hypoxia. Cancer Lett 2009; 279: 108–17. [DOI] [PubMed] [Google Scholar]
- 16. Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV‐1 infection in human T cells by lentiviral‐mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA 2003; 100: 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun HL, Liu YN, Huang YT et al YC‐1 inhibits HIF‐1 expression in prostate cancer cells: contribution of Akt/NF‐κB signaling to HIF‐1α accumulation during hypoxia. Oncogene 2007; 26: 3941–51. [DOI] [PubMed] [Google Scholar]
- 18. Aubert S, Fauquette V, Hémon B et al MUC1, a new hypoxia inducible factor target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer Res 2009; 69: 5707–15. [DOI] [PubMed] [Google Scholar]
- 19. Kizaka‐Kondoh S, Inoue M, Harada H, Hiraoka M. Tumor hypoxia: a target for selective cancer therapy. Cancer Sci 2003; 94: 1021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu X, Kang Y. Hypoxia and hypoxia‐inducible factors: master regulators of metastasis. Clin Cancer Res 2010; 16: 5928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev 2010; 29: 285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bosco MC, Puppo M, Santangelo C et al Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune‐related genes and identification of CC‐chemokine ligand 20 as a new hypoxia‐inducible gene. J Immunol 2006; 177: 1941–55. [DOI] [PubMed] [Google Scholar]
- 23. Bosco MC, Reffo G, Puppo M, Varesio L. Hypoxia inhibits the expression of the CCR5 chemokine receptor in macrophages. Cell Immunol 2004; 228: 1–7. [DOI] [PubMed] [Google Scholar]
- 24. Mancino A, Schioppa T, Larghi P et al Divergent effects of hypoxia on dendritic cell functions. Blood 2008; 112: 3723–34. [DOI] [PubMed] [Google Scholar]
- 25. Copple BL, Bai S, Burgoon LD, Moon JO. Hypoxia‐inducible factor‐1alpha regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int 2011; 31: 230–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Opdenakker G, Van Damme J. The countercurrent principle in invasion and metastasis of cancer cells. Recent insights on the roles of chemokines. Int J Dev Biol 2004; 48: 519–27. [DOI] [PubMed] [Google Scholar]
- 27. Gijsbers K, Geboes K, Van Damme J. Chemokines in gastrointestinal disorders. Curr Drug Targets 2006; 7: 47–64. [DOI] [PubMed] [Google Scholar]
- 28. Biswas SK, Gangi L, Paul S et al A distinct and unique transcriptional program expressed by tumor‐associated macrophages: defective NF‐κB and enhanced IRF‐3/STAT1 activation. Blood 2006; 107: 2112–22. [DOI] [PubMed] [Google Scholar]
- 29. Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 2008; 371: 771–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and Methods.
Fig. S1. Western blotting for CCR5 protein in MDA‐MB‐231 and MDA‐MB‐435 cells transfected with pcDNA3.1‐CCR5.
Fig. S2. Knockdown of CCR5 by short hairpin RNA specifically designed against human CCR5.
Fig. S3. Knockdown of hypoxia‐inducible factor‐1α (HIF‐1α) by short hairpin RNA specifically designed against human HIF‐1α.
Fig. S4. Western blotting for hypoxia‐inducible factor‐1α (HIF‐1α) protein in MDA‐MB‐231 cells transfected with pcDNA3.1‐ HIF‐1α.