

Abstract
MicroRNA (miRNA) genes, located in intergenic or intragenic non‐coding regions of the genome, are transcribed and processed to small non‐protein‐coding RNA of approximately 22 nucleotides negatively regulating gene expression. Some miRNA have already been reported for their genetic alterations, aberrant expression and oncogenic or tumor‐suppressive functions. After 2008, there has been a striking increase in the number of publications reporting tumor‐suppressive miRNA (TS‐miRNA) silenced epigenetically in various types of cancers, suggesting important clinical applications for miRNA‐based molecular diagnosis and therapy for cancers. Here, we introduce a correlation of the gene silencing of TS‐miRNA through CpG island hypermethylation with the genomic distances between intergenic and intragenic miRNA genes or protein‐coding host genes and CpG islands located around these genes. Furthermore, we also discuss the potential of miRNA replacement therapy for cancers using double‐stranded RNA mimicking TS‐miRNA. (Cancer Sci 2012; 103: 837–845)
MicroRNA (miRNA) are small, non‐coding and single‐strand RNA of 19–22 nucleotides with a primary role in post‐transcriptional silencing generally through imperfect pairing with the 3′‐UTR of protein‐coding transcripts.1 Approximately 98% of the human genome is known to be non‐coding DNA harboring a large number of intergenic and intronic miRNA genes.1, 2, 3 Intergenic miRNA genes are located in the non‐coding regions between genes, while intragenic miRNA genes, or intronic miRNA genes, are harbored within introns of their protein‐coding host genes. In normal cells, some of these endogenous RNA play crucial roles in many processes, such as proliferation, development, differentiation and apoptosis.4, 5, 6, 7 In cancer cells, several studies demonstrate the deregulation of miRNA expression and the genetic aberration of a few miRNA genes within amplified or deleted regions,8, 9, 10 showing that miRNA can contribute to the multistep processes of carcinogenesis as oncogenes or tumor suppressor genes (TSG).6, 11, 12, 13 Recently, several miRNA genes have also been demonstrated to have copy number variations (CNV), although whether CNV affect miRNA genes in human cancers remains unclear.14
DNA hypermethylation of CpG sites within CpG islands is known as an epigenetic aberration leading to the inactivation of tumor‐suppressive miRNA (TS‐miRNA) in cancer cells,15 in the same manner as that of many classical TSG.16 In fact, the expression of several miRNA is generally downregulated in malignant tissues compared with corresponding non‐malignant tissues. Recent studies, including our own, clearly demonstrate DNA methylation‐mediated downregulation of TS‐miRNA gene expression in various types of cancers.17, 18, 19, 20, 21, 22, 23, 24 Although the genomic distances between the 5′‐end of intergenic miRNA genes or host genes harboring intronic miRNA and their proximal CpG islands vary, these distances might provide more important information for the understanding of silencing of TS‐miRNA genes through DNA hypermethylation. However, few studies have focused on these genomic distances.
The many achievements in the field of TS‐miRNA discovery and in vitro/in vivo delivery technology may offer the possibility of new therapeutic approaches for cancer. Because one miRNA can target many messenger RNA (mRNA) of protein‐coding genes, the in vivo applications of miRNA for cancer therapies are considered better than those of short interfering RNA (siRNA). In addition, among miRNA‐based in vivo delivery approaches, including the use of DNA plasmids or viral vectors, miRNA replacement therapy using double‐stranded RNA (dsRNA) mimicking TS‐miRNA is one of the most promising, offering hope for new cancer therapies.25
We recently evaluated the genomic distribution of 1523 miRNA genes and their CpG islands, and then determined the genomic distance between these genes and CpG islands located within 10 kb upstream of miRNA gene using the miRBase database (Release 18: November 2011) and the UCSC Genome Browser on Human February 2009 Assembly (hg19). In this review, based on information from these databases, we provide insights into the relationship of the gene silencing of TS‐miRNA through aberrant DNA hypermethylation with the genomic distances between TS‐miRNA genes or protein‐coding host genes harboring intragenic miRNA and related CpG islands. We also discuss the potential of these TS‐miRNA as therapeutic agents for cancer.
Distribution of Micro RNA Genes and Their Related CpG Islands in the Human Genome
We examined the genomic distribution of the 1523 genes registered as human miRNA genes in the miRBase database (Release 18: November 2011), (Fig. 1A). Interestingly, 20.0% (304/1523) of these miRNA genes are located on chromosomes 14, 19 and X. Notably, some of these genes are concentrated at 14q32.31, 19q13.42 and Xq27.3, and lie within limited regions of 44, 122 and 33 kb, respectively. In 19q13.42 and Xq27.3, the chromosome 19 miRNA cluster (C19MC) and chromosome X miRNA cluster were revealed as primate‐specific.26 Some C19MC miRNA were described as expressed at a very low level in most human tissues.27 A correlation between their expression patterns and the methylation status of a distal CpG‐rich region located approximately 17.6 kb upstream of the C19MC region has also been demonstrated in gastric cancer cells.28
Figure 1.
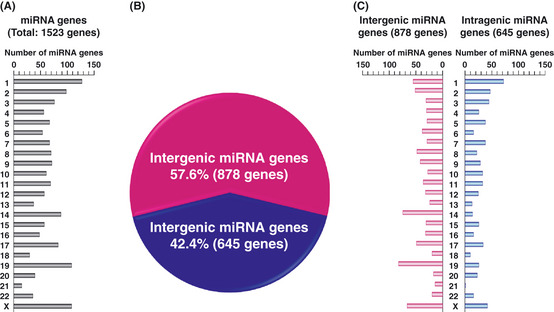
Genomic feature of micro RNA (miRNA) genes. (A) Genomic distribution of 1523 human miRNA genes registered in the miRBase database (Release 18: November 2011). (B) Ratio of intergenic and intragenic miRNA genes in 1523 human miRNA genes. (C) Genomic distribution of 878 intergenic (left) and 645 intragenic (right) miRNA genes. These data were obtained from the miRBase database (http://www.mirbase.org/index.shtml) and UCSC Genome Browser on Human February 2009 Assembly (hg19) (http://genome.ucsc.edu/cgi-bin/hgGateway). In our database analyses, miRNA genes, which were located within introns of protein‐coding host genes and considered to be transcribed in the same direction as those of their host genes, were analyzed as intragenic miRNA genes.
Tumor‐specific downregulation of subsets of miRNA has been generally observed in various types of human cancer,11 suggesting that some of these miRNA act as TSG. Because the downregulation of some TS‐miRNA has been shown to be tightly linked to CpG island hypermethylation, the aberrant DNA hypermethylation of CpG islands located around TS‐miRNA genes, similar to various protein‐encoding TSG, has been recognized as one of the main epigenetic alterations in cancer cells.17, 18, 19, 20, 21, 22, 23, 24 The most recent study indicated 11.6% (122/1048) of miRNA to be epigenetically regulated in 23 cancer types,29 and 19.5% (26/133) of the 133 miRNA genes transcribing these 122 miRNA to have a CpG island within 5 kb upstream. In addition, 14.2% (19/133) of these miRNA genes were also demonstrated to reside within CpG islands. Few studies have examined the relationship of the transcriptional regulation of intergenic, intragenic miRNA genes or host genes harboring intronic miRNA genes through proximal CpG island hypermethylation with genomic distances between these individual genes and their related CpG islands. We discuss these relationships in the following sections.
Intergenic Tumor‐Suppressive Micro RNA Genes Silenced by CpG Island Hypermethylation in Cancer Cells
A recent study showed that RNA polymerase II (Pol II) promoters driving miRNA expression contained most of the features of the protein‐coding gene promoters and that intergenic and some intragenic miRNA were transcribed by RNA Pol II at a distance that could be as large as 40 kb from the miRNA genes.30 In addition, a computational approach demonstrated that 81.9% (59/72) of predicted promoters of intergenic miRNA genes (37 miRNA clusters among 46 miRNA clusters) contained or overlapped with at least one CpG island.31
In our database analyses, intergenic and intragenic miRNA genes made up 57.6% (878/1523) and 42.4% (645/1523), respectively, of human miRNA genes (Fig. 1B,C). Among intergenic miRNA genes examined in our database analyses, 15.3% (134/878) were located within 500 bp downstream of CpG islands (Fig. 2A). These 134 intergenic miRNA genes included 40 genes whose name contained a number lower than 700. Several research groups, including ours, have been investigating some of intergenic miRNA since the early days of miRNA study, and 47.5% (19/40) of these genes are well‐known TS‐miRNA (Table 1). Moreover, gene silencing of these known TS‐miRNA has already been reported to be related with DNA hypermethylation in several types of cancers.
Figure 2.
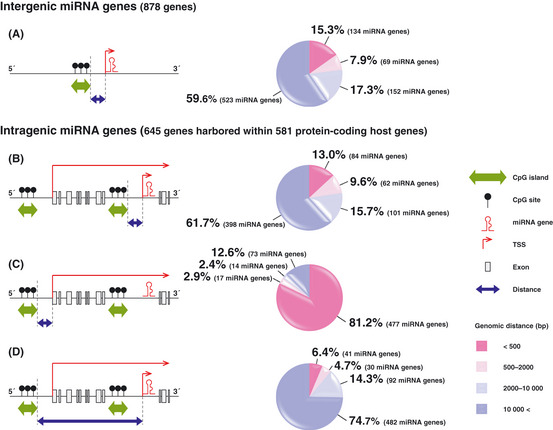
Genomic distances between intergenic and intragenic micro RNA (miRNA) genes or protein‐coding host genes and their related CpG islands. Each map indicates the relationship between miRNA genes or host genes and CpG islands located around these genes on the genome. All intragenic miRNA genes examined in our database analyses were considered to be transcribed in the same direction as those of their protein‐coding host genes. TSS, transcription start site. Each pie graph shows results of our database analyses for genomic distances between CpG islands and the 5′‐end of intergenic (A) or intragenic miRNA genes (B and D) or protein‐coding host genes (C). These data were obtained from the miRBase database (Release 18: November 2011) (http://www.mirbase.org/index.shtml) and UCSC Genome Browser on Human February 2009 Assembly (hg19) (http://genome.ucsc.edu/cgi-bin/hgGateway).
Table 1.
Intergenic and intragenic tumor‐suppressive micro RNA (TS‐miRNA) genes (miRNA number in a name < 700): These intergenic TS‐miRNA genes and protein‐coding host genes harboring intragenic TS‐miRNA genes are located within 500 bp downstream of CpG islands
| miRNA genes (ID < 700) | Loci | Distances from 5′ end of miRNA genes to 3′ end of their CpG islands (bp) | Targets | References indicating tumor suppressive functions of miRNA | Host gene | Distances from TSS of host genes to 3′ end of their CpG islands (bp) | Distances from 5′ end of miRNA genes to 3′ end of CpG islands around TSS of host genes (bp) |
|---|---|---|---|---|---|---|---|
| Intergenic miRNA genes | |||||||
| hsa‐mir‐9‐3 | 15q26.1 | 0 | NF‐kappaB1, Androgen receptor (AR) | Guo, 2009; Ostling, 2011 | None | — | — |
| hsa‐mir‐34b | 11q23.1 | 0 | CDK6, c‐MYC, E2F3, MET, CCNE2, CDK4, CAV1, MYB, SFRS2, CREB | Corney, 2007t; Kozaki et al.19; Toyota et al.20 | None | — | — |
| hsa‐mir‐34c | 11q23.1 | 272 | CDK6, c‐MYC, E2F3, MET, CCNE2, CDK4, CAV1, MYB, SFRS2, Androgen receptor (AR) | Corney, 2007; Toyota et al.20; Ostling, 2011 | None | — | — |
| hsa‐mir‐92b | 1q22 | 0 | PRMT5 | Pal, 2007 | None | — | — |
| hsa‐mir‐124‐1 | 8p23.1 | 0 | CDK6, C/EBPα, SMYD3, VIM, IQGAP1, IGFBP7 | Silber, 2008; Furuta, 2010 | None | — | — |
| hsa‐mir‐124‐3 | 20q13.33 | 0 | CDK6, C/EBPα, SMYD3, VIM, IQGAP1, IGFBP7 | Silber, 2008; Furuta, 2010 | None | — | — |
| hsa‐mir‐127 | 14q32.2 | 0 | BCL6 | Saito et al.17 | None | — | — |
| hsa‐mir‐129‐2 | 11p11.2 | 0 | SOX4 | Dyrskjot, 2009; Huang, 2009 | None | — | — |
| hsa‐mir‐137 | 1p21.3 | 0 | CDK6, MITF, Cdc42 | Kozaki et al.19; Bemis, 2008; Liu et al.65 | None | — | — |
| hsa‐mir‐148a | 7p15.2 | 406 | DNMT‐1, TGIF2 | Braconi et al.60 | None | — | — |
| hsa‐mir‐193a | 17q11.2 | 0 | E2F6, c‐kit | Kozaki et al.19; Gao, 2011 | None | — | — |
| hsa‐mir‐196b | 7p15.2 | 0 | c‐myc | Bhatia, 2010 | None | — | — |
| hsa‐mir‐203 | 14q32.33 | 0 | ABCE1, ABL | Kozaki et al.19; Bueno, 2008; Furuta, 2010 | None | — | — |
| hsa‐mir‐210 | 11p15.5 | 0 | FGFRL1 | Camps, 2008; Tsuchiya, 2011 | None | — | — |
| hsa‐mir‐212 | 17p13.3 | 0 | MeCP2, PED | Incoronato, 2010; Wada, 2010 | None | — | — |
| hsa‐mir‐375 | 2q35 | 0 | PDK1, 14‐3‐3zeta, RASD1, YAP | Tsukamoto, 2010; Liu, 2010; de Souza Rocha Simonini, 2010 | None | — | — |
| hsa‐mir‐409 | 14q32.31 | 0 | RDX | Zheng, 2011 | None | — | — |
| hsa‐mir‐424 | Xq26.3 | 0 | PLAG1 | Pallasch, 2009 | None | — | — |
| hsa‐mir‐663 | 20p11.1 | 0 | JunB, JunD, TGFβ1, p21/Waf1/Cip1 | Pan, 2010; Tili, 2010; Tili, 2010 | None | — | — |
| Intragenic miRNA genes | |||||||
| hsa‐mir‐1‐1 | 20q13.33 | 3 | HDAC4, FoxP1, MET | Datta et al.44 | C20orf166 | 0 | 3726 |
| hsa‐mir‐15b | 3q25.33 | 3498 | BCL2 | Cimmino, 2005; Xia, 2008 | SMC4 | 0 | 3498 |
| hsa‐mir‐16‐2 | 3q25.33 | 3655 | BCL2 | Cimmino, 2005; Xia, 2008 | SMC4 | 0 | 3655 |
| hsa‐mir‐23b | 9q22.32 | 554 | GLS, uPA, c‐Met | Gao, 2009; Salvi, 2009 | C9orf3 | 0 | 358 318 |
| hsa‐mir‐26a‐1 | 3p22.2 | 24 730 | EZH2, cyclins D2, cyclin E2 | Lu et al.48; Sander, 2008; Kota et al.47 | CTDSPL | 0 | 106 856 |
| hsa‐mir‐26a‐2 | 12q14.1 | 20 365 | EZH2, cyclins D2, cyclin E2 | Sander, 2008; Kota et al.47 | CTDSP2 | 0 | 20 365 |
| hsa‐mir‐26b | 2q35 | 1813 | SLC7A11 | Ma, 2010 #133; Liu et al.65 | CTDSP1 | 0 | 1813 |
| hsa‐mir‐27b | 9q22.32 | 317 | CYP1B1 | Tsuchiya, 2006 | C9orf3 | 0 | 358 555 |
| hsa‐mir‐30c‐1 | 1p34.1 | 26 046 | BCL2‐like 11 (BIM) | Garofalo, 2011 | NFYC | 0 | 64 981 |
| hsa‐mir‐33a | 22q13.2 | 8601 | Pim‐1 | Thomas, 2011 | SREBF2 | 0 | 66 964 |
| hsa‐mir‐95 | 4p16.1 | 65 175 | SNX1 | Huang, 2011 | ABLIM2 | 0 | 152 620 |
| hsa‐mir‐101‐2 | 9p24.1 | 45 517 | EZH2, COX‐2, Mcl‐1 | Varambally, 2008; Strillacci, 2009; Su, 2009 | RCL1 | 0 | 56 744 |
| hsa‐mir‐107 | 10q23.31 | 51 653 | CDK6, PLAG1, HIF‐1β, PKCɛ | Lee, 2009; Pallasch, 2009; Yamakuchi, 2010; Datta, 2011 | PANK1 | 281 | 51 653 |
| hsa‐mir‐126 | 9q34.3 | 0 | IRS‐1, SLC7A5, SOX2, VEGFA, PIK3R2 | Tavazoie et al.34; Zhang et al.35 | EGFL7 | 0 | 4049 |
| hsa‐mir‐128‐1 | 2q21.3 | 76 097 | E2F3a, Bmi‐1, NTRK3 | Zhang, 2009; Godlewski, 2008; Guidi, 2010 | R3HDM1 | 0 | 133 252 |
| hsa‐mir‐133a‐2 | 20q13.33 | 10 216 | FSCN1 | Chiyomaru et al.55; Kano, 2010 | C20orf166 | 0 | 14 332 |
| hsa‐mir‐139 | 11q13.4 | 24 361 | ROCK2 | Wong, 2011 | PDE2A | 0 | 27 048 |
| hsa‐mir‐149 | 2q37.3 | 0 | Akt1, E2F1 | Lin et al.27 | GPC1 | 0 | 19 098 |
| hsa‐mir‐152 | 17q21.32 | 0 | DNMT1, E2F3, MET, Rictor | Huang et al.61; Das, 2010; Tsuruta et al.24 | COPZ2 | 93 | 0 |
| hsa‐mir‐153‐1 | 2q35 | 516 | Bcl‐2, Mcl‐1 | Xu, 2010 | PTPRN | 12 | 14 948 |
| hsa‐mir‐153‐2 | 7q36.3 | 2006 | Bcl‐2, Mcl‐1 | Xu, 2010 | PTPRN2 | 0 | 1 012 214 |
| hsa‐mir‐185 | 22q11.21 | 11 562 | Six1, RhoA, Cdc42, Androgen receptor (AR) | Imam, 2010; Liu et al.65; Ostling, 2011 | C22orf25 | 0 | 11 562 |
| hsa‐mir‐186 | 1p31.1 | 13 087 | P2X7 | Zhou, 2008 | ZRANB2 | 189 | 13 087 |
| hsa‐mir‐198 | 3q13.33 | 45 984 | c‐MET | Tan, 2011 | FSTL1 | 0 | 54 626 |
| hsa‐mir‐218‐1 | 4p15.31 | 171 810 | ECOP, IKK‐b, LASP1, PXN, Robo1, BIRC5, GJA1, Rictor | Martinez, 2008; Wu et al.56; Alajez et al.49; Uesugi et al.23 | SLIT2 | 0 | 273 030 |
| hsa‐mir‐218‐2 | 5q34 | 188 233 | ECOP, IKK‐b, LASP1, PXN, Robo1, BIRC5, GJA1, Rictor | Martinez, 2008; Wu et al.56; Alajez et al.49; Uesugi et al.23 | SLIT3 | 0 | 532 169 |
| hsa‐mir‐326 | 11q13.4 | 15 994 | Notch, MRP‐1/ABCC1 | Kefas, 2009; Liang, 2010 | ARRB1 | 0 | 15 994 |
| hsa‐mir‐335 | 7q32.2 | 2841 | SOX4 | Tavazoie et al.34 | MEST | 0 | 2841 |
| hsa‐mir‐338 | 17q25.3 | 2999 | SMO | Huang, 2011 | AATK | 0 | 39 752 |
| hsa‐mir‐340 | 5q35.3 | 55 894 | MITF, c‐Met | Goswami, 2010; Wu, 2011 | RNF130 | 0 | 55 894 |
| hsa‐mir‐346 | 14q32.2 | 1172 | Androgen receptor (AR) | Grady et al.43 | GRID1 | 0 | 98 379 |
| hsa‐mir‐449a | 5q11.2 | 2333 | HDAC‐1, E2F1, Androgen receptor (AR) | Noonan, 2009; Yang, 2009; Lize, 2009; Ostling, 2011 | CDC20B | 0 | 2333 |
| hsa‐mir‐449b | 5q11.2 | 2213 | E2F1, Androgen receptor (AR) | Yang, 2009; Lize, 2009; Ostling, 2011 | CDC20B | 0 | 2213 |
| hsa‐mir‐486 | 8p11.21 | 6311 | OLFM4 | Oh, 2011 | ANK1 | 0 | 235 314 |
| hsa‐mir‐488 | 1q25.2 | 134 811 | Androgen receptor (AR) | Sikand, 2010 | ASTN1 | 178 | 134 811 |
| hsa‐mir‐489 | 7q21.3 | 90 754 | PTPN11 | Kikkawa, 2010 | CALCR | 43 | 90 754 |
| hsa‐mir‐548d‐1 | 8q24.13 | 47 722 | ERBB2 | Chen, 2009; Heyn, 2011 | ATAD2 | 0 | 47 722 |
| hsa‐mir‐559 | 2p21 | 7558 | ERBB2 | Chen, 2009 | EPCAM | 343 | 7558 |
| hsa‐mir‐562 | 2q37.1 | 178 223 | EYA1 | Drake, 2009 | DIS3L2 | 0 | 210 527 |
| hsa‐mir‐591 | 7q21.3 | 101 711 | – | Shohet, 2011 | SLC25A13 | 0 | 101 711 |
| hsa‐mir‐593 | 7q32.1 | 22 182 | PLK1 | Ito, 2010 | SND1 | 0 | 429 362 |
| hsa‐mir‐634 | 17q24.2 | 47 907 | Androgen receptor (AR) | Ostling, 2011 | PRKCA | 0 | 483 573 |
TSS, transcription start site. These data were obtained from the miRBase database (Release 18: November 2011) (http://www.mirbase.org/index.shtml), UCSC Genome Browser on Human February 2009 Assembly (hg19) (http://genome.ucsc.edu/cgi-bin/hgGateway) and PubMed (http://www.ncbi.nlm.nih.gov/pubmed). All intragenic miRNA genes, indicated in this table, are transcribed in the same direction as those of their host genes.
Pioneer studies of these intergenic TS‐miRNA demonstrated that miR‐127 was decreased by aberrant DNA methylation and histone modification in bladder cancer cells17 and that miR‐124 was inactivated by CpG island hypermethylation in several types of cancers.18 These studies suggest DNA hypermethylation to be an important molecular mechanism for downregulation of miRNA expression in cancers. Previously, we identified four intergenic TS‐miRNA silenced through DNA hypermethylation of CpG islands located within 500 bp upstream in oral squamous cell carcinoma (OSCC) and hepatocellular carcinoma (HCC), and also reported their targets (Table 1). We first identified miR‐137 and miR‐193a as an intergenic TS‐miRNA frequently silenced by tumor‐specific DNA hypermethylation in OSCC using expression‐based screening with a series of sequential analyses of expression profiles of 148 miRNA, DNA hypermethylation status of selected candidates, and their tumor‐suppressive activities in a panel of 18 OSCC cell lines and 11 primary tumors of OSCC with paired normal oral mucosa.19 Our study also revealed that miR‐137 and miR‐193 might induce cell cycle arrest at the G1‐S checkpoint and apoptosis, respectively, through direct binding to their target mRNA, CDK6 and E2F6, respectively, in OSCC cell lines. We performed methylation‐based screening, comparing methylation and expression status for 39 miRNA located at 43 loci containing CpG islands within 500 bp upstream of these miRNA genes in a panel of 19 HCC cell lines and 41 primary HCC tumors with corresponding non‐tumorous tissue, resulting in the discovery of hypermethylation‐mediated silencing of miR‐124 and miR‐203 genes as a relatively frequent molecular event in HCC.21 In this study, miR‐124 and miR‐203 were elucidated to exert cell growth‐inhibitory effects on HCC cell lines through the induction of cell cycle arrest at the G1‐S checkpoint and apoptosis, respectively, with the downregulation of protein expression of their targets, CDK6,VIM,SMYD3 and IQGAP1 or ABCE1, respectively. Based on these observations, we believe that the tumor‐specific DNA hypermethylation of CpG islands located immediately 5′‐upstream of intergenic miRNA genes is a useful landmark to explore novel TS‐miRNA silenced epigenetically in cancer cells, similar to classical TSG.
Intragenic Tumor‐Suppressive Micro RNA Genes and Their Host Genes Silenced by CpG Island Hypermethylation in Cancer Cells
Sizable regions of genomic DNA are recognized as introns harboring many intragenic miRNA genes, referred to as intronic miRNA genes. Approximately 37% of mammalian miRNA genes appear to be intronic miRNA genes.32 A recent computational approach demonstrated that 94.2% (49/52) of predicted promoter regions of intronic miRNA genes overlapped with promoters of their host genes.31 In addition, the majority of mammalian intronic miRNA genes are found to be frequently coexpressed with their protein‐coding host genes under the promoter‐driven regulation of the host gene, while nearly 26% of intronic miRNA genes are described to be transcribed from their own promoters.3, 30, 33 In our database analyses using the miRBase database (Release 18: November 2011), intragenic miRNA genes accounted for 42.4% (645/1523) of human miRNA genes (Fig. 1B). Notably, among these 645 intragenic miRNA genes, 13.0% (84/645) were located within 500 bp downstream of CpG islands (Fig. 2B). In contrast, the data on the genomic distance between the 5′‐end of protein‐coding host genes harboring intragenic miRNA genes and the 3 + +′‐end of CpG islands located at the 5′‐side of host genes show that 81.2% (477/581) of these host genes, containing 645 intragenic miRNA genes within their introns, were located within 500 bp of CpG islands (Fig. 2C), whereas only 6.4% (41/645) of intragenic miRNA genes were located within 500 bp downstream of these CpG islands (Fig. 2D). In addition, These 477 host genes harbored 534 intragenic miRNA genes, including 176 genes assigned a name with a number below 700. Among these 176 genes, no more than 23.9% (42/176) of intragenic miRNA genes have been identified as TS‐miRNA (Table 1).
Previous studies have shown tumor‐suppressive activities of miR‐126 and miR‐335 located within introns of the EGFL7 and MEST genes, respectively, in breast cancer,34 and report IRS‐1,35 SLC7A5,36 SOX2,37 VEGFA,38, 39 and PIK3R2 39 or SOX4 34 to be their targets, respectively. Coexpression of miR‐126 and EGFL7 is reported to be downregulated through histone modification and CpG island hypermethylation in the EGFL7 gene promoter in T24, HeLa and MCF7 cells and primary bladder and prostate tumors.40 Transcription of miR‐335 is also demonstrated to be coregulated with MEST by promoter hypermethylation in breast cancer cells.41 miR‐342 located within intron of the EVL gene is described as acting as an TS‐miRNA by targeting DNMT1 in colorectal cancer.42 CpG island hypermethylation upstream of EVL is indicated to suppress both EVL and miR‐342 expression.43 As regards to miR‐1‐1 located within the C20orf166 gene, miR‐1 is described as a TS‐miRNA targeting HDAC4,FOXP1 and MET, and is inactivated by DNA methylation of CpG island in HCC.44 However, these four host genes have never been examined for tumor‐suppressive activities.
Physiological or pathophysiological functions of host genes harboring intragenic TS‐miRNA mostly remain unclear, whereas miR‐26a and miR‐218 are unique intragenic TS‐miRNA located within introns of known TSG. miR‐26a‐1 located within an intron of CTDSPL/SCP3/HYA22/RBSP3 having a tumor‐suppressive function45 is reported to be silenced through CpG island hypermethylation.46 Cyclin D2 and E2 or EZH2 are described as miR‐26a targets in liver cancer47 and nasopharayngeal carcinoma (NPC),48 respectively. Regarding miR‐218, downregulation of its expression was demonstrated to be associated with DNA methylation of CpG islands at the 5′‐ends of SLIT2 and SLIT3 harboring miR‐218‐1 and miR‐218‐2, respectively, in NPC.49 The SLIT2 and SLIT3 genes are indicated to act as TSG through SLIT‐Robo signaling in breast cancer cell lines,50 and to be inactivated by their promoter hypermethylation in several types of cancers51, 52. BIRC5,49 ECOP,53 GJA1 49, IKK‐β,54 LASP1 55, PXN 56 and Robo1 57 are reported as targets of miR‐218. Recently, to explore TS‐miRNA having potential for miRNA replacement therapy, we performed function‐based screening using OSCC and endometrial cancer (EC) cell lines (Fig. 3A), and identified miR‐218 and miR‐152 as intragenic TS‐miRNA frequently silenced through tumor‐specific DNA hypermethylation in OSCC and EC, respectively.23, 24 COPZ2 is a protein‐coding host gene harboring the miR‐152 gene and has recently been shown to display no tumor‐suppressive activities, but, rather, to protect tumor cells from apoptosis induced by COPZ1‐knockdown.58 Moreover, our studies elucidated that Rictor was a novel direct target of these two TS‐miRNA. Rictor, together with mTOR, forms mTOR complex 2 (mTORC2), and the Rictor‐mTOR complex directly regulates the phosphorylation of Akt at Ser‐473, resulting in cell growth.59 In our study, miR‐218 was clearly demonstrated to act as a suppressor of the TOR‐Akt pathway, independently of the PI3K‐Akt pathway, in an OSCC cell line without a genetic alteration of EGFR,PIK3CA and PTEN (Fig. 3B).23 We also identified E2F3 and MET as direct targets of miR‐152 other than DNMT1, which had been reported previously.60, 61 A correlation between aberrant DNA methylation of CpG island of miR‐152 and a poor clinical outcome is reported in breast cancer62 and MLL‐rearranged acute lymphoblastic leukemia.63 These observations strongly support our notion that the tumor‐specific DNA hypermethylation of CpG islands located around protein‐coding host genes harboring intragenic miRNA genes, as well as intergenic miRNA genes, is a useful landmark to explore novel TS‐miRNA silenced epigenetically in cancer cells.
Figure 3.
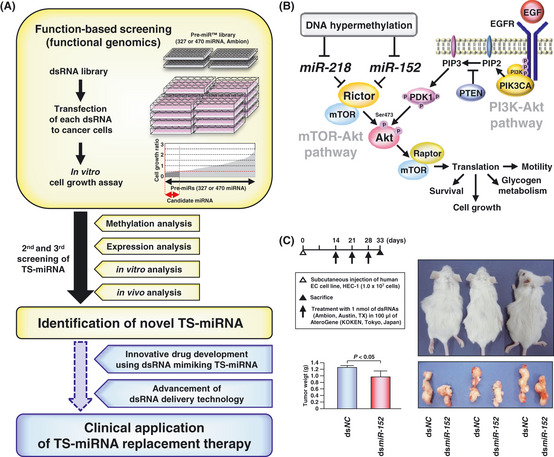
Function‐based screening of tumor‐suppressive micro RNA (TS‐miRNA) for miRNA replacement therapy as a cancer treatment. (A) Strategy of our function‐based approach to the identification of epigenetically silenced TS‐miRNA in cancer cells. Previously, to identify novel TS‐miRNA having the great potential for miRNA replacement therapy, we performed function‐based screening combined with methylation and expression analyses in oral squamous cell carcinoma (OSCC) and endometrial cancer (EC) cell lines according to this strategy shown in this figure, resulting in identification of novel TS‐miRNA,miR‐218 and ‐152, respectively, directly targeting Rictor.23, 24 (B) A model summarizing the molecular mechanism of miR‐218,miR‐152 and their direct target Rictor in the TOR‐Akt signaling pathway. Our previous studies demonstrated that these TS‐miRNA acted as suppressors of the TOR‐Akt signaling pathway, independently of the PI3K‐Akt signaling pathway and that methylation‐mediated silencing of these TS‐miRNA might contribute to the pathogenesis of OSCC and EC through the activation of this signaling pathway. (C) Therapeutic effects of dsRNA mimicking miR‐152 (dsmiR‐152) or control non‐specific miRNA (dsNC) on tumor growth in vivo. Left panel: a schema indicating the protocol of in vivo analysis (upper) and bar graph showing effects of dsmiR‐152 on tumor growth in three SCID mice (lower). Right panel: photograph shows tumor‐bearing SCID mice (upper) and their subcutaneous tumors (lower) at the end of in vivo analysis. These findings strongly support the great potential of dsRNA mimicking miR‐152 to be applied to miRNA replacement therapy for cancers.
Future Perspectives on Tumor‐Suppressive Micro RNA Silenced by Tumor‐Specific DNA Hypermethylation in Cancer Research
Taking miRNA‐induced effects on normal cells into consideration, TS‐miRNA, the endogenous expression of which is sufficiently activated in normal cells and remarkably reduced in cancer cells, are assumed to have great potential for miRNA replacement therapy.25 The concept behind this therapy is a restoration of loss of function in cancer cells by exogenous expression of TS‐miRNA. A few TS‐miRNA, such as miR‐34a 64, 65 and let‐7,66 have already demonstrated therapeutic effects on tumor formation in vivo following the replacement of these miRNA using dsRNA mimicking their mature forms. Because dsRNA are unlike proteins, are substantially smaller than DNA plasmids or viral vectors, and have the ability to enter the cytoplasm of target cells and to be delivered systemically by technologies that have been used for siRNA,25 a dsRNA‐based approach may be better than other approaches for exogenous expression of miRNA in vivo. TS‐miRNA silenced through tumor‐specific DNA hypermethylation might be better suited as prime candidates for this therapy. Recently, we successfully showed for the first time that dsRNA mimicking miR‐152 administered with atelocollagen to SCID mice could suppress the in vivo growth of an EC cell line (Fig. 2C),24 leading us to consider the possibility of miRNA replacement therapy for cancer using dsRNA mimicking TS‐miRNA silenced by tumor‐specific DNA hypermethylation.
While the function‐based approach is a powerful tool for exploring the use of dsRNA with tumor‐suppressive effects, including TS‐miRNA and siRNA, as therapeutic agents for cancers, the tumor‐suppressive functions of miRNA eventually identified in our studies have been reexamined using two or three kinds of dsRNA purchased from independent companies to take into consideration the off‐target effects associated with dsRNA.23, 24 Although such effects have been known to complicate the interpretation of phenotypic effects in gene‐silencing experiments using siRNA,67 dsRNA mimicking miRNA might potentially cause these unwanted actions, similar to siRNA. These unpredictable target‐independent effects should be addressed during data interpretation in all dsRNA‐based studies related to functional genomics, drug target discovery and dsRNA‐therapeutics. In addition, these dsRNA have mainly been used at 1.0–50.0 nM in dsRNA‐based studies, and overexpression above their physiological concentrations might lead to toxic effects in normal cells with an accumulation of these exogenous dsRNA. In contrast, although atelocollagen24, 64 or lipid‐based delivery agents65, 66 have been used for in vivo dsRNA delivery, these systems are inadequate for clinical applications to fully restore downregulated miRNA in cancer cells. Therefore, the development of solutions attenuating the nonspecific off‐target effects associated with dsRNA mimicking TS‐miRNA and the advancement of dsRNA‐delivery technology may yield a new field of miRNA‐based cancer therapy.
Concluding Remarks
In this review, we focused on genomic distances between intergenic TS‐miRNA genes or protein‐coding host genes harboring intragenic miRNA genes and their related CpG islands, and illustrated how the tumor‐specific DNA hypermethylation of CpG islands located immediately 5′‐upstream of intergenic miRNA genes and host genes, as well as classical protein‐encoding TSG, might be a useful epigenetic marker for exploration of TS‐miRNA, cancer diagnosis and prognosis. Moreover, we also discussed the potential of miRNA replacement therapy for cancers using dsRNA mimicking TS‐miRNA silenced epigenetically in cancer cells. Further studies of molecular mechanisms of TS‐miRNA and significant advancement of dsRNA‐delivery technology are essential for the actualization of TS‐miRNA replacement therapy for several types of cancers.
Disclosure Statement
The authors declare no financial or commercial conflicts of interest.
Acknowledgments
Our studies were supported in part by a Grant‐in‐Aid for Scientific Research (A), (B) and (C) and for Scientific Research on Priority Areas and Innovative Areas, and by the Global Center of Excellence Program for International Research Center for Molecular Science in Tooth and Bone Diseases of the Ministry of Education, Culture, Sports, Science, and Technology, Japan, as well as by a Health and Labour Sciences Research Grant by the Ministry of Health, Labour and Welfare, Japan.
References
- 1. Ambros V. The functions of animal microRNAs. Nature 2004; 431: 350–5. [DOI] [PubMed] [Google Scholar]
- 2. Mattick JS. RNA regulation: a new genetics? Nat Rev Genet 2004; 5: 316–23. [DOI] [PubMed] [Google Scholar]
- 3. Brown JW, Marshall DF, Echeverria M. Intronic noncoding RNAs and splicing. Trends Plant Sci 2008; 13: 335–42. [DOI] [PubMed] [Google Scholar]
- 4. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 5. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522–31. [DOI] [PubMed] [Google Scholar]
- 6. Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev 2005; 15: 563–8. [DOI] [PubMed] [Google Scholar]
- 7. Harfe BD. MicroRNAs in vertebrate development. Curr Opin Genet Dev 2005; 15: 410–5. [DOI] [PubMed] [Google Scholar]
- 8. Calin GA, Dumitru CD, Shimizu M et al Frequent deletions and down‐regulation of micro‐ RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002; 99: 15524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–66. [DOI] [PubMed] [Google Scholar]
- 10. Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med 2006; 12: 580–7. [DOI] [PubMed] [Google Scholar]
- 11. Lu J, Getz G, Miska EA et al MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–8. [DOI] [PubMed] [Google Scholar]
- 12. Esquela‐Kerscher A, Slack FJ. Oncomirs: microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–69. [DOI] [PubMed] [Google Scholar]
- 13. Osada H, Takahashi T. MicroRNAs in biological processes and carcinogenesis. Carcinogenesis 2007; 28: 2–12. [DOI] [PubMed] [Google Scholar]
- 14. Marcinkowska M, Szymanski M, Krzyzosiak WJ, Kozlowski P. Copy number variation of microRNA genes in the human genome. BMC Genomics 2011; 12: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res 2007; 61: 24R–9R. [DOI] [PubMed] [Google Scholar]
- 16. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349: 2042–54. [DOI] [PubMed] [Google Scholar]
- 17. Saito Y, Liang G, Egger G et al Specific activation of microRNA‐127 with downregulation of the proto‐oncogene BCL6 by chromatin‐modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–43. [DOI] [PubMed] [Google Scholar]
- 18. Lujambio A, Ropero S, Ballestar E et al Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res 2007; 67: 1424–9. [DOI] [PubMed] [Google Scholar]
- 19. Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor‐suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res 2008; 68: 2094–105. [DOI] [PubMed] [Google Scholar]
- 20. Toyota M, Suzuki H, Sasaki Y et al Epigenetic silencing of microRNA‐34b/c and B‐cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68: 4123–32. [DOI] [PubMed] [Google Scholar]
- 21. Furuta M, Kozaki K, Tanaka S, Arii S, Imoto I, Inazawa J. miR‐124 and miR‐203 are epigenetically silenced tumor‐suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2009; 31: 766–76. [DOI] [PubMed] [Google Scholar]
- 22. Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta 2010; 1799: 10–2. [DOI] [PubMed] [Google Scholar]
- 23. Uesugi A, Kozaki K, Tsuruta T et al The tumor suppressive microRNA miR‐218 targets the mTOR component Rictor and inhibits AKT phosphorylation in oral cancer. Cancer Res 2011; 71: 5765–78. [DOI] [PubMed] [Google Scholar]
- 24. Tsuruta T, Kozaki K, Uesugi A et al miR‐152 is a tumor suppressor microRNA that is silenced by DNA hypermethylation in endometrial cancer. Cancer Res 2011; 71: 6450–62. [DOI] [PubMed] [Google Scholar]
- 25. Bader AG, Brown D, Winkler M. The promise of microRNA replacement therapy. Cancer Res 2010; 70: 7027–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bentwich I, Avniel A, Karov Y et al Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 2005; 37: 766–70. [DOI] [PubMed] [Google Scholar]
- 27. Lin S, Cheung WK, Chen S et al Computational identification and characterization of primate‐specific microRNAs in human genome. Comput Biol Chem 2010; 34: 232–41. [DOI] [PubMed] [Google Scholar]
- 28. Tsai KW, Kao HW, Chen HC, Chen SJ, Lin WC. Epigenetic control of the expression of a primate‐specific microRNA cluster in human cancer cells. Epigenetics 2009; 4: 587–9. [DOI] [PubMed] [Google Scholar]
- 29. Kunej T, Godnic I, Ferdin J, Horvat S, Dovc P, Calin GA. Epigenetic regulation of microRNAs in cancer: an integrated review of literature. Mutat Res 2011; 717: 77–84. [DOI] [PubMed] [Google Scholar]
- 30. Corcoran DL, Pandit KV, Gordon B, Bhattacharjee A, Kaminski N, Benos PV. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS ONE 2009; 4: e5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang G, Wang Y, Shen C et al RNA polymerase II binding patterns reveal genomic regions involved in microRNA gene regulation. PLoS ONE 2010; 5: e13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffiths‐Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006; 34: D140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 2005; 11: 241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tavazoie SF, Alarcon C, Oskarsson T et al Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008; 451: 147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J, Du YY, Lin YF et al The cell growth suppressor, mir‐126, targets IRS‐1. Biochem Biophys Res Commun 2008; 377: 136–40. [DOI] [PubMed] [Google Scholar]
- 36. Miko E, Margitai Z, Czimmerer Z et al miR‐126 inhibits proliferation of small cell lung cancer cells by targeting SLC7A5. FEBS Lett 2011; 585: 1191–6. [DOI] [PubMed] [Google Scholar]
- 37. Otsubo T, Akiyama Y, Hashimoto Y, Shimada S, Goto K, Yuasa Y. MicroRNA‐126 inhibits SOX2 expression and contributes to gastric carcinogenesis. PLoS ONE 2011; 6: e16617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu B, Peng XC, Zheng XL, Wang J, Qin YW. MiR‐126 restoration down‐regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 2009; 66: 169–75. [DOI] [PubMed] [Google Scholar]
- 39. Zhu N, Zhang D, Xie H et al Endothelial‐specific intron‐derived miR‐126 is down‐regulated in human breast cancer and targets both VEGFA and PIK3R2. Mol Cell Biochem 2011; 351: 157–64. [DOI] [PubMed] [Google Scholar]
- 40. Saito Y, Friedman JM, Chihara Y, Egger G, Chuang JC, Liang G. Epigenetic therapy upregulates the tumor suppressor microRNA‐126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun 2009; 379: 726–31. [DOI] [PubMed] [Google Scholar]
- 41. Png KJ, Yoshida M, Zhang XH et al MicroRNA‐335 inhibits tumor reinitiation and is silenced through genetic and epigenetic mechanisms in human breast cancer. Genes Dev 2011; 25: 226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang H, Wu J, Meng X et al MicroRNA‐342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 2011; 32: 1033–42. [DOI] [PubMed] [Google Scholar]
- 43. Grady WM, Parkin RK, Mitchell PS et al Epigenetic silencing of the intronic microRNA hsa‐miR‐342 and its host gene EVL in colorectal cancer. Oncogene 2008; 27: 3880–8. [DOI] [PubMed] [Google Scholar]
- 44. Datta J, Kutay H, Nasser MW et al Methylation mediated silencing of MicroRNA‐1 gene and its role in hepatocellular carcinogenesis. Cancer Res 2008; 68: 5049–58. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Kashuba VI, Li J, Wang F et al RBSP3 (HYA22) is a tumor suppressor gene implicated in major epithelial malignancies. Proc Natl Acad Sci USA 2004; 101: 4906–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sinha S, Singh RK, Alam N, Roy A, Roychoudhury S, Panda CK. Frequent alterations of hMLH1 and RBSP3/HYA22 at chromosomal 3p22.3 region in early and late‐onset breast carcinoma: clinical and prognostic significance. Cancer Sci 2008; 99: 1984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kota J, Chivukula RR, O'Donnell KA et al Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009; 137: 1005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu J, He ML, Wang L et al MiR‐26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res 2011; 71: 225–33. [DOI] [PubMed] [Google Scholar]
- 49. Alajez NM, Lenarduzzi M, Ito E et al MiR‐218 suppresses nasopharyngeal cancer progression through downregulation of survivin and the SLIT2‐ROBO1 pathway. Cancer Res 2011; 71: 2381–91. [DOI] [PubMed] [Google Scholar]
- 50. Marlow R, Strickland P, Lee JS et al SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res 2008; 68: 7819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dallol A, Da Silva NF, Viacava P et al SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res 2002; 62: 5874–80. [PubMed] [Google Scholar]
- 52. Dickinson RE, Dallol A, Bieche I et al Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br J Cancer 2004; 91: 2071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao C, Zhang Z, Liu W, Xiao S, Gu W, Lu H. Reduced microRNA‐218 expression is associated with high nuclear factor kappa B activation in gastric cancer. Cancer 2010; 116: 41–9. [DOI] [PubMed] [Google Scholar]
- 54. Song L, Huang Q, Chen K et al miR‐218 inhibits the invasive ability of glioma cells by direct downregulation of IKK‐beta. Biochem Biophys Res Commun 2010; 402: 135–40. [DOI] [PubMed] [Google Scholar]
- 55. Chiyomaru T, Enokida H, Kawakami K et al Functional role of LASP1 in cell viability and its regulation by microRNAs in bladder cancer. Urol Oncol 2010; doi: 10.1016/j.urolonc.2010.05.008 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 56. Wu DW, Cheng YW, Wang J, Chen CY, Lee H. Paxillin predicts survival and relapse in non‐small cell lung cancer by microRNA‐218 targeting. Cancer Res 2010; 70: 10392–401. [DOI] [PubMed] [Google Scholar]
- 57. Tie J, Pan Y, Zhao L et al MiR‐218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet 2010; 6: e1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shtutman M, Baig M, Levina E et al Tumor‐specific silencing of COPZ2 gene encoding coatomer protein complex subunit zeta 2 renders tumor cells dependent on its paralogous gene COPZ1. Proc Natl Acad Sci USA 2011; 108: 12449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3‐L1 adipocytes. J Biol Chem 2005; 280: 40406–16. [DOI] [PubMed] [Google Scholar]
- 60. Braconi C, Huang N, Patel T. MicroRNA‐dependent regulation of DNA methyltransferase‐1 and tumor suppressor gene expression by interleukin‐6 in human malignant cholangiocytes. Hepatology 2010; 51: 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang J, Wang Y, Guo Y, Sun S. Down‐regulated microRNA‐152 induces aberrant DNA methylation in hepatitis B virus‐related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology 2010; 52: 60–70. [DOI] [PubMed] [Google Scholar]
- 62. Lehmann U, Hasemeier B, Christgen M et al Epigenetic inactivation of microRNA gene hsa‐mir‐9‐1 in human breast cancer. J Pathol 2008; 214: 17–24. [DOI] [PubMed] [Google Scholar]
- 63. Stumpel DJ, Schotte D, Lange‐Turenhout EA et al Hypermethylation of specific microRNA genes in MLL‐rearranged infant acute lymphoblastic leukemia: major matters at a micro scale. Leukemia 2010; 25: 429–39. [DOI] [PubMed] [Google Scholar]
- 64. Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor‐suppressive miR‐34a induces senescence‐like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 2007; 104: 15472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu C, Kelnar K, Liu B et al The microRNA miR‐34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 2011; 17: 211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Trang P, Medina PP, Wiggins JF et al Regression of murine lung tumors by the let‐7 microRNA. Oncogene 2010; 29: 1580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Svoboda P. Off‐targeting and other non‐specific effects of RNAi experiments in mammalian cells. Curr Opin Mol Ther 2007; 9: 248–57. [PubMed] [Google Scholar]