

Abstract
An improved understanding of biomechanical factors that control tumor development, including angiogenesis, could explain why few of the promising treatment strategies discovered via in vitro models translate well into in vivo or clinical studies. The ability to manipulate and in real-time study the multiple independent biomechanical properties on cellular activity has been limited, primarily due to limitations in traditional in vitro platforms or the inability to manipulate such factors in vivo. We present a novel microfluidic platform that mimics the vascularized tumor microenvironment with independent control of interstitial flow and mechanical strain. The microtissue platform design isolates mechanically-stimulated angiogenesis in the tumor microenvironment, by manipulating interstitial flow to eliminate soluble factors that could drive blood vessel growth. Our studies demonstrate that enhanced mechanical strain induced by cancer-associated fibroblasts (CAFs) promotes angiogenesis in microvasculature models, even when preventing diffusion of soluble factors to the growing vasculature. Moreover, small but significant decreases in micro-strains induced by inhibited CAFs were sufficient to reduce angiogenesis. Ultimately, we believe this platform represents a significant advancement in the ability to investigate biomechanical signals while controlling for biochemical signals, with a potential to be utilized in fields beyond cancer research.
Introduction
In the cancer microenvironment, angiogenesis becomes necessary when the growing tumor reaches a critical size and passive diffusion of nutrients is no longer sufficient for sustained growth.1–4 In light of this, anti-angiogenic therapies have been utilized as anti-cancer strategies since the 1980s albeit with limited success.5,6 Currently, these mainly include small molecule inhibitors or monoclonal antibodies designed to interrupt the signaling pathway of vascular endothelial growth factor (VEGF) and its primary receptor, VEGFR2.7–10 Within the tumor microenvironment not only are there changes in cytokine and growth factor production but also changes in the biomechanical properties of the ECM. During angiogenesis, a quiescent endothelial cell is stimulated to become a tip cell, which actively migrates and remodels neighboring ECM to lead new vessel growth.11–17 Whether tip cell formation, growth, and migration is also regulated by mechanical signals is poorly understood, despite evidence suggesting that mechanical strain can promote this type of phenotypic shift in endothelial cells.18,19 This could be an important consideration as present strategies clinically targeting angiogenesis to prevent cancer growth do not consider the biomechanics of the tumor microenvironment.
Cancer-associated fibroblasts (CAFs) in the tumor microenvironment are activated, myofibroblast-like cells that secrete soluble factors including VEGF that impact tumor progression.10,20,21 Additionally, CAFs demonstrate mechanosensing and mechanotransduction properties, remodeling the ECM in such a way as to promote metastatic activities.22,23 Compared to normal fibroblasts, CAFs exhibit upregulated expression of alpha smooth muscle actin (αSMA) and transcriptional regulators SNAIL1 and YAP.23,24 The prominence of CAFs as regulators of angiogenesis and metastasis has been demonstrated using in vivo models of breast and other cancers.25,26 Recently, our group demonstrated that the mechanotransducing functions of CAFs alone can drive vascular growth via mechanical perturbations in an in vitro model.27 Furthermore, CAF remodeling of the tumor ECM can drive tumor progression through secretion of cytokines, matrix components and matrix remodeling enzymes, or direct physical remodeling of collagen fibers, all of which lead to an increase in matrix stiffness.28–41 Forces due to increased interstitial flow from “leaky” tumor blood vessels can also drive tumor progression, through integrins or growth factor receptors.35,42–49 Elucidating how biomechanical properties of CAFs affect tumor angiogenesis requires advanced in vitro models that permit isolation of biomechanical and biochemical factors.
To address this, we have developed a novel microfluidic model with independent control over multiple mechanical parameters. Microfluidic models are highly customizable systems for creating novel in vitro models for studying biological processes particularly within cancer research.34,50–57 This includes designs that test experimental setups in both 2- and 3D, which can be tailored to mimic normal or diseased tissues. Additionally, microfluidic models can be utilized to examine behaviors of a single cell type or be configured for multiple cell types in a co-culture system. An important advantage of in vitro microfluidic systems over in vivo studies is the ability to control and observe changes in biomechanical environments. Currently, most systems describe control or investigation of individual biomechanical parameters. For example, models designed to determine effects of shear stress from interstitial flow on endothelial cells may not investigate interacting effects from ECM composition or stiffness.34,55,57 Efforts to study how cell-generated forces impact processes such as angiogenesis are confounded by the need to separate effects of biochemical and biomechanical stimuli. Recent studies from Abe et al. demonstrate the crosstalk of VEGF signaling and interstitial flow, highlighting how these factors interact and affect angiogenesis.58 Previous work in our lab generated microtissues with self-assembled vascular networks composed of endothelial colony forming endothelial cells and a stromal cell.43,51,52,59–61 The objective of the current study was to develop and optimize a multi-tissue chamber model with independent control over multiple mechanical factors to investigate angiogenesis associated with tumor progression.
Materials & methods
Device design, modeling, and synthesis
A multi-tissue chamber device with independent fluidic lines was designed using AutoCAD (2015). The goal of the device design was to create a model with multiple microtissues that are mechanically coupled. This includes the ability for strains to be transferred between microtissues, along with user-control over the direction and magnitude of interstitial flow between tissues. The latter parameters provide control over communication via soluble mediators by convective (interstitial) flow. Furthermore, the aspect ratio of the rectangular-shaped compartments encouraged lateral alignment of stromal cells (Fig. 1a). Molds were made using standard soft lithography protocols.43,53,54 Briefly, a silicon wafer was spin coated with SU-2075 at a thickness of 100 μm and then exposed to UV light via a mask aligner. Molds were rinsed with SU8 developer for 30 min before final cleaning with methanol and acetone. Polydimethyl siloxane (Sylgard 184, Dow Corning) was cast on each mold at a base to curing agent ratio of 10: 1, degassed for 30 min prior to curing at 65 °C for a minimum of 3 h. Excised devices were plasma bonded to glass slides and sterilized via autoclave. COMSOL Multiphysics (5.4) Fluid Flow and Heat Transfer Module, specifically the fluid flow through porous media and transport of diluted species, was used to determine parameters for flow regimes that would either prevent crosstalk between chambers or control directionality of flow between chambers. In these studies, tissue chambers were loaded with fibrin, which was modeled as a linear elastic solid with Young’s modulus of 300 Pa and Poisson ration of 0.49; media flow was modeled as an incompressible, single-phase fluid with laminar flow characteristics.43 Additionally, the Structural Mechanics and Acoustics Module in COMSOL was used to model transfer of mechanical strain between chambers, using 10 μm diameter spheres to model cells located randomly throughout the tissue chamber near the interface. Forces of 100 nN and 1000 nN were utilized to represent the differences in normal fibroblasts and CAFs, respectively.62–64 These forces were applied in a uniaxial direction away from the interface between microtissue chambers, as a simplified model to determine if strains propagate through the communication ports.
Fig. 1.
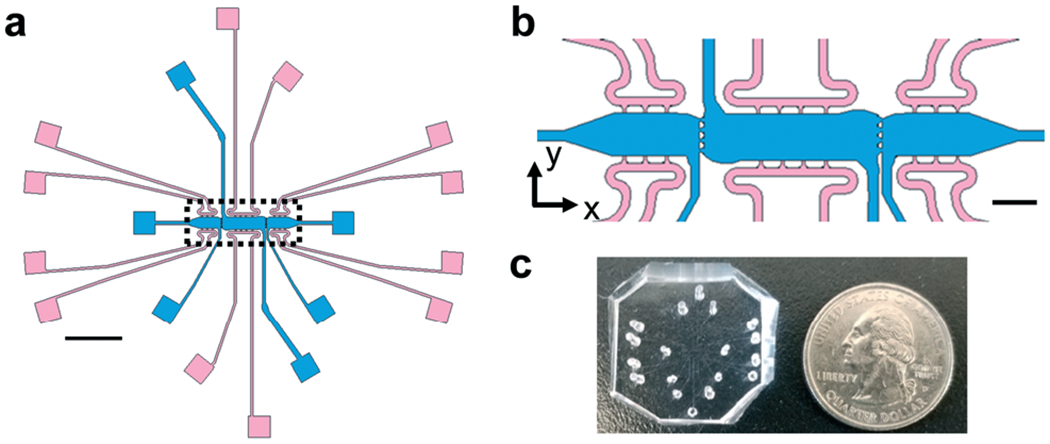
Schematic of microtissue platform for biomechanical investigations. (a) A schematic of the full microtissue device showing three tissue chambers (blue) and media lines (pink) for each chamber. Each tissue chamber can be loaded independently of adjacent chambers; the individual media lines for each tissue chamber allows for control over interstitial flow directionality and magnitude between microtissues. Scale bar = 2 mm. (b) Inset from A (black dotted lines) to show communication ports (20 μm) between microtissues. Ports are sized to allow for mechanical crosstalk between microtissues. Scale bar = 500 μm. (c) Photograph of PDMS microtissue platform with holes punched for cell/gel loading and feeding ports.
Flow studies
To validate flow regime parameters determined in COMSOL, we loaded the devices with fibrin gels (10 mg mL−1) and utilized either FITC- or Rhodamine B tagged-dextran (10 kDa, 20 kDa, or 70 kDa) at 1.5 mg mL−1 in DPBS as the feeding media. These sizes of dextran were chosen as they encapsulate and approximate a wide range of secreted factors including 8 kDa SDF and 46 kDa (homodimer form) VEGF. Devices were fed in either a top-to-bottom regime, where flow exclusively occurred in only the y-direction in an individual compartment (Fig. 1b), or outward flow regime where higher pressures were loaded into the center chamber compared to side chambers to generate flow in the x-direction (Fig. 1b). Devices were imaged after a minimum of 18 h to determine equilibrium conditions; relative fluorescent intensities were measured on a Nikon microscope equipped with an environmental chamber maintained at 37 °C at 5% CO2. Images were stitched together using FIJI Stitching Plug-In and fluorescent intensity was measured through the entire length of an imaged device.65 A minimum of three devices were analyzed per condition. In outward flow studies, devices previously loaded with the top-to-bottom flow regime had fluorescent dextran solutions removed from feeding reservoirs and replaced with DPBS. Devices were again imaged after 18 h to determine relative fluorescent intensities in individual chambers over time.
Cell culture
To create 3D vasculature, human endothelial cells derived from umbilical cord blood (ECs) and normal human lung fibroblasts (NHLFs, Lonza) were collected and used as previously described.52–54 Human CAFs and normal breast fibroblasts (NBFs) were derived previously and cultured in DMEM containing 10% non-heat inactivated FBS, with 1% each l-glutamine, Penn/Strep, non-essential amino acids and 2% sodium pyruvate.66 These cells originated from a breast cancer patient, were immortalized via hTERT and constitutively express GFP.66 Previous characterization of NBFs and CAFs show that the CAFs have significantly higher levels of YAP and Snail1 compared to NBFs. Additionally, CAFs express high amounts of αSMA, while the NBFs do not.67 Additionally, we have described CAFs as more mechanically active than NBFs through use of an in-house bead displacement algorithm.27 Mechanically inhibited CAFs were generated by utilizing shRNA against YAP and mechanically activated NBFs were generated by incorporating a constitutively active Rho.27 Briefly, constitutively active RhoA(Q63L)-Flag tagged cDNA or shRNAi against YAP were subcloned into the pLVX-hygro vector. HEK293T cells were used to produce lentiviruses; NBFs and CAFs were transduced with lentivirus, then subsequently selected in 200 μg mL−1 hygromycin. Empty vectors (EV) were utilized for controls in both NBFs and CAFs. ECs were grown in EGM (Lonza) with either 2 or 10% heat inactivated FBS. All cells were kept at 37 °C and 5% CO2 in a fully humidified incubator. For all experimental setups, the concentration of the stromal cells in side chambers was 1 × 107 cells per mL in 10 mg mL−1 fibrin gels. For center chambers, a total cell concentration of 2 × 107cells per mL in 10 mg mL−1 fibrin gels, with a 1 : 1 ratio ECs and NHLFs, was used. Side chambers were loaded on day 0, approximately 5–15 min after center chambers were loaded, except where otherwise noted. For loading, cells were harvested and resuspended in fibrinogen, mixed with 5 U mL−1 thrombin, and injected into the chambers. Devices were incubated for 30 min after cell loading to allow for full fibrin gelation, before feeding with EGM with 2% FBS; media was changed every 24 h. A set of studies were completed where side chambers were loaded via the same protocol, except on day 4 after center chambers had been loaded; these experiments were utilized to determine if there was a difference in angiogenic growth from a well-established vasculature network. For all cell studies, the side chambers of devices were loaded in multiple configurations to prevent artifacts due to loading protocols. For example, to study CAF versus NBF angiogenic potential, half of the samples were loaded with CAFs in the left side chamber and NBFs in the right chamber, while the other half of samples were loaded in the opposite configuration. For studies testing NBFs, CAFs, cell-free chambers, and genetically-modified fibroblasts, the outward flow regime was used. For studies testing blebbistatin inhibition, the top-to-bottom flow regime was utilized. For these devices, both side chambers received vehicle media for days 0–3; on day 4, one side chamber was selected to receive blebbistatin treatment for the remainder of the study. Blebbistatin concentration was selected by analyzing αSMA expression in CAFs via Western blots.
Bead displacement
To validate COMSOL models of strain propagation, bead displacement studies were utilized to determine how cell-induced strains were propagated between chambers and if this mechanical activity correlated to angiogenesis. To measure mechanical activity of fibroblasts embedded in the side chambers of devices, 1 μm blue fiducial markers were included with the NHLFs/ECs/fibrin mixture during loading into the center chamber. The markers were present in all communication ports at the interface of all chambers. Devices were subjected to 3D live cell imaging (Nikon Ti-E, 40; controlled temperature, humidity, and oxygen (20% O2) and carbon dioxide (5% CO2)) to measure bead displacement over 1 h in a 50 × 50 × 25 μm region of multiple communication ports in each device and configuration. Displacement values correspond to deformations in the ECM generated by fibroblasts. Both direction and magnitude of bead deformations were analyzed using a custom-built Matlab program.27 The resulting displacement values represent dynamic changes introduced by cell movements during the course of 1 h of bead tracking. In some experiments, side chambers were given either 50 μm blebbistatin or vehicle media. Devices were imaged for bead displacement on day 7, then fixed and analyzed for blood vessel growth on day 8.
Western blots
To verify knockdown or enhancement of contractility pathways in CAFs and NBFs, as well as efficacy of blebbistatin on CAFs used in the devices, Western blot analyses were completed. Cell lysates were collected in 1X RIPA buffer with protease inhibitors, and standard protocols were used to process the samples. Antibody concentrations can be found in Table S1.†
Immunofluorescence
Blood vessel growth was quantified via immunofluorescence. To ensure diffusion of all reagents through the microtissue chambers, the outward flow regime was used to administer each solution. During each step of the protocol, the device and solutions were incubated for 48 h at 4 °C, with a switch of flow in the y-direction (Fig. 1b) after 24 h. Devices were then fixed on day 8 with 10% formalin. For antibody staining, microtissues were blocked with 2% BSA in PBS + 0.1% Tween-20, then stained with CD31 or pMLC antibodies in block solution. Afterwards devices were washed with PBS + 0.1% Tween-20 for at least 24 h and then incubated with secondary antibodies. See ESI† Table S1 for antibodies and concentrations. Devices were imaged with a Nikon inverted epifluorescent microscope and images were stitched together using FIJI before analysis of blood vessel growth with AngioTool.65,68 Blood vessel growth was quantified by measuring total blood vessel length in each chamber, and side chamber values were normalized to the total length of vessels in the center chamber of a specific device. For pMLC measurements, fluorescent intensity of pMLC staining in a 50 μm wide region near the interface between chambers was normalized to fluorescent intensity of GFP reporter inserted into NBFs and CAFs in the same region.
Statistical analysis
Except where otherwise stated, all results are averages plus or minus the standard error of the mean for the number of devices analyzed for each condition. A minimum of three devices were utilized for interstitial flow and all bead displacement studies. A minimum of four devices were utilized for all angiogenesis studies. For bead displacement studies, sample size is considered number of unique devices analyzed. A one-way ANOVA was run on all data using Sigmaplot, with post hoc Holm–Sidak tests as necessary.
Results
Microfluidic models for biomechanical investigations
The device design contains three microtissue chambers (blue regions, Fig. 1a and b) which can be loaded independently and are mechanically- and chemically-coupled via communication ports (~20 μm minimum diameter) between adjacent compartments. Each tissue chamber has a loading port, allowing the chambers to be loaded with different matrices and cell populations. In addition, there are multiple independent media feeding lines for each tissue chamber (pink regions, Fig. 1a and b). The overall length scales of the device allows for tissue-mimics on the order of 0.5–2 mm, with a thickness of 100 μm; fluidic line ports are spatially arranged to permit unobstructed views of tissue chambers during experiments (Fig. 1c). The design was developed to allow for generation of vascularized microtissues in the center tissue chamber, allowing for experimental conditions to be tested in the side tissue chambers (Fig. 1b). To achieve this, center chambers were loaded with NHLFs and ECs, while side chambers were loaded with stromal cells (NBFs or CAFs) or cell-free fibrin gels (controls). Unless otherwise noted, the data presented in this study represent devices that had tissue chambers loaded within 15 min of each other and flow was initiated after full fibrin gelation (~45 min after cell loading). To assess the contributions of interstitial flow, we first employed computational modeling (COMSOL) to achieve a design suitable for controlling interstitial flow between chambers. Two regimes are described and were used in subsequent studies: top-to-bottom flow (Fig. 2a) and outward flow (Fig. 2b). In the top-to-bottom flow regime, convective flow between chambers was negligible (Fig. 2c, grey arrows) and interstitial flow occurred predominantly from the upper fluidic lines to the lower fluidic lines for each individual chamber. In the outward flow regime, convection occurs predominantly from the center chamber towards the side chambers, with peak flow velocity occurring within the pores that connect the center and side chambers (Fig. 2d, grey arrows).
Fig. 2.
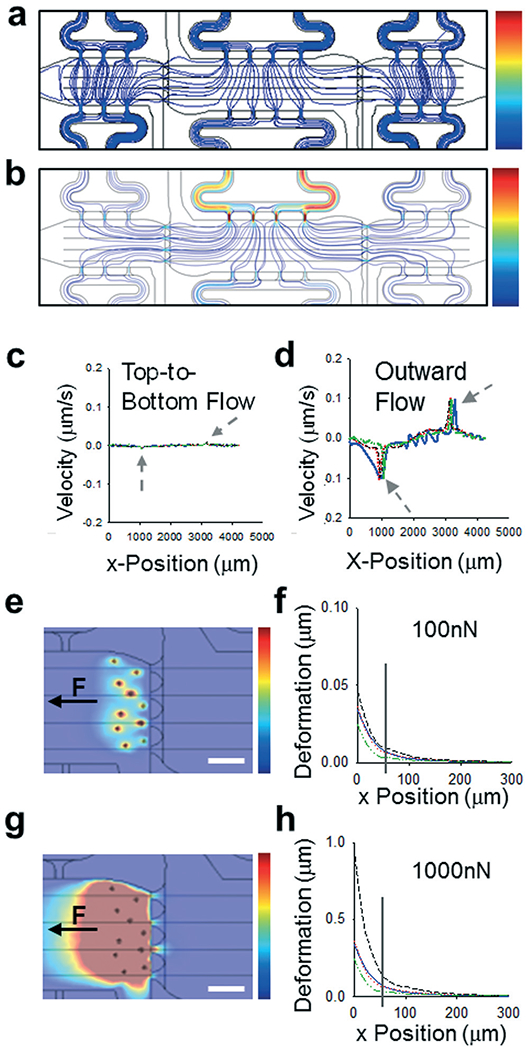
Modeling control over biomechanical parameters. (a and b) Streamline maps of flow regimes in microtissue models showing (a) top-to-bottom flow with no crosstalk between chambers and (b) outward flow with flow from center chamber to side chambers. Color scale bars from 0–0.2 μm s−1. (c and d) Profiles of velocities plotted versus x-position through length of devices, along lines through center of each communication port. Grey arrows indicate chamber interfaces. (e) Color map showing deformations induced by “cell” models with “normal” fibroblast forces of 100nN. Color scale map = 0–0.5 μm. Black arrow denotes direction of force applied. Scale bar = 50 μm. (f) Plot of deformation versus position for map shown in (e), with position 0 representing the leftmost edge of the communication port, closest to the “cells”. The grey drop line indicates the rightmost edge of the communication port. (g) Color map showing deformations induced by “cell” models with CAF-like induced forces of 1000 nN. Color scale map = 0–0.5 μm. (h) Plot of deformation versus position for map shown in (g), with position 0 representing the leftmost edge of the communication port, closest to the “cells”. The grey drop line indicates the rightmost edge of the communication port.
To determine if the microtissues in adjacent compartments were mechanically-coupled, we utilized our computational model to predict how randomly embedded “cells” would transmit forces through a fibrin gel and if these forces propagate between chambers via the communication ports. Deformation of the matrix was modelled along each center line of the communication ports and plotted as a function of distance from the edge of the communication port between side chambers (Fig. 2e–h). As a proof of principle, all “cells” were given a uniform force value and a uniaxial directionality, then a range of forces were used to represent the differences between normal fibroblasts (Fig. 2e and f) and much more mechanically active CAFs (Fig. 2g and h). While most of the displacement shown in the model occurred in the side chamber containing the “cells”, deformations (0.01–0.1 μm for NBFs and 0.1–1 μm for CAFs) of the fibrin matrix also propagated through the communication ports and into the first ~50 μm of the center chamber. These modeling studies indicated that our platform could be used to determine the effects of small mechanical perturbations emanating from side chambers on endothelial cells and blood vessels located in the center chamber.
Control over interstitial flow and diffusion of soluble factors
Predicted interstitial flow patterns were validated using fluorescently-tagged dextrans in DPBS. For top-to-bottom flow studies, there was limited diffusion of 70 kDa FITC-dextran from side chambers to the center chamber and no significant diffusion of RhodamineB-dextran from the center chamber into the side chambers (Fig. 3a and b). Stark demarcations were apparent between tissue chambers in devices loaded in the top-to-bottom flow regime, representing an effective isolation of the chambers from crosstalk by soluble mediators. In a second series of studies, the equilibrated devices from the top-to-bottom studies were subjected to the outward flow regime with DPBS only. The outward flow regime flushed all FITC-tagged dextran from the side chambers, demonstrating that diffusion of factors from these chambers into the center chamber should not occur (Fig. 3c). Lower molecular weight dextrans demonstrated the same behaviors (Fig. S1†).
Fig. 3.
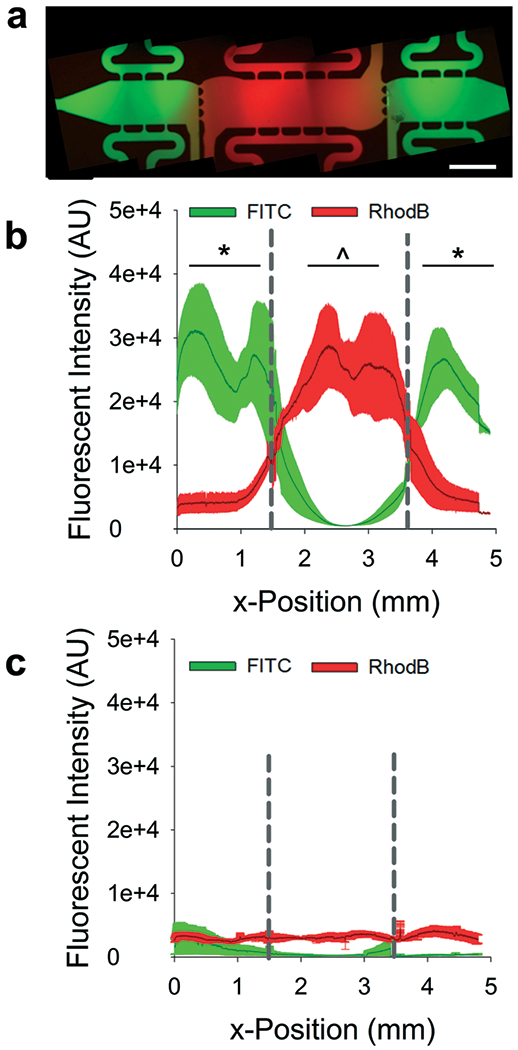
Validation of flow regimes. (a) Representative fluorescent image of multi-tissue chamber microfluidic device loaded with FITC- and RhodamineB-tagged dextrans (70 kDa) after 18 h of top-to-bottom flow. Scale bar = 500 μm. (b) Line tracings of fluorescent intensities across multi-tissue chambers loaded as described in (a) dark line represents average intensity with ± SEM shown in the lighter color. Dashed drop lines represent tissue interfaces. Markers of significance represent comparisons between overall average fluorescent values for separate chambers. *p < 0.01 versus FITC intensity in center chamber; ^p < 0.01 versus RhodamineB intensity in side chambers. (c) Line tracings of fluorescent intensity after devices were subjected to outward flow for 18 h; the same color scheme is used as in (b). For (b) and (c) n = three devices.
Angiogenesis is promoted by mechanical properties of CAFs
Using the microtissue model described above, we determined the mechanical effects of normal fibroblasts or CAFs within the side chambers on angiogenesis from a blood vessel network in the center chamber. Within the center chamber, normal human lung fibroblasts (NHLFs) and human umbilical cord blood derived endothelial cells (ECs) embedded in fibrin gels were utilized to generate a self-assembled vascular network (Fig. 4a).27,52–54 Blood vessel growth in the center chamber and in side chambers was determined after 8 days in culture by staining for CD31. Significantly more blood vessels grew towards chambers containing CAFs compared to NBFs (Fig. 4b). The same trend was observed if side chambers were loaded on day 4 of the experiment (Fig. S2†). For bead displacement studies, results showed that significantly larger deformations occurred at the interface of the CAFs and NHLF/EC chambers compared to NBF interfaces (Fig. 4c). When displacements were segregated by direction, defined as movements towards the center chamber or towards the side chamber, the average magnitude of displacement or deformation was larger towards CAFs than towards NBFs (Fig. S3a and b†). There was no significant difference or preference for bead displacement direction towards the side chambers containing NBFs or CAFs versus towards the center chamber (Fig. S3c†). Control devices with cell-free side chambers showed minimal bead displacements at the interfaces (Fig. S4†). In the control devices with cell-free side chambers, more than 50% of all bead displacements occurred towards the center chamber.
Fig. 4.
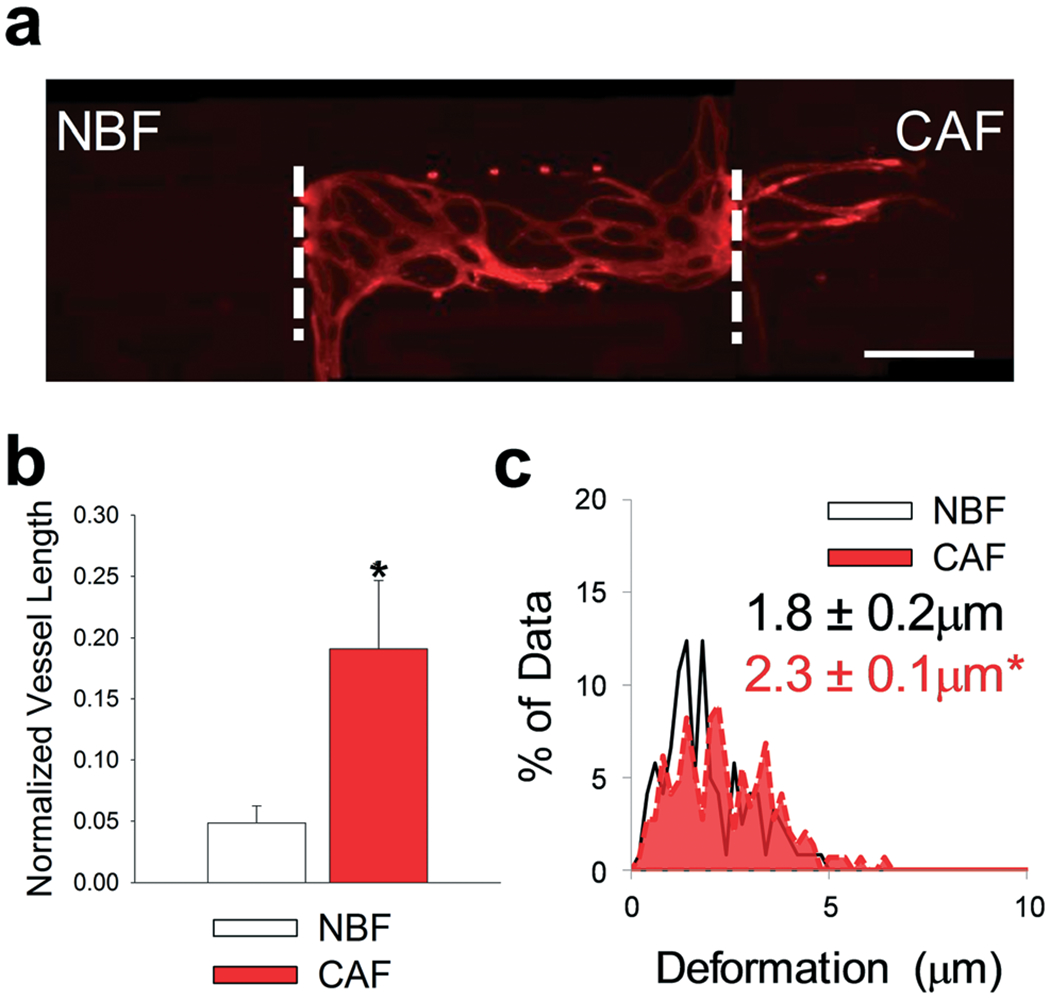
CAFs promoted angiogenesis in microtissue models. (a) Representative fluorescent image of multi-tissue chamber device loaded with NBFs in the left chamber, ECs and NHLF in the center chamber, and CAFs in the right chamber. Devices have been stained for CD31 after 8 days. White dashed lines represent interfaces between chambers. Scale bar = 500 μm. (b) Quantification of vessel growth in side chambers with different fibroblast populations, normalized to total vessel growth in center chamber. *p < 0.01 versus NBF chambers. n = four devices. (c) Histograms showing bead displacement tracked in the communication ports at the chamber interfaces. Inset numbers represent average bead deformation ± SEM. *p < 0.01 versus NBF. n = six devices. See Fig. S9a† for box plots and 95% CI for bead deformation data.
Mechanical inhibition prevents angiogenesis
To determine if the observed preferential angiogenesis was indeed due to mechanical activity of CAFs, a series of experiments were designed to selectively inhibit the mechanotransductive pathways in the stromal cells loaded into the side chamber. First, CAFs were treated with blebbistatin, a soluble inhibitor of actomyosin cytoskeletal contractility. Both side chambers were loaded with CAFs and the devices were fed via the top-to-bottom flow regime to isolate the chambers with respect to interstitial flow. One side chamber (control) received vehicle media, while the other received 50 μM blebbistatin (Fig. 5a). There was a significant decrease in the blood vessel growth into CAF-containing chambers that had been treated with blebbistatin compared to vehicle controls in the same device (Fig. 5b). Vehicle (veh) treated chambers also demonstrated significantly higher ECM deformations induced by CAFs compared to blebbistatin (blebb) treated chambers (Fig. 5c). Control devices received vehicle media in both side chambers (Fig. 5d). No differences in vessel growth or bead deformation magnitudes were observed in vehicle only control devices (Fig. 5e and f). Furthermore, no differences were observed in displacement directionality (Fig. S5†). Side chambers were stained for pMLC to demonstrate inhibition of mechanical characteristics of the stromal cell-containing chambers with respect to angiogenic growth (Fig. 5g and h). There was significantly lower expression of pMLC in cell in the blebbistatin treated chambers when normalized to GFP expression. Furthermore, after device fixation, sequential CD31 and pMLC staining was completed. In other words, pMLC staining occurred 14–16 days after CD31 staining, or approximately 28 days from the time devices were fixed. Debris present in these devices (Fig. 5g and h) likely represents particulate accumulation in devices during either the first or secondary staining protocols, as both were completed in non-sterile conditions.
Fig. 5.
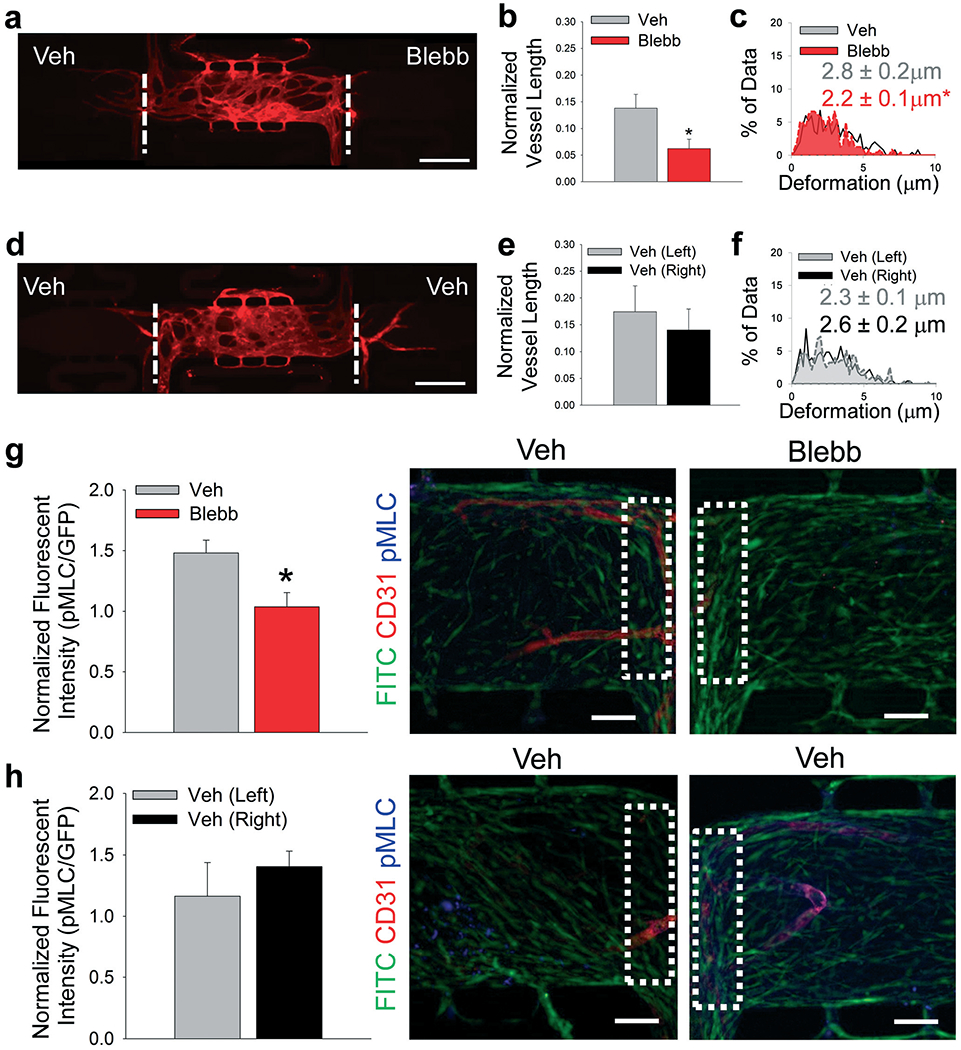
Inhibition of CAF contractility. (a) Representative fluorescent image of multi-tissue chamber devices with CAFs loaded in both side chambers with NHLFs and ECs in center chamber, stained for CD31 after 8 days. Side chambers received either vehicle (Veh) or 50 μM blebbistatin (Blebb) in media in the top-to-bottom flow regime. Scale bar = 500 μm. (b) Quantification of blood vessel growth into side chambers treated with Veh or Blebb media, normalized to total vessel length in center chamber. *p < 0.05 versus Veh. n = 11 devices. (c) Histogram showing deformations induced in the ECM present in communication ports between chambers. Black/white – interface of vehicle-treated and center chambers; red – interface of blebbistatin-treated and center chambers. Inset numbers are average deformation magnitudes ± SEM. *p < 0.05 versus Veh. n = nine devices. (d) Representative fluorescent image of multi-tissue chamber devices with CAFs loaded in both side chambers with NHLFs and ECs in center chamber, stained for CD31 after 8 days. Side chambers were both treated with Veh media in top-to-bottom flow regime. Scale bar = 500 –m. (e) Quantification of blood vessel growth into side chambers treated with Veh media, normalized to total vessel length in center chamber. n = 14 devices. (f) Histogram showing deformations induced in the ECM present in communication ports between chambers. Grey – interface of left vehicle-treated and center chambers; black/white – Interface of right vehicle-treated and center chambers. Inset numbers show average deformation magnitudes ± SEM. n = eight devices. (g and h) Quantification of pMLC staining in control and blebbistatin-treated devices. Inset boxes (white dots) represent ROI analyzed for pMLC staining. Green – CAFs, red – CD31, blue – pMLC. Scale bar = 50 μm. *p < 0.05 versus Veh. n = three devices. See Fig. S9bi and bii† for box plots and 95% CI for bead deformation data.
In another approach to circumvent possible nonspecific inhibition of mechanical activity by blebbistatin, we added cells to side chambers in which cell-intrinsic mechanical properties were manipulated by expression of either constitutively active Rho in NBFs (caRho) or depleted of YAP with shRNA-expressing lentiviruses in CAFs (CAF-shYAP). Angiogenesis from the center chamber was significantly decreased in the side chambers containing CAF-shYAP cells compared to control CAFs modified with an empty vector (CAF–EV) cells (Fig. 6a and b). An analysis of ECM deformations of cells embedded in the microtissue device indicated that the mechanical activity of CAF-shYAP was significantly lower than empty vector (EV) controls (Fig. 6c). Segregated directional data suggested that CAF-shYAP cells were responsible for the decrease in ECM deformations (Fig. S6†). On the other hand, increased angiogenic blood vessel growth (Fig. 6d and e) was observed with NBF-caRho compared to NBF-EV samples. Furthermore, increased mechanical activity, as denoted by increased average magnitude bead displacement, was observed in NBF-caRho compared to NBF-EV samples (Fig. 6f) with no differences in displacement directionality (Fig. S6†), and this resulted in enhanced angiogenesis. All modified cells were had protein levels verified by Western blot (Fig. S7†). These results correlate with previous in vitro 3D models utilized by our lab that measured mechanical properties of these genetically-modified cells in a vasculogenesis assay.27
Fig. 6.
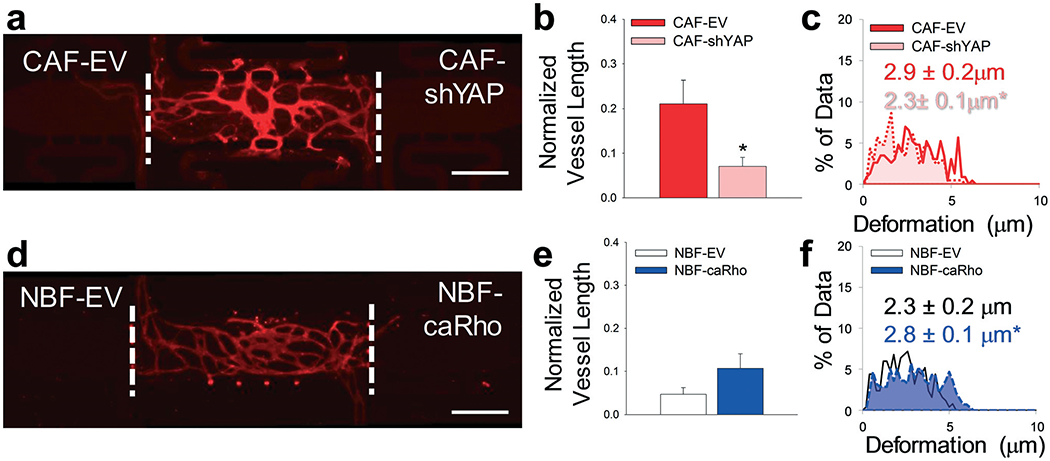
Modified fibroblast phenotype regulation of angiogenesis. (a) Representative fluorescent image of multi-tissue chamber with NHLFs and ECs in the center chamber and genetically-modified fibroblasts including CAF with empty vector (EV) control and CAF-shYAP in the side chambers. Devices were stained for CD31 on day 8. Scale bar = 500 μm. (b) Quantification of blood vessel growth into side chambers containing CAF–EV and CAF-shYAP cells. *p < 0.01 versus CAF–EV. n = 16 devices. (c) Histogram showing deformations induced in the ECM present in communication ports between chambers. Solid line/red – interface of CAF–EV and center chambers. Dotted line/pink – interface of CAF-shYAP and center chambers. Inset numbers show average deformation magnitudes ± SEM. *p < 0.05 versus CAF–EV. n = seven devices. (d) Representative fluorescent image of multi-tissue chamber with NHLFs and ECs in the center chamber and genetically-modified fibroblasts including NBF with empty vector (EV) control and NBF-caRho. Devices were stained for CD31 on day 8. Scale bar = 500 μm. (e) Quantification of blood vessel growth into side chambers containing NBF-EV and NBF-caRho cells. n = 10 devices. (f) Histogram showing deformations in ECM present in communication ports. Black/white – interface of NBF-EV and center chambers; blue – interface of NBF-caRho and center chambers. Inset numbers show average deformation magnitudes ± SEM. *p < 0.05 versus NBF-EV. n = eight devices. See Fig. S9ci and cii† for box plots and 95% CI for bead deformation data.
Discussion
The ability to monitor in real-time the biomechanical activity within a tumor microenvironment in vivo is currently not feasible. To address current limitations in in vitro platforms and further our understanding of the biomechanical regulation of angiogenesis in the tumor microenvironment, we developed a microfluidic-based model of vascularized microtissues. Our model contains three separate microtissue chambers (blue regions, Fig. 1b) that can be loaded independently, providing spatiotemporal control over the initial ECM composition loaded as well as control over number and type of cells embedded in the microtissues. Fluidic lines for each chamber (pink regions, Fig. 1b) are also independent of one another, providing user-control over direction and magnitude of interstitial flow throughout each chamber and the device as a whole. The smallest dimension present in the platform is 20 μm at the opening of the communication ports between microtissues and is approximately 2× the diameter of the average cell. Cell migration from side chambers into the center chambers was not quantified, as almost no GFP-positive cells, CAFs or NBFs, appeared in any center chambers regardless of experimental configuration (data not shown). The overall system exhibits left-to-right symmetry that permits direct comparison of microtissues on either side of a center chamber, thus providing built-in control for all experimental conditions. The microfluidic platform allows for control of multiple mechanical factors, including interstitial flow, tensile forces in the ECM (and thus mechanical strain), and matrix composition or stiffness, representing a novel in vitro platform for biomechanical investigations.
We developed two flow regimes that exhibit control over interstitial flow, limiting crosstalk of soluble fibroblast-secreted factors between microtissue chambers. Experiments utilizing either regime permit isolation and study of biomechanical characteristics of stromal cells present at the microtissue interfaces. Additionally, the outward flow regime demonstrates peak interstitial velocity flow magnitudes of ~0.1 μm s−1, within the normal physiological range, further highlighting the biological relevance of the model.43,51 Average flow rates less than 0.05 μm s−1 are present throughout each microtissue, providing sufficient convection for growth media to diffuse through the entire chamber. Experimental results demonstrated clear demarcations between microtissues loaded with fluorescently-tagged dextrans, validating that diffusion of soluble factors between chambers can be readily controlled. The sizes of dextrans (10–70 kDa) used in these studies covers a range representing many of the different growth factors commonly recognized as angiogenic regulators such as VEGF. Together, the two sets of flow parameters provided efficient methods to control crosstalk from soluble factors between microtissues.
Our microtissue model demonstrated that CAFs drive increased angiogenesis compared to NBFs, consistent with previously reported in vivo data.69 Furthermore, as the outward flow regime prevented diffusion of soluble factors from CAFs or NBFs towards the vascular bed in the center chamber, the increased angiogenesis can be attributed to increased levels of CAF mechanical activity. Interfaces between CAF-loaded chambers and the center chambers had larger average bead displacement magnitudes compared to NBF-loaded chamber interfaces. No strong preference for direction of bead displacement was observed in these studies; bead displacement towards the side chamber is equally as likely as displacement towards the center chamber, regardless of stromal cell type. Since no differences were observed in deformation directionality, some of the bead displacements measured may be due to mechanical activity of NHLFs in the communication ports, and not strictly due to the CAFs or NBFs. This is further supported by control devices with no stromal cells in the side chambers exhibiting minimal bead displacements measured in the communication ports but no angiogenesis. Throughout the experiments, little to no fibrin gel collapse was observed in any chamber, regardless of cells present (Fig. S8†). Second harmonic generation (SHG) imaging was utilized to determine if CAFs deposited and subsequently organized collagen during experiments; however, any collagen present was below the detection threshold of the imaging system used (data not shown). While some ECM remodeling is expected in the microtissues, these studies indicate that gel integrity remains intact for the duration of experiments while permitting angiogenesis and subsequent analyses.
To further characterize how biomechanical characteristics of stromal cells regulate angiogenesis in our model, we utilized a series of experiments to inhibit or enhance cell biomechanical behaviors. For blebbistatin-treated CAFs, angiogenic potential is significantly decreased. Since blebbistatin is non-specific, it could affect multiple cell types in the microtissues as demonstrated by decreased ECM deformations in both directions (towards the side and center chambers) at blebbistatin-treated interfaces. Regardless, results showed that mechanical inhibition of cells in microtissue models suppressed blood vessel growth. Furthermore, there may be significant effects on other aspects of cell behavior induced by blebbistatin treatment; therefore we utilized genetically-modified fibroblasts to further support our argument that cell biomechanical activity correlates to angiogenic potential. For genetically-modified cells, levels of mechanical activity directly correlated with angiogenesis, with CAF-shYAP supporting decreased blood vessel growth and NBF-caRho enhanced growth. By selecting these two proteins for biomechanical activity studies, we have shown that multiple mechanotransduction regulators alter stromal cell supported blood vessel growth. Since these studies were conducted in the outward flow regime, where factors secreted by cells in side chambers were effectively washed away from the chamber interfaces, the results demonstrate that angiogenesis from microvasculature in the center chamber is independently regulated by mechanical properties of stromal cells.
Small variations of angiogenic growth were observed in separate experiments utilizing CAFs. This could be due to different endothelial cell donors or different passage numbers. To ensure validity and reproducibility, all experimental results presented include multiple biological and technical replicates. For vessel measurement studies, “biological replicates” were defined as separate devices and “technical replicates” as experiments started on different dates. Biological replicates utilized the same cell donors and same passage numbers for stromal cells. For all studies, a minimum of two technical replicates were performed. Biological replicate numbers are included in the figure captions. Additionally, variations in average magnitudes of deformations induced by CAFs were observed across different experimental setups. This may be due to heterogeneity of the CAF line or subtle differences in fibrin architecture in individual devices. The CAF–EV cells are genetically-modified control cells infected with an empty lenti-viral vector and selected via hygromycin. Therefore it is unsurprising that there are minor differences in Fig. 6c for CAF–EV bead deformation magnitudes compared to unmodified CAF distributions shown in Fig. 4c and 5c and f. To permit comparison of the inherent variation of CAF biomechanical activity, we have presented average data, distribution data, and 95% CI (Fig. S9†). Importantly, the distributions for CAFs and CAF–EV controls are remarkably similar for all experimental setups.
Finally, it is particularly interesting to note that the vascular networks are exquisitely sensitive to changes in mechanical strain. Increasing or decreasing by ~ 0.5 μm (approximately 1/20th the diameter of a typical cell) resulted in significantly enhanced or suppressed blood vessel growth. This was observed with CAFs, relative to NBFs, as well as blebbistatin-treatment and genetically-modified stromal cells. This result possibly indicates that endothelial cells in vascular networks have a threshold level of strain that initiates angiogenesis and mechanical perturbations below this threshold will allow the cells to remain quiescent. While deformations in the ECM were observed occurring towards side chambers and the center chambers, some of the movements traced may represent mechanical activity of the NHLFs present in the communication ports. However, as these cells are present at both interfaces their biomechanical behaviors do not appear to be sufficient to promote angiogenesis towards microtissues with mechanically-inhibited stromal cells. Additionally, as these studies utilized genetic manipulation techniques targeted to specific cells and mechanotransductive elements, the model demonstrates how biomechanical behaviors of stromal cells explicitly regulate angiogenesis.
Conclusions
In this study, we present experimental models that explore the impact of mechanical strain on angiogenesis using CAFs that are normally present in the tumor microenvironment. Our results demonstrate that we can eliminate diffusion of soluble factors between neighboring microtissues using interstitial flow, and thus isolate the effects of ECM deformations generated by the mechanical properties of cells. Additionally, our studies demonstrated the usefulness of the symmetric model design for built-in controls, as the same vascularized microtissue in the center chamber can be exposed to experimental and control conditions. Consistent with previous in vivo and human data, our model demonstrates that CAFs increase angiogenesis in a directed fashion. Increases in the mechanical behavior of stromal cells correspond to small (0.5 μm) increases in local strains near neighboring blood vessels and subsequent increases in blood vessel growth. Whether endothelial cells respond to mechanical forces by forming tip cells that lead angiogenic vessel growth was not determined, but the described microtissue model should permit such investigations in the future. Our multi-tissue microfluidic device represents a unique and novel opportunity to investigate biomechanical regulation of physiological processes with the ability to independently control multiple mechanical factors as well as measure biomechanical behaviors in real-time.
Supplementary Material
Acknowledgements
The authors wish to thank the following funding sources: K99/R00-CA230202 (M. K. S. L.), R01-CA170879 (S. C. G.), R01-CA223758 (G. D. L.), and U54-CA210173 (G. D. L.). We would like to thank Daphne Cornish, Dr. Priscilla Hwang, and Dr. Audrey Brenot for assistance with experimental setups.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00145g
Data availability
All data that support findings in this study are available from the corresponding author at reasonable request.
Conflicts of interest
The Longmore laboratory receives funding from Pfizer-CTI, San Diego CA and the Centene Corporation of St. Louis MO. However, none of these funds were used to directly support these studies.
References
- 1.Hanahan D and Weinberg RA, Cell , 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J, Engl N . J. Med, 1971, 285, 1182–1186. [DOI] [PubMed] [Google Scholar]
- 3.Ingberand DE, Folkman J, J. Cell Biol, 1989, 109, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weidner N, Semple JP, Welch WR and Folkman J, N. Engl. J. Med, 1991, 324, 1–8. [DOI] [PubMed] [Google Scholar]
- 5.Al-Marrawi MY, Rini BI, Harshman LC, Bjarnason G, Wood L, Vaishampayan U, MacKenzie M, Knox JJ, Agarwal N, Al-Harbi H, Kollmannsberger C, Tan MH, Rha SY, Donskov FN, North S, Choueiri TK, Heng DY and International RCCDC, Target Oncol., 2013, 8, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duda DG, Batchelor TT, Willett CG and Jain RK, Trends Mol. Med, 2007, 13, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross MJ and Claesson-Welsh L, Trends Pharmacol. Sci, 2001, 22, 201–207. [DOI] [PubMed] [Google Scholar]
- 8.Chen TT, Luque A, Lee S, Anderson SM, Segura T and Iruela-Arispe ML, J. Cell Biol, 2010, 188, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warren CM, Ziyad S, Briot A, Der A and Iruela-Arispe ML, Sci. Signaling, 2014, 7, ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A and Maggiolini M, Breast Cancer Res, 2013, 15, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H and Betsholtz C, Nature, 2007, 445, 776–780. [DOI] [PubMed] [Google Scholar]
- 12.Boareto M, Jolly MK, Ben-Jacob E and Onuchic JN, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, E3836–E3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Bock K, Georgiadou M and Carmeliet P, Cell Metab., 2013, 18, 634–647. [DOI] [PubMed] [Google Scholar]
- 14.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M and Carmeliet P, Cell, 2013, 154, 651–663. [DOI] [PubMed] [Google Scholar]
- 15.Sauteur L, Krudewig A, Herwig L, Ehrenfeuchter N, Lenard A, Affolter M and Belting HG, Cell Rep, 2014, 9, 504–513. [DOI] [PubMed] [Google Scholar]
- 16.Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, Weis SM, Byzova TV, Ruggeri ZM, Iruela-Arispe ML and Cheresh DA, Blood, 2012, 119, 2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Smet F, Segura I, De Bock K, Hohensinner PJ and Carmeliet P, Arterioscler., Thromb., Vasc. Biol, 2009, 29, 639–649. [DOI] [PubMed] [Google Scholar]
- 18.Gjorevski N, Boghaert E and Nelson CM, Cancer Microenviron., 2012, 5, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sewell-Loftin MK, Delaughter DM, Peacock JR, Brown CB, Baldwin HS, Barnett JV and Merryman WD, Biomaterials, 2014, 35, 2809–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA and Orimo A, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 20009–20014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL and Weinberg RA, Cell, 2005, 121, 335–348. [DOI] [PubMed] [Google Scholar]
- 22.Bayer SV, Grither WR, Brenot A, Hwang PY, Barcus CE, Ernst M, Pence P, Walter C, Pathak A and Longmore GD, eLife, 2019, 8, e45508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang K, Grither WR, Van Hove S, Biswas H, Ponik SM, Eliceiri KW, Keely PJ and Longmore GD, J. Cell Sci, 2016, 129, 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G and Sahai E, Nat. Cell Biol, 2012, 15, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Wever O, Van Bockstal M, Mareel M, Hendrix A and Bracke M, Semin. Cancer Biol, 2014, 25, 33–46. [DOI] [PubMed] [Google Scholar]
- 26.Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH and Diamandis EP, Mol. Cancer Res, 2012, 10, 1403–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sewell-Loftin MK, Bayer SVH, Crist E, Hughes T, Joison SM, Longmore GD and George SC, Sci. Rep, 2017, 7, 12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ananthanarayanan B, Kim Y and Kumar S, Biomaterials, 2011, 32, 7913–7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S and Weaver VM, Cancer Metastasis Rev., 2009, 28, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pathak A and Kumar S, Integr. Biol, 2011, 3, 267–278. [DOI] [PubMed] [Google Scholar]
- 31.Butcher DT, Alliston T and Weaver VM, Nat. Rev. Cancer, 2009, 9, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dvorak HF, Weaver VM, Tlsty TD and Bergers G, J. Surg. Oncol, 2011, 103, 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H, Mouw JK and Weaver VM, Trends Cell Biol., 2011, 21, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park YK, Tu TY, Lim SH, Clement IJM, Yang SY and Kamm RD, Cell. Mol. Bioeng, 2014, 7, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polacheck WJ and Kamm RD, Biophys. J, 2013, 104, 322a.23442854 [Google Scholar]
- 36.Zaman MH, Trapani LM, Siemeski A, MacKellar D, Gong HY, Kamm RD, Wells A, Lauffenburger DA and Matsudaira P, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 10889–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, Mencia-Trinchant N, Negron Abril YL, Hassane DC, Bonassar LJ, Butcher JT, Weiss RS and Reinhart-King CA, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 492–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carey SP, Kraning-Rush CM, Williams RM and Reinhart-King CA, Biomaterials, 2012, 33, 4157–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carey SP, Martin KE and Reinhart-King CA, Sci. Rep, 2017, 7, 42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lintz M, Munoz A and Reinhart-King CA, J. Biomech. Eng, 2017, 139, 0210051–0210059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC and Reinhart-King CA, Convergent Sci. Phys. Oncol, 2017, 3, 044001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akbari E, Spychalski GB, Rangharajan KK, Prakash S and Song JW, Lab Chip, 2018, 18, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shirure VS, Lezia A, Tao A, Alonzo LF and George SC, Angiogenesis, 2017, 20, 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semino CE, Kamm RD and Lauffenburger DA, Exp. Cell Res, 2006, 312, 289–298. [DOI] [PubMed] [Google Scholar]
- 45.Otranto M, Sarrazy V, Bonte F, Hinz B, Gabbiani G and Desmouliere A, Cell Adhes. Migr, 2012, 6, 203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbari E, Spychalski GB, Rangharajan KK, Prakash S and Song JW, Micromachines, 2019, 10, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song JW and Munn LL, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 15342–15347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain RK, Nat. Med, 2003, 9, 685–693. [DOI] [PubMed] [Google Scholar]
- 49.Jouanneau J, Moens G, Montesano R and Thiery JP, Growth Factors, 1995, 12, 37–47. [DOI] [PubMed] [Google Scholar]
- 50.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alonzo LF, Moya ML, Shirure VS and George SC, Lab Chip, 2015, 15, 3521–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moya M, Tran D and George SC, Stem Cell Res. Ther, 2013, 4(Suppl 1), S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moya ML, Alonzo LF and George SC, Methods Mol. Biol, 2014, 1202, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moya ML, Hsu YH, Lee AP, Hughes CC and George SC, Tissue Eng., Part C, 2013, 19, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V and Kamm RD, Lab Chip, 2009, 9, 269–275. [DOI] [PubMed] [Google Scholar]
- 56.Funamoto K, Zervantonakis IK, Liu Y, Ochs CJ, Kim C and Kamm RD, Lab Chip, 2012, 12, 4855–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB and Kamm RD, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 13515–13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abe Y, Watanabe M, Chung S, Kamm RD, Tanishita K and Sudo R, APL Bioeng, 2019, 3, 036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC and George SC, Integr. Biol, 2014, 6, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heylman C, Sobrino A, Shirure VS, Hughes CC and George SC, Exp. Biol. Med, 2014, 239, 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghajar CM, Suresh V, Peyton SR, Raub CB, Meyskens FL Jr., George SC and Putnam AJ, Mol. Cancer Ther, 2007, 6, 552–561. [DOI] [PubMed] [Google Scholar]
- 62.Alcoser TA, Bordeleau F, Carey SP, Lampi MC, Kowal DR, Somasegar S, Varma S, Shin SJ and Reinhart-King CA, Cell. Mol. Bioeng, 2015, 8, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, Washington MK, Shi C, Franco OE, Weaver AM, Hayward SW, Li D and Webb DJ, J. Cell Biol, 2017, 216, 3799–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Undyala VV, Dembo M, Cembrola K, Perrin BJ, Huttenlocher A, Elce JS, Greer PA, Wang YL and Beningo KA, J. Cell Sci, 2008, 121, 3581–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Preibisch S, Saalfeld S and Tomancak P, Bioinformatics, 2009, 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica-Worms D, Monahan J, Novack DV, McAllister SS and Stewart SA, Cancer Discovery, 2014, 4, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corsa CA, Brenot A, Grither WR, Van Hove S, Loza AJ, Zhang K, Ponik SM, Liu Y, DeNardo DG, Eliceiri KW, Keely PJ and Longmore GD, Cell Rep., 2016, 15, 2510–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zudaire E, Gambardella L, Kurcz C and Vermeren S, PLoS One, 2011, 6, e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erez N, Truitt M, Olson P, Arron ST and Hanahan D, Cancer Cell, 2010, 17, 135–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.