

Summary
The cellular origin of sporadic pancreatic neuroendocrine tumors (PNETs) is obscure. Hormone expression suggests that these tumors arise from glucagon-producing alpha cells or insulin-producing β cells, but instability in hormone expression prevents linage determination. We utilize loss of hepatic glucagon receptor (GCGR) signaling to drive alpha cell hyperproliferation and tumor formation to identify a cell of origin and dissect mechanisms that drive progression. Using a combination of genetically engineered Gcgr knockout mice and GCGR-inhibiting antibodies, we show that elevated plasma amino acids drive the appearance of a proliferative population of SLC38A5+ embryonic progenitor-like alpha cells in mice. Further, we characterize tumors from patients with rare bi-allelic germline GCGR loss-of-function variants and find prominent tumor-cell-associated expression of the SLC38A5 paralog SLC7A8 as well as markers of active mTOR signaling. Thus, progenitor cells arise from adult alpha cells in response to metabolic signals and, when inductive signals are chronically present, drive tumor initiation.
Keywords: GCGR, Glucagon, serum amino-acids, mTOR, tumorigenesis, pancreatic neuroendocrine tumor, PNET, SLC38A5, SLC38A7
Graphical Abstract
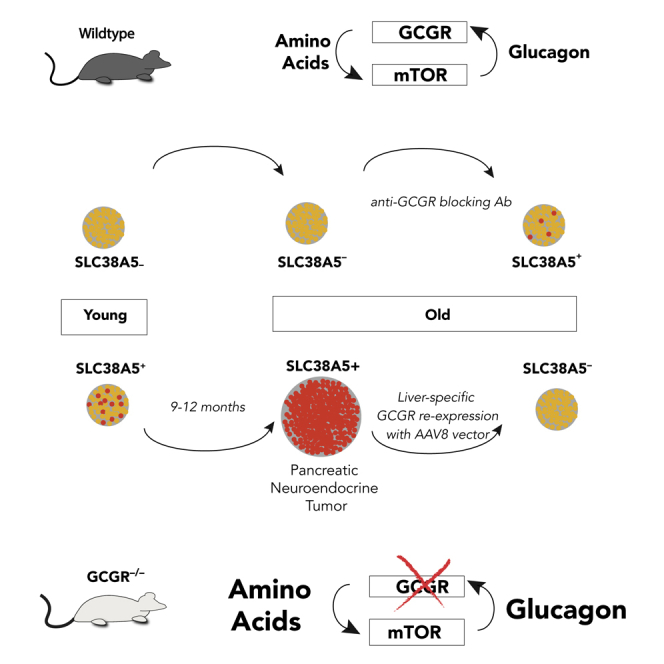
Highlights
GCGR inhibition induces an SLC38A5+ alpha cell population in aged mice
An SLC38A5+ alpha cell subpopulation initiates pancreatic tumors in aged Gcgr−/− mice
Tumors exhibit low mutational burden and response to mTOR inhibition by rapamycin
Tumors in GCGR loss-of-function models lack immune cell infiltration
Smith et al. demonstrate that chronic mTOR signaling, driven by elevated serum amino acids, in Gcgr knockout mice promotes tumor initiation from an SLC38A5+ alpha cell subpopulation and supports tumor maintenance. Furthermore, they provide evidence that an SLC7A8+ alpha cell population contains the progenitors for human tumors in GCGR-mutant patients.
Introduction
Pancreatic neuroendocrine tumors (PNETs) are a relatively rare form of cancer with an incidence of approximately two cases per one million people.1 Although these tumors are often relatively indolent, they are usually identified only after progression, with metastases to the liver a common factor that complicates tumor resection. Although 10% of tumors are associated with familial endocrine tumor syndromes and germline loss of the tumor suppressors MEN1, TSC, VHL, or NF-1, most tumors are sporadic, with exome sequencing finding somatic MEN1 mutations in a significant fraction of these cases.2 This suggests that mechanisms may be shared between familial and sporadic tumorigenesis. Additionally, mutations in DAXX, ATRX, or mTOR pathway components have been identified, indicating that distinct mechanisms are also at work.3
Effective clinical therapies for PNETs have been challenging to identify. Somatostatin or alpha-interferon treatment can counteract the effects of hormone hypersecretion and may have some effect on tumor growth.4 Peptide receptor radionuclide therapy delivers cytotoxic radiolabeled somatostatin directly to tumor cells and may also affect tumor progression.5 Streptozocin is a DNA-alkylating agent that is selectively transported into GLUT2-expressing tumor cells with minimal efficacy.6 More recently, the tyrosine kinase inhibitor sunitinib and the mammalian target of rapamycin (mTOR) inhibitor everolimus were found to improve progression-free survival and dramatically changed the treatment landscape for PNET.7, 8, 9 The effectiveness of everolimus, in light of the fact that mutations in the mTOR pathway commonly arise in PNETs, suggests that a better understanding of the mechanisms behind PNET initiation might open new avenues for treatment.
The cellular origin and molecular etiology of PNETs have been difficult to define. Cell lineage in pancreatic islets is closely tied to the pattern of hormone expression, but it is clear that hormone expression can be substantially dysregulated during tumorigenesis, preventing any clear inference regarding the cell of origin. Most tumors, around 90%, do not produce hormone hyper-secretion syndromes, 6% express multiple hormones, and 4% have been found to change the pattern of hormonal secretion during treatment.10 Patients with familial endocrine tumor syndromes have hyperplastic lesions or microadenomas in their pancreas. Although such plausibly preneoplastic lesions have not been associated with sporadic tumors, hyperplastic changes are found in as many as 10% of normal autopsies, where they may represent a precursor state that leads to frank neoplasia in the small number of individuals who present with sporadic PNETs. Profiling of surgically resected human PNETs has identified subpopulations of tumorigenic cells with stem-like marker gene expression, but these studies have yet to identify a cell of origin that is relatable to a mechanism for tumor initiation.11,12
We previously identified a reciprocal feedback loop involving glucagon secreted by pancreatic alpha cells and hepatic catabolism of amino acids.13 This signaling circuit relies on hepatic glucagon receptor (GCGR) to down-modulate plasma amino acid concentrations in response to glucagon binding. Loss of hepatic GCGR results in elevated plasma amino acids and mTOR-driven alpha cell hyperplasia.14, 15, 16, 17, 18, 19, 20, 21 Ultimately, chronic disruption of GCGR signaling results in gross PNET formation in aged mice and humans.22, 23, 24, 25, 26, 27
Taking advantage of this defined genetic model, we systematically characterized PNET progression using genetically engineered Gcgr knockout mice and identified a proliferative SLC38A5+ cell of origin in mTOR-dependent, immune-restricted adenomas harboring low mutational burden. Importantly, alpha cells of young and aged adult wild-type mice retain transcriptional plasticity and adopt a similar proliferative SLC38A5+ progenitor-like identity in response to elevated plasma amino acids. Characterization of human GCGR-mutated tumors revealed elevated mTOR signaling, minimal immune infiltration, and upregulation of alternative amino acid transporters, including SLC7A8.
Results
An Inducible SLC38A5+ Alpha Cell Subpopulation Drives PNET Initiation
Young adult Gcgr knockout mice exhibit broadly elevated concentrations of free plasma amino acids.13 To understand whether this phenotype persists throughout the period leading up to the appearance of tumors, we isolated blood plasma from 80 heterozygous and homozygous Gcgr knockout mice ranging from 2 weeks to 12 months of age (Figure 1A). We observed sustained increases in plasma amino acid concentration across all ages as well as some amino acids—such as arginine, serine, and threonine—exhibiting concentration increases in late life (Figure S1A).
Figure 1.

Plasticity of Alpha Cell Identity in Aged Wild-Type and Gcgr Knockout Mice Reveals an SLC38A5+ Subpopulation Responsible for Tumor Initiation
(A) Time-course analysis of blood amino acid concentrations in heterozygous and homozygous Gcgr knockout mice corresponding to the mice shown in (B).
(B) Time-course analysis of relative alpha cell area in heterozygous and homozygous Gcgr knockout mice. Insets show representative islets from each genotype at 4 months. Five biological replicates per genotype at each time point. Scale bars, 50 μm.
(C) Analysis of bulk RNA-seq data from FACS-isolated alpha cells from homozygous Gcgr knockout and heterozygous control mice.
(D) Quantification of SLC38A5 immunostaining in 4-month-old mouse pancreas tissues. Five biological replicates per genotype. Scale bars, 50 μm
(E) tSNE visualization of single-cell RNA-seq data from dispersed Gcgr heterozygous islets with alpha cell-specific Gcg and Slc38a5 expression plots.
(F) T-distributed stochastic neighbor embedding (tSNE) visualization of single-cell RNA sequencing data from dispersed Gcgr homozygous knockout islets with alpha cell-specific Gcg and Slc38a5 expression plots.
(G) Time-course analysis of alpha cell area and SLC38A5 expression in mice treated with mAb7 or A9 anti-GCGR antibodies for 2 or 21 days. Five biological replicates per condition at each time point. Scale bars, 50 μm.
(H) Whole-slide H&E staining of pancreas tissue from a Gcgr homozygous knockout mouse with multiple PNETs. Scale bar, 2.5 mm.
(I) SLC38A5 and phosphorylated S6 immunostaining of pancreatic tumor tissue from a Gcgr homozygous knockout mouse. Scale bars, 50 μm.
We next quantified the relative alpha cell area in pancreatic islets over the lifespan of these mice. Hyper-proliferation of alpha cells in knockout mice was detectable as early as 2 weeks of age and persisted throughout adulthood (Figure 1B). Although alpha cells were over-represented in islets of Gcgr knockout mice throughout life, complete loss of growth regulation and escape toward neoplasia were not observed until 9–12 months of age. To better understand the transition from preneoplastic alpha cells to frank tumors, we first used fluorescence-activated cell sorting (FACS) to purify alpha cells from tamoxifen-treated, 3-month-old Gcg-Cre-ERT2;tdTomato mice that were Gcgr heterozygous controls or Gcgr homozygous knockouts and then bulk-sequenced mRNA. In agreement with recent work,17 we observed upregulation of Dapl1, Cd2, and the sodium-dependent neutral amino acid transporter Slc38a5 (Figure 1C). We functionally validated this RNA-based observation by SLC38A5 immunostaining and, although no expression in control tissues was detected, we observed distinct membrane-localized expression in a subset of alpha cells from Gcgr knockout islets (Figure 1D).
To capture the diversity of individual alpha cells within the islets of these mice, we performed single-cell RNA sequencing of dissociated mouse islets and identified diverse islet-associated cell types (Figures 1E and 1F). As expected, islets from Gcgr knockout mice had proportionally higher numbers of alpha cells and, interestingly, an emergent population of pancreatic polypeptide (PP) cells was detectable that could not be clearly identified in control islets (Figure 1F). To assess the heterogeneity of alpha cells, we plotted the Gcg and Slc38a5 unique molecular identifiers (UMIs) for each cell from both datasets. This confirmed limited Slc38a5 expression in control islets and elevated Slc38a5 expression in knockout islets. Further graph-based clustering of alpha cells from Gcgr knockout mice revealed two distinct populations, and, importantly, this clustering was driven not only by Slc38a5 but also by other genes, including Vgf and Dapl1 (Figure S1B).
In light of this alpha cell diversity, we asked whether Slc38a5-expressing alpha cells in knockout mice persisted as residual, incompletely differentiated embryonic alpha cell precursors or represented an inherent mechanism of alpha cell plasticity that enabled proliferation in response to metabolic flux.28 To answer this question, we dosed 2-, 4-, 8-, and 12-month-old wild-type mice with mAb7 (3C7v44) or the A9 anti-GCGR-blocking antibody and then quantified alpha cell area and SLC38A5 expression. As anticipated, alpha cell mass increased following treatment with both antibodies in wild-type mice of all ages over a 3-week period (Figure 1G). More interestingly, a subset of alpha cells from anti-GCGR antibody-treated mice expressed membrane-localized SLC38A5 at each evaluated age (Figure 1G). SLC38A5 was not observed in mice treated with an isotype control antibody. This gene expression plasticity suggests that alpha cells are responsive to increased extracellular plasma amino acids at all ages, independent of the driving mechanism, and in Gcgr knockout mice, this response is likely chronic. We next asked whether chronic induction of this inherent plasticity pathway might be responsible for the transition of preneoplastic alpha cells to adenoma or adenocarcinoma cells of aged mice (Figure 1H). We immunostained PNETs from aged Gcgr knockout mice to assess SLC38A5 expression and observed strong membrane-localized fluorescence in 21 of 23 tumors (Figure 1I). Further, phosphorylated S6 in these cells indicated active mTOR signaling within SLC38A5-positive tumor cells. These observations encouraged transcriptome-wide profiling of tumors to understand how young SLC38A5-expressing alpha cells differ from aged SLC38A5-expressing tumor cells.
Proliferation Signature in SLC38A5+ Tumor Cells Is Amino Acid Dependent
With transcriptomics data from alpha cells in hand, we microdissected PNETs and neighboring control tissue from aged Gcgr knockout mice and extracted mRNA for bulk sequencing. These tumors had a bulk expression profile remarkably similar to alpha cells from young knockout mice and expressed Gcg, Dapl1, Vgf, and Slc38a5 (Figure 2A). To identify possible heterogeneity within tumors, we performed single-cell RNA sequencing of four PNETs and identified small populations of proliferation-enriched glucagon- and SLC38A5-expressing cells (Figure 2B). Although these proliferating tumor cells represent less than 2% of the total tumor volume, they have the strongest proliferation signal of any cells in the dataset, expressing Mki67, Cd168, Cdk1, and numerous other cell cycle regulators (Figure 2C). We validated this observation by co-immunostaining SLC38A5 and MKI67 in fixed PNET tissues (Figure 2D). With the knowledge that SLC38A5-expressing cells are the major proliferative component in tumors, we next asked whether these cells were dependent on supra-physiological plasma amino acid concentrations for continued proliferation and tumor progression. To evaluate this question, we aimed to normalize plasma amino acid concentrations by expression of GCGR in the liver of aged tumor-bearing knockout mice using AAV8 and the liver-specific thyroxine-binding globulin (TBG) promoter. We first defined the optimal titer of AAV8 using a control virus with a GFP reporter. Immunostaining of transduced liver tissues showed that 5 × 1010 units yielded less than 10% expression efficiency, 5 × 1011 units yielded 40% ± 6% efficiency, and 5 × 1012 units reproducibly yielded more than 90% efficiency (Figure 2E) and no GFP expression in pancreatic or tumor tissues. Based on this titer study, we intravenously administered 5 × 1012 units of AAV8 encoding a TBG-regulated mouse Gcgr coding sequence to seven aged tumor-bearing Gcgr knockout mice. We sampled blood plasma before treatment, 2 weeks after administration, 4 weeks after administration, and 6 weeks after administration and then evaluated total amino acid concentration by mass spectrometry. This time series revealed that, although amino acids remained consistently high for control knockout mice, re-expression of Gcgr quickly normalized amino acid concentrations, and all seven mice showed blood amino acid concentrations in a physiological range comparable with aged heterozygous mice (Figure 2F). Simultaneously, we monitored tumor volumes in these mice using live pancreatic ultrasound twice per week for 6 weeks (Figure 2G). Remarkably, restoration of physiological amino acid levels in the blood was associated with significantly reduced tumor volumes in all mice and with complete responses in four of seven mice. Further immunohistochemical analysis of pancreatic tumor tissues from these mice showed attenuated islet hyperplasia, loss of SLC38A5 expression among alpha cells localized to islets, and general loss of SLC38A5 and MKI67 expression within the remaining tumors (Figure 2H).
Figure 2.

PNETs from Gcgr Knockout Mice Are Amino Acid Dependent and Dominantly Composed of Proliferative SLC38A5+ Tumor Cells
(A) Analysis of bulk RNA-seq data from tumors and neighboring pancreatic control tissue from aged homozygous Gcgr knockout mice.
(B) tSNE visualization of single-cell RNA-seq data from dispersed tumors, with insets showing graph-based clustering, Gcg UMI counts, and proliferation signatures for tumor cells.
(C) Differential expression analysis of tumor cells classified as having a proliferative signature based on hallmark expression of MKI67 and cyclin-dependent kinases.
(D) SLC38A5 and MKI67 co-immunostaining in mouse PNET tissue. Scale bars, 100 μm.
(E) GFP immunostaining of liver tissues for quantification of AAV8 titer efficiency. The liver tissue section represents a 5 × 1012 unit titer. Three biological replicates per virus concentration. Scale bar, 1 mm.
(F) Time-course analysis of blood amino acid concentrations in aged homozygous Gcgr knockout mice transduced with AAV8-Gcgr (colored symbols) and aged Gcgr knockout control mice (black and white symbols).
(G) Ultrasound quantification of tumor volume in aged homozygous Gcgr knockout mice transduced with AAV8-Gcgr.
Data point colors correspond to unique mice across (F) and (G).
(H) Whole-slide glucagon, insulin, and SLC38A5 immunostaining of pancreas tissues from tumor-bearing Gcgr homozygous knockout mice that were transduced with AAV8 engineered for liver-specific expression of Gcgr. Scale bars, 1.5 mm.
Dysregulated mTOR Signaling Promotes Low Mutational Burden Tumor Progression
The significant reduction in pancreatic tumor volumes following liver-specific, AAV8-mediated Gcgr expression in aged knockout mice suggests that dysregulated mTOR signaling might be the primary driver of tumor progression because of responsiveness to re-regulation of plasma amino acids. To test this hypothesis, we first evaluated the genomic mutation burden in 11 tumors by exome sequencing (Figure 3A). Although tumors from one mouse exhibited a significant number of mutations, including Pten loss, the remaining mice showed relatively few protein-coding mutations. Further, the few identified mutations in these tumors were found in genes without a clear role in growth regulation and did not disrupt genes in common between mice or even tumors from the same mouse (Figure 3A). The overall pattern of mutation and the lack of obvious driver mutations suggests that acquired mutations or loss of oncogenic repressors are not likely to drive tumor initiation in these mice.
Figure 3.

Alpha Cell-Derived PNETs Are mTOR Dependent but Generally Harbor a Low Exomic Mutational Burden
(A) Analysis of exome sequencing data generated from 11 pancreatic tumors isolated from six independent mice. Bar length represents the total number of protein-altering mutations per tumor, accompanied by mutation state and copy number, represented by circle size, for specific genes of interest. The rightmost network plot shows mutations shared across tumors, with node size representing the relative number of mutations.
(B) Quantification of relative alpha cell area in wild-type mice treated with anti-GCGR antibody and homozygous Gcgr knockout mice treated with rapamycin. Five biological replicates per condition at each time point.
(C) Ultrasound quantification of tumor volume over time in aged homozygous Gcgr knockout mice treated with vehicle or rapamycin. Relative volume change is shown as a percentage of the starting tumor volume.
(D) Cleaved caspase-3 staining of tumor tissues isolated from Gcgr knockout mice treated with vehicle or rapamycin. Scale bars, 25 μm.
We next treated mice with rapamycin to understand the role of mTOR signaling throughout mouse islet development, alpha cell hyperplasia, and tumor onset. First, wild-type mice were treated with an anti-GCGR antibody or anti-GCGR antibody and rapamycin for 21 days (Figure 3B). Rapamycin treatment clearly blunted the expansion of alpha cell area in antibody-treated mice; however, it did not have a substantial effect on alpha cell mass in Gcgr knockout mice from early development through mid-life (Figure 3B). Interestingly, rapamycin did significantly affect alpha cell dysplasia in 12-month-old mice. Because this initial time-course analysis used terminal quantification of alpha cell area from fixed tissues and excluded gross PNETs to minimize group variation, we performed live tumor volume quantification by ultrasound concurrent with twice weekly rapamycin dosing. This revealed that most large tumors showed a steep initial response to rapamycin, followed by a slow continued decrease in tumor volume (Figure 3C). Smaller tumors with volumes of less than 50 mm3, on the other hand, generally exhibited a plateau in response by the third week of treatment. Tumor tissues from these mice treated with rapamycin for 3 weeks were stained for cleaved caspase 3, which confirmed increased intratumoral cell death in treated versus untreated knockout mice (Figure 3D).
Expression Analysis of Human Loss-of-Function GCGR Tumors Identifies SLC7A8 and Lack of Immune Infiltration
Alpha cell hyperplasia and pancreatic tumors have been identified in six human patients carrying GCGR loss-of-function mutations.23, 24, 25,27 Tumors from three of these patients with hyperglucagonemia were analyzed (patients 1–3).25 To understand whether the molecular underpinnings of these cases parallel mouse PNET progression, we performed bulk mRNA sequencing using formalin-fixed, paraffin-embedded (FFPE) tumor specimens from two patients. This analysis identified significant tumor-specific upregulation of GCG, CHGA, VGF, and other anticipated alpha cell-expressed genes (Figure 4A). Although adult mouse and adult human pancreatic alpha cells do not express Slc38a5 under normal physiological amino acid concentrations, we found that elevated plasma amino acid concentrations induce Slc38a5 expression in tumor-initiating alpha cells. Unlike mouse tumors, we did not identify SLC38A5 transcript upregulation or SLC38A5 expression by immunostaining. To determine whether a paralogous amino acid transporter might fulfill the role of amino acid transport in human tumors, we plotted solute carrier gene expression in these tumors and found upregulation of four amino acid transporters: SLC38A4, SLC7A2, SLC7A8, and SLC7A14 (Figure 4B). Immunostaining of tissue sections from three tumors, including those used in RNA sequencing (RNA-seq) analyses, revealed robust, tumor-specific, cell membrane-localized expression of the sodium-independent amino acid transporter SLC7A8 in all three specimens (Figure 4C). In contrast, we observed low signal intensity for SLC38A4, SLC7A2, and SLC7A8 with no specific cellular localization or specificity to tumor cells. In agreement with recent work,22 we observed modest SLC38A4 transcript upregulation (3.1-fold tumor 1, 1.6-fold tumor 2); however, we observed no consequential protein expression in tumor cell membranes or lysosomes (Figure S2A). Having demonstrated that mouse SLC38A5-expressing tumor cells are the dominant proliferative population, we next co-immunostained SLC7A8 and MKI67 in each human specimen to evaluate whether SLC7A8-expressing tumor cells are proliferative. As expected of indolent PNETs, MKI67 expression was infrequent but could be robustly detected in a small population of SLC7A8-expressing tumor cells (Figure 4C).
Figure 4.

Human GCGR-Mutant, Alpha Cell-Derived PNETs Dominantly Express the Amino Acid Transporter SLC7A8+ and Exhibit an Immune Desert-like Phenotype
(A) Analysis of bulk RNA-seq data from two human tumor biopsies obtained from patients harboring premature stop codons or missense mutations in both GCGR alleles. Scale bars, 5 mm.
(B) Reads per kilobase of transcript, per million mapped reads (RPKM) expression analysis and fold change for human solute carrier genes. Amino acid transporters are highlighted by dashed boxes.
(C) Whole-slide SLC7A8 immunostaining of three GCGR mutant human tumor biopsies. Scale bars, 2.5 mm and 250 μm.
(D) Multiplexed digital spatial profiling of mTOR signaling-related and immune cell-related markers using two FFPE human tumor sections.
(E) Whole-slide CD3 immunostaining of mouse pancreas tissue harboring multiple tumors with an in-tissue pancreatic lymph node as a positive control. Scale bars, 250 μm.
PNETs are notoriously challenging to eradicate, and, after an initial transient response to mTOR inhibition, tumors often progress in spite of continued therapy.26,29 To better understand whether recent advances in immune-based therapies might be applicable to this specific PNET subtype, we profiled the immune infiltrate and mTOR pathway signatures in two human tumors using Nanostring multiplexed digital spatial profiling (Figure 4D). As anticipated, glucagon and mTOR activity were detected within tumor cell populations, whereas almost no immune signature was detectable in either specimen. This suggests that alpha cell-derived PNETs in GCGR loss-of-function patients are unlikely to respond to immune-based therapies in the absence of sensitization to an immune-mobilizing event. To confirm this observation in our animal model, we immunostained for CD3 using pancreas tissue containing multiple tumors and pancreatic lymph nodes as a positive control (Figure 4E). We similarly observed minimal T cell infiltration, suggesting that Gcgr knockout mice might serve as an interesting in vivo model for follow-up low-mutational-burden, immune-excluded, and immune-desert tumor studies.
Discussion
In this work, we investigate the cell of origin and signaling pathways that drive initiation and progression of PNETs in Gcgr knockout mice. Loss of hepatic GCGR results in disrupted glucagon signaling13 and elevated free amino acids in circulating blood. Recent work has demonstrated that some alpha cells in young GCGR knockout mice express the amino acid transporter SLC38A5,14,17 which has been observed previously only in the embryonic pancreas.28 This prompted us to investigate the plasticity of alpha cell gene expression in response to metabolic changes in adult mice as well as determine whether alpha cell-derived tumors might originate from embryonic precursor-like alpha cells. Pancreatic islets of wild-type adult mice treated with an anti-GCGR antibody for 3 weeks, but not control animals, contained alpha cells expressing SLC38A5, much like engineered GCGR knockout animals. This revealed that adult alpha cells retain transcriptional plasticity and, in response to metabolic challenge, such as serum amino acid saturation, can transition to a proliferative mTOR-active state.
This plasticity encouraged a deeper sequencing-based study of the relationship between SLC38A5-expressing cells from young adult knockout mice and aged mice with SLC38A5-expressing tumor cells. Interestingly, these cells have very similar transcriptional profiles with markedly higher proliferation signatures in the tumor population. Here we implicate alpha cell plasticity in tumor initiation and show that constitutive activation of an amino acid-responsive pathway in an alpha cell subpopulation drives tumor progression.
Extensive genomic characterization of PNETs with an alpha cell signature identified ATRX, DAXX, and MEN1 mutations as causative drivers of tumorigenesis as well as enhancer activation driving the lineage-restricted transcription factors ARX and PDX1;30,31 however, PNETs originating specifically from Gcgr knockout mice generally show few exonic mutations, indicating that signaling-driven mechanisms are likely responsible for tumor growth. Further, support for this concept came from RNA-seq profiling and tumor response studies following adeno-associated virus (AAV)-mediated hepatic expression of Gcgr in knockout mice and rapamycin treatment of tumor-bearing mice. In both studies, tumor volume significantly decreased following treatment, indicating that tumor progression is strictly reliant on continued supply of serum amino acids and mTOR activity. In agreement with prior work, it has been shown that loss of mTORC1 signaling alters adult pancreatic alpha cell mass13 and that everolimus, an mTOR inhibitor, is effective for treatment of advanced PNETss.26
Building on results from our mouse model studies, we analyzed resected human tumor biopsies from Gcgr-mutant patients by Nanostring digital spatial profiling. These tumors exhibit an alpha cell character, elevated mTOR signaling, and lack of immune infiltration similar to mouse tumors. Interestingly, human tumors do not express SLC38A5 and, instead, strongly express the amino acid transporter SLC7A8, a sodium-independent neutral amino acid transporter that is upregulated in tumors of diverse origin.32 Recent studies have identified transcription evidence of expression of SLC38A4 in these tumors;22 however, we have been unable to verify protein expression by immunostaining. Much like the amino acid transporters SLC1A5, SLC7A5, and SLC7A11, which are upregulated in numerous cancer types, SLC7A8 represents an interesting target for modulating tumor cell metabolism.33
Collectively, this work represents a significant advance in our understanding of the origin of alpha cell-derived PNETs and defines signaling pathways that can potentially be targeted to prevent disease progression.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| A9 anti-GCGR | Genentech | A9 |
| 3C7v44 anti-GCGR | Genentech | mAb7 |
| Guinea Pig anti-insulin | Abcam | Cat#:AB7842 |
| Human anti-GP120 | Genentech | GP120 |
| Mouse anti-glucagon | Sigma Aldrich | Cat#:G2654 |
| Rabbit anti-CD3 | ThermoFisher | Cat#:RM9107S |
| Rabbit anti-GFP | Abcam | Cat#:AB183734 |
| Rabbit anti-glucagon | Abcam | Cat#:AB92517 |
| Rabbit anti-pS6 | CST | Cat#:5364S |
| Rabbit anti-SLC38A4 | LifeSpan Biosciences | Cat#:LS-C31936 |
| Rabbit anti-SLC38A5 | Abcam | Cat#:AB72717 |
| Rabbit anti-SLC7A8 | Origene | Cat#:UM500058 |
| Rat anti-MKI67 | Invitrogen | Cat#:14-5698-82 |
| Secondary Alexa 488, 555, 594, 647 labeled antibodies | Invitrogen | Multiple |
| Bacterial and Virus Strains | ||
| AAV8-GCGR | Virovek | N/A |
| AAV8-GFP | Vector Biolabs | Cat#:7061 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accumax | Innovative Cell Technologies | Cat#:AM105 |
| Histopaque 1077 | Sigma Aldrich | Cat#:10771 |
| Hoechst 33342 | Life Technologies | Cat#:H21492 |
| Liberase TL | Roche | Cat#:5401020001 |
| MACS Tumor Dissociation Kit | Miltenyi Biotec | Cat#:130-096-730 |
| Rapamycin | SelleckChem | Cat#:S1039 |
| Tamoxifen | Sigma Aldrich | Cat#:T5648 |
| Deposited Data | ||
| Bulk RNA-seq | This Paper | GSE142232 |
| Exome-seq | This Paper | GSE142231 |
| Single Cell RNA-seq | This Paper | GSE142233 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Gcgr knockout | Genentech | N/A |
| Mouse: C57/BL6J | Jackson Labs | Cat#:000664 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Peterson (andpete@gmail.com)
Materials Availability
This study did not generate new unique reagents that have not been previously published in scientific literature.
Data and Code Availability
All bulk RNA-seq (GSE142232), single cell RNA-seq (GSE142233), and exome-seq (GSE142231) datasets have been made available under NCBI GEO series accession number GSE142234.
Experimental Model and Subject Details
Mouse Models
Animal husbandry and veterinary care was provided by the Department of Laboratory Animal Resources at Genentech. Experimental procedures were reviewed and approved by Genentech’s Institutional Animal Care and Use Committee, South San Francisco, CA USA.
Adult mice with equal numbers of male and female mice were used in all studies. In the case of transgenic reporter labeling of alpha cells, animals from 1 week to 3 weeks of age were used in some experiments. The animals ranged in age from 1 week to 18 months of age with the specific ranges of the animals used for each experimental approach described in the section on experimental details below. The number of mice used for each experiment was based on prior characterization of the mice as previously described.13.
Gcgr knockout and transgenic reporter (GcgCreERT2;tdTomato) mice were generated as previously described.13 Gcgr knockout experimental cohorts were generated from heterozygous breeding pairs. C57BL/6J mice were from The Jackson Laboratory. Animals were maintained in a pathogen-free animal facility under 12hr light/12hr dark cycle with access to a standard rodent chow and water ad libitum.
Human Subjects
Individuals homozygous for germ-line loss of function mutations in the GCGR gene were ascertained at the University of Tübingen Hospital following presentation with glucacgonomas as previously described.25 Informed consent for the donation of tumor tissue and its use in research were reviewed and approved by the Institutional Review Board for Human Subjects at the University of Tübingen Hospital, Tübingen Germany. The tumor material was obtained during tumor re-section for medical reasons and our studies did not involve treatment of the patients. Material from 3 donors was used: Male, Age 25; Female, Age 43; Male, Age 58.
Method Details
Mouse Model Experimental Details
The induction of GcgCreERT2;tdTomato transgenic alleles was performed using tamoxifen (Sigma Aldrich, #T5648) in corn oil (40 mg/ml) administered at 200 mg/kg every day for 5 to 7 days by intraperitoneal (IP) injection into animals ranging from 1 week to 12 months of age. Both anti-GCGR antibodies (Genentech) and GP120 control antibody (Genentech) were administered by IP injection at 10mg/kg either once (2-day time point) or once every seven days for three weeks (21-day time point) into animals from 2 months to 1 year of age. Rapamycin was administered by IP injection at 10 mg/kg from 3 weeks to 18 months of age; twice weekly for tumor volume quantification by ultrasound, once every seven days for three weeks for anti-GCGR experiments, and once every four days for 12 total days for Gcgr knockout time course analysis. AAV8 expressing either mouse GCGR or GFP was administered by intravenous tail vein injection into mice from 12 to 18 months of age at titers specified in Figure 2. For tumor volume quantification, mice were anesthetized with sevofluorane and abdominal imaging performed with Vevo2100 (Visual Sonics). Tumor volumes at baseline were assessed and animals were assigned to groups to provide a similar distribution of tumor volumes in both treatment and cantrol groups.
Plasma Amino Acid Concentration Analysis
Blood was collected at specified time points by retroorbital bleed or terminal cardiac puncture. EDTA plasma samples were analyzed by mass spectrometry (UC Davis West Coast Metabolomics Center) as previously described.13
Immunohistology and Islet Cell Area Quantification
Immunostaining was performed on 5- to 7-mm formalin-fixed, paraffin- embedded (FFPE) mouse tissue sections using citrate-buffered boiling epitope retrieval. The following antibodies were used: guinea pig anti-Insulin (Abcam, AB7842), mouse anti-Glucagon (Sigma-Aldrich, G2654), rabbit anti-CD3 (ThermoFisher, RM9107S), rabbit anti-GFP (Abcam, AB183734), rabbit anti-Glucagon (Abcam, AB92517), rabbit anti-pS6 (Cell Signaling Technology, 5364S), rabbit anti-SLC38A4 (LifeSpan Biosciences, LS-C31936), rabbit anti-SLC38A5 (Abcam, AB72717), rabbit anti-SLC7A8 (Origene, UM500058), rat anti-MKI67 (Invitrogen, 14-5698-82), and appropriate Alexa Fluor 488-, 555-, 594-, or 647-labeled secondary antibodies (Invitrogen). Nuclei were stained with Hoechst 33342 (Life Technologies, #H21492). Immunofluorescence was quantified from images of pancreatic sections using MATLAB software (version R2012b, Mathworks).
RNA Sequencing: Islet Cells
Single islet cells were obtained by euthanizing mice, inserting a needle along the bile duct, and inflating the pancreas with ice-cold Liberase TL (Roche, 5401020001). The pancreas tissue was removed by dissection, placed in 5 mL DMEM-Liberase TL solution, and incubated for 10 minutes at 37°C. Following incubation, tissue was dissociated by 10 repetitions of gentle shaking and addition of 20 mL of quench buffer (25 mM HEPES pH 7.2, 1.8 mM CaCl2, 10% fetal bovine serum) followed by gentle trituration and incubation at room temperature for 10 minutes. Once the dissociated tissues were settled, 10 mL Histopaque 1077 (Sigma Aldrich, 10771) was added to the bottom of the conical tube followed by centrifugation to trap islets at the medium-Histopaque interface. Islets at the interface were collected by pipet, transferred to a clean tube, and pelleted by centrifugation. Pelleted islets were dissociated into single cells by incubation with Accumax (Innovative Cell Technologies, AM105) for 15 minutes at room temperature and clarified by passing through a sterile filter cap tube. For single cell studies, unsorted single islet cells were prepared with Chromium Single Cell 3′ Library and Gel Bead Kit V2 (10X Genomics). The cell density and viability were quantified using a Vi-Cell XR cell counter (Beckman Coulter) with all processed samples have > 95% viability. Cell density was used to calculate the volume of single cell suspension needed in the reverse transcription master mix for library generation (target 6,000 cells, 10X Genomics). Libraries were profiled by Bioanalyzer High Sensitivity DNA Kit (Agilent Technologies) and quantified using a Kapa Library Quantification Kit (Kapa Biosystems). Each library was sequenced using one HiSeq4000 lane (Illumina) following 10X Genomics recommendations. Data analysis was performed using Partek and pancreatic islet cell types were initially classified based on established hallmark gene expression such as glucagon and insulin.
RNA Sequencing: Bulk Alpha Cells and Tumors
Islets were isolated as described above and single cells were tdTomato-fluorescence sorted, bulk RNA isolated with Trizol, and purified. Libraries were prepared using a TruSeq RNA Sample Preparation kit (Illumina). The libraries were multiplexed and sequenced on Illumina HiSeq 2500 and HiSeq 4000 (1x50bp). Reads were aligned to the human genome (GRCm38) using GSNAP. Expression counts per gene were obtained by counting the number of reads aligned concordantly within a pair and uniquely to each gene locus as defined by NCBI and Ensembl gene annotations and RefSeq mRNA sequences. Differential gene expression analysis was performed using edgeR and DESeq2 was used to compute the variance stabilized expression values. For tumor studies, gross pancreatic tumors were hand dissected, cleaned of surrounding non-tumor tissues in PBS, and dissociated using the MACS Tumor Dissociation Kit (Miltenyi Biotec, 130-096-730) with program 37C_m_TDK_1. Strained cells were treated with red blood cell lysis solution (Miltenyi Biotec, 130-094-183) then subjected to library preparation, sequencing, and data analysis as described above.
RNA Sequencing: FFPE Tumor Tissue
Human FFPE-tumor tissue samples were obtained from a study approved by the Ethics Committees of the Universities of Kiel and Tübingen where all living patients and healthy relatives (1-3) gave an informed consent. Total RNA was extracted from FFPE slides using QIAGEN RNeasy FFPE kit (QIAGEN). RNA was quantified using Qubit 2.0 Fluorometer RNA assay (Invitrogen) and RNA integrity was checked with Agilent 4200 TapeStation (Agilent Technologies). Ribosomal RNA was depleted using Ribo-Zero Gold Kit (Human/Mouse/Rat probe) (Illumina). Multiplexed RNA libraries were prepared NEBNext Ultra RNA Library Prep Kit (Illumina), clustered on three flow cell lanes, and sequenced on an Illumina HiSeq instrument (2x150 paired end configuration). Sequence reads were trimmed to remove adaptor sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens GRCh38.87 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. Unique gene hit counts were calculated using feature counts from the Subread package v.1.5.2. The gene hit counts table was used for downstream differential expression analysis using edgeR. The QL F-test was used for dispersion estimation and hypothesis testing to generate p values and log2 fold changes. Genes with adjusted p values less than 0.05 and absolute log2 fold changes greater than 1 were called as differentially expressed genes for each comparison.
Exome Sequencing
Gross pancreatic tumors were hand dissected and cleaned of surrounding non-tumor tissues in PBS while control tissue was isolated from non-tumor regions of the same mouse. Tissues were disaggregated as described above and exome capture was performed using the Agilent SureSelect Mouse All Exome kit. Exome capture libraries were sequenced on HiSeq 2500 (Illumina) to generate 2 × 75 bp paired-end data. Sequencing reads were mapped to UCSC mouse genome (GRCm38) using BWA software set to default parameters. Local realignment, duplicate marking, and raw variant calling were performed according to GATK best practices. Somatic variant calling on tumor and its matched normal BAM file was performed using Strelka.
Nanostring Digital Spatial Profiling
An automated setup capable of imaging and sample collection was developed by modifying a standard microscope. For protein detection, a multiplexed cocktail of primary antibodies, each with a unique, UV photocleavable indexing oligo, and three fluorescent antibody markers was applied to a slide-mounted FFPE tissue section. The tissue slide was placed on the stage of an inverted microscope. A custom gasket was then clamped onto the slide, allowing the tissue to be submerged in 1.5 mL of buffer solution. The microcapillary tip is connected to a syringe pump primed with buffer solution, allowing for accurate aspiration of small volumes (< 2.5 μL). Under the microscope, wide field fluorescence imaging was performed with epi-illumination from visible LED light engine. The tissue area of interest was then located using fluorescence imaging with a CMOS camera and 20x images (650μm x 650μm) were stitched together to yield a high-resolution image of the tissue area of interest. The regions of interest (ROIs) were then selected based on the fluorescence information and sequentially processed by the microscope automation. The steps performed for each ROI by the microscope automation were as follows. First, the microcapillary tip was washed by dispensing clean buffer out the capillary and into a wash station. Next, the tissue slide was bulk washed by exchanging the buffer solution on the slide via the inlet and outlet wash ports on the gasket clamp. The microcapillary tip was then moved into position directly over the ROI with a distance of 50 μm from the tissue. The local region of tissue around the ROI was washed by dispensing 100 μL of buffer solution from the microcapillary. The area of tissue within the ROI was selectively illuminated with UV light to release the indexing oligos. UV LED light was collimated to be reflected from the DMD surface into the microscope objective, and focused at the sample tissue. Each micromirror unit in the DMD corresponds to ∼1μm2 area of sample and reflects the UV light in controlled pattern based on the ROI selection in the image. Following each UV illumination cycle, the eluent was collected from the local region via microcapillary aspiration and transferred to an individual well of a microtiter plate. Once all ROIs were processed, indexing oligos were hybridized to NanoString optical barcodes for ex situ digital counting and subsequently analyzed with an nCounter Analysis System.
Quantification and Statistical Analysis
Statistical analysis was performed using relevant DNA- or RNA-sequencing software for each experiment as described in Method Details. Statistical significance for in vivo experiments was considered at a threshold of p < 0.05 by Student’s t test and experimental data are presented as mean ± SEM. The replicate and statistical details of experiments can be found in figure legends.
Acknowledgments
We thank our mouse genome engineering and colony management colleagues for generation and genotyping of mice, our colleagues in necropsy and pathology for assistance with tissue collection and sample preparation, our colleagues in antibody engineering for production of anti-GCGR reagents, and our colleagues in the next-generation sequencing facility for sample preparation and data analysis. We also acknowledge Genewiz, Genscript, Nanostring Technologies, UC Davis West Coast Metabolomics Center, and Virovek for reagent or experimental service contributions to this work.
Author Contributions
D.K.S., L.K., S.D., N.P., E.S., N.K., and A.S.P. designed and conducted the studies. B.S. provided human tumor biopsy tissues. D.K.S., S.D., E.S., O.F., M.J.S., B.B.A., and A.S.P. analyzed the data. D.K.S. and A.S.P. wrote the manuscript.
Declarations of Interest
The authors declare no competing interests.
Published: July 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100058.
Supplemental Information
References
- 1.Halfdanarson T.R., Rabe K.G., Rubin J., Petersen G.M. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008;19:1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ro C., Chai W., Yu V.E., Yu R. Pancreatic neuroendocrine tumors: biology, diagnosis,and treatment. Chin. J. Cancer. 2013;32:312–324. doi: 10.5732/cjc.012.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiao Y., Shi C., Edil B.H., de Wilde R.F., Klimstra D.S., Maitra A., Schulick R.D., Tang L.H., Wolfgang C.L., Choti M.A. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bajetta E., Procopio G., Ferrari L., Catena L., Del Vecchio M., Bombardieri E. Update on the treatment of neuroendocrine tumors. Expert Rev. Anticancer Ther. 2003;3:631–642. doi: 10.1586/14737140.3.5.631. [DOI] [PubMed] [Google Scholar]
- 5.Sharma N., Naraev B.G., Engelman E.G., Zimmerman M.B., Bushnell D.L., Jr., OʼDorisio T.M., OʼDorisio M.S., Menda Y., Müller-Brand J., Howe J.R., Halfdanarson T.R. Peptide receptor radionuclide therapy outcomes in a North American cohort with metastatic well-differentiated neuroendocrine tumors. Pancreas. 2017;46:151–156. doi: 10.1097/MPA.0000000000000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCollum A.D., Kulke M.H., Ryan D.P., Clark J.W., Shulman L.N., Mayer R.J., Bartel S., Fuchs C.S. Lack of efficacy of streptozocin and doxorubicin in patients with advanced pancreatic endocrine tumors. Am. J. Clin. Oncol. 2004;27:485–488. doi: 10.1097/01.coc.0000135343.06038.eb. [DOI] [PubMed] [Google Scholar]
- 7.Ge W., Zhou D., Zhu L., Song W., Wang W. Efficacy and safety of Everolimus plus somatostatin analogues in patients with neuroendocrine tumors. J. Cancer. 2018;9:4783–4790. doi: 10.7150/jca.25908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raymond E., Dahan L., Raoul J.-L., Bang Y.-J., Borbath I., Lombard-Bohas C., Valle J., Metrakos P., Smith D., Vinik A. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 9.Yao J.C., Shah M.H., Ito T., Bohas C.L., Wolin E.M., Van Cutsem E., Hobday T.J., Okusaka T., Capdevila J., de Vries E.G.E., RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crona J., Norlén O., Antonodimitrakis P., Welin S., Stålberg P., Eriksson B. Multiple and secondary hormone secretion in patients with metastatic pancreatic neuroendocrine tumors. J. Clin. Endocrinol. Metab. 2016;101:445–452. doi: 10.1210/jc.2015-2436. [DOI] [PubMed] [Google Scholar]
- 11.Krampitz G.W., Norton J.A., Poultsides G.A., Visser B.C., Sun L., Jensen R.T. Lymph nodes and survival in pancreatic neuroendocrine tumors. Arch. Surg. 2012;147:820–827. doi: 10.1001/archsurg.2012.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krampitz G.W., George B.M., Willingham S.B., Volkmer J.-P., Weiskopf K., Jahchan N., Newman A.M., Sahoo D., Zemek A.J., Yanovsky R.L. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA. 2016;113:4464–4469. doi: 10.1073/pnas.1600007113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solloway M.J., Madjidi A., Gu C., Eastham-Anderson J., Clarke H.J., Kljavin N., Zavala-Solorio J., Kates L., Friedman B., Brauer M. Glucagon couples hepatic amino acid catabolism to mTOR-dependent regulation of α-cell mass. Cell Rep. 2015;12:495–510. doi: 10.1016/j.celrep.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 14.Dean E.D., Li M., Prasad N., Wisniewski S.N., Von Deylen A., Spaeth J., Maddison L., Botros A., Sedgeman L.R., Bozadjieva N. Interrupted glucagon signaling reveals hepatic α cell axis and role for L-glutamine in α cell proliferation. Cell Metab. 2017;25:1362–1373.e5. doi: 10.1016/j.cmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galsgaard K.D., Winther-Sørensen M., Ørskov C., Kissow H., Poulsen S.S., Vilstrup H., Prehn C., Adamski J., Jepsen S.L., Hartmann B. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab. 2018;314:E93–E103. doi: 10.1152/ajpendo.00198.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelling R.W., Du X.Q., Dichmann D.S., Rømer J., Huang H., Cui L., Obici S., Tang B., Holst J.J., Fledelius C. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J., Okamoto H., Huang Z., Anguiano G., Chen S., Liu Q., Cavino K., Xin Y., Na E., Hamid R. Amino acid transporter Slc38a5 controls glucagon receptor inhibition-induced pancreatic α cell hyperplasia in mice. Cell Metab. 2017;25:1348–1361.e8. doi: 10.1016/j.cmet.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M., Dean E.D., Zhao L., Nicholson W.E., Powers A.C., Chen W. Glucagon receptor inactivation leads to α-cell hyperplasia in zebrafish. J. Endocrinol. 2015;227:93–103. doi: 10.1530/JOE-15-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longuet C., Robledo A.M., Dean E.D., Dai C., Ali S., McGuinness I., de Chavez V., Vuguin P.M., Charron M.J., Powers A.C., Drucker D.J. Liver-specific disruption of the murine glucagon receptor produces alpha cell hyperplasia. Diabetes. 2013;62:1196–1205. doi: 10.2337/db11-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vuguin P.M., Kedees M.H., Cui L., Guz Y., Gelling R.W., Nejathaim M., Charron M.J., Teitelman G. Ablation of the glucagon receptor gene increases fetal lethality and produces alterations in islet development and maturation. Endocrinology. 2006;147:3995–4006. doi: 10.1210/en.2005-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J., MacDougall M.L., McDowell M.T., Xi L., Wei R., Zavadoski W.J., Molloy M.P., Baker J.D., Kuhn M., Cabrera O., Treadway J.L. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: implications on anti-glucagon therapies for diabetes. BMC Genomics. 2011;12:281. doi: 10.1186/1471-2164-12-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J., Dominguez Gutierrez G., Xin Y., Cavino K., Sung B., Sipos B., Kloeppel G., Gromada J., Okamoto H. Increased SLC38A4 amino acid transporter expression in human pancreatic αcells after glucagon receptor inhibition. Endocrinology. 2019;160:979–988. doi: 10.1210/en.2019-00022. [DOI] [PubMed] [Google Scholar]
- 23.Larger E., Albrechtsen N.J.W., Hansen L.H., Gelling R.W., Capeau J., Deacon C.F., Madsen O.D., Yakushiji F., Meyts P.D., Holst J.J., Nishimura E. Pancreatic α cell hyperplasia and hyperglucagonemia due to a glucagon receptor splice mutation. Endocrinol. Diabetes Metab. Case Rep. 2016;2016:16-0081. doi: 10.1530/EDM-16-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller H.C., Kidd M., Modlin I.M., Cohen P., Dina R., Drymousis P., Vlavianos P., Klöppel G., Frilling A. Glucagon receptor gene mutations with hyperglucagonemia but without the glucagonoma syndrome. World J. Gastrointest. Surg. 2015;7:60–66. doi: 10.4240/wjgs.v7.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sipos B., Sperveslage J., Anlauf M., Hoffmeister M., Henopp T., Buch S., Hampe J., Weber A., Hammel P., Couvelard A. Glucagon cell hyperplasia and neoplasia with and without glucagon receptor mutations. J. Clin. Endocrinol. Metab. 2015;100:E783–E788. doi: 10.1210/jc.2014-4405. [DOI] [PubMed] [Google Scholar]
- 26.Yao J.C., Phan A.T., Jehl V., Shah G., Meric-Bernstam F. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res. 2013;73:1449–1453. doi: 10.1158/0008-5472.CAN-12-3923. [DOI] [PubMed] [Google Scholar]
- 27.Zhou C., Dhall D., Nissen N.N., Chen C.-R., Yu R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas. 2009;38:941–946. doi: 10.1097/MPA.0b013e3181b2bb03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanescu D.E., Yu R., Won K.-J., Stoffers D.A. Single cell transcriptomic profiling of mouse pancreatic progenitors. Physiol. Genomics. 2017;49:105–114. doi: 10.1152/physiolgenomics.00114.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandamme T., Beyens M., de Beeck K.O., Dogan F., van Koetsveld P.M., Pauwels P., Mortier G., Vangestel C., de Herder W., Van Camp G. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br. J. Cancer. 2016;114:650–658. doi: 10.1038/bjc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cejas P., Drier Y., Dreijerink K.M.A., Brosens L.A.A., Deshpande V., Epstein C.B., Conemans E.B., Morsink F.H.M., Graham M.K., Valk G.D. Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat. Med. 2019;25:1260–1265. doi: 10.1038/s41591-019-0493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarpa A., Chang D.K., Nones K., Corbo V., Patch A.M., Bailey P., Lawlor R.T., Johns A.L., Miller D.K., Mafficini A., Australian Pancreatic Cancer Genome Initiative Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. doi: 10.1038/nature21063. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q., Holst J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015;5:1281–1294. [PMC free article] [PubMed] [Google Scholar]
- 33.Bhutia Y.D., Babu E., Ramachandran S., Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75:1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All bulk RNA-seq (GSE142232), single cell RNA-seq (GSE142233), and exome-seq (GSE142231) datasets have been made available under NCBI GEO series accession number GSE142234.