

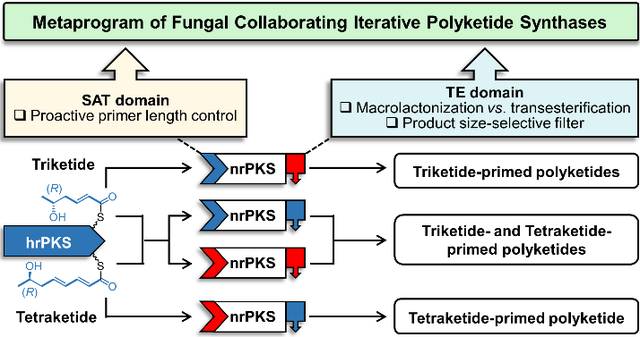
Abstract
Combinatorial biosynthesis with fungal polyketide synthases (PKSs) promises to produce unprecedented bioactive “unnatural” natural products (uNPs) for drug discovery. Genome mining of the dothideomycete Rhytidhysteron rufulum uncovered a collaborating highly reducing PKS (hrPKS) – nonreducing PKS (nrPKS) pair. These enzymes produce trace amounts of rare S-type benzenediol macrolactone congeners with a phenylacetate core in a heterologous host. However, subunit shuffling and domain swaps with voucher enzymes demonstrated that all PKS domains are highly productive. This contradiction led us to reveal novel programming layers exerted by the starter unit acyltransferase (SAT) and the thioesterase (TE) domains on the PKS system. First, macrocyclic vs. linear product formation is dictated by the intrinsic biosynthetic program of the TE domain. Next, the chain length of the hrPKS product is strongly influenced in trans by the off-loading preferences of the nrPKS SAT domain. Last, TE domains are size-selective filters that facilitate or obstruct product formation from certain priming units. Thus, the intrinsic programs of the SAT and TE domains are both part of the extrinsic program of the hrPKS subunit, and modulate the observable metaprogram of the whole PKS system. Reconstruction of SAT and TE phylogenies suggests that these domains travel different evolutionary trajectories, with the resulting divergence creating potential conflicts in the PKS metaprogram. Such conflicts often emerge in chimeric PKSs created by combinatorial biosynthesis, reducing biosynthetic efficiency or even incapacitating the system. Understanding the points of failure for such engineered biocatalysts is pivotal to advance the biosynthetic production of uNPs.
Graphical Abstract
INTRODUCTION
Polyketides constitute a large group of natural products with enormous structural diversity. In their natural contexts, they act as toxins, pigments, virulence factors, and effectors of communication and competition.1 Many polyketides, together with their semisynthetic derivatives and analogs have been developed into selective chemical probes and prominent therapeutics.2 In particular, fungal polyketide scaffolds have provided blockbuster drugs such as the anticholesteremic agent lovastatin, the immunosuppressant mycophenolic acid, and the antifungal griseofulvin.3 Fungal polyketides are biosynthesized from short chain carboxylic acid monomers (typically, acetyl or malonyl-coenzyme A) through recursive, decarboxylative thio-Claisen condensations by multidomain monomodular megasynthases similar to a single module of the bacterial Type I modular PKSs.4 However, unlike most modular PKSs, the fungal enzymes iteratively use a single set of ketoacyl synthase (KS), malonyl acyltransferase (AT) and acyl carrier protein (ACP) domains to catalyze all chain extension cycles (iterative PKSs).3 Based on their domain content, fungal iterative PKSs are classified into three groups. Highly reducing PKSs (hrPKSs) harboring ketoacyl reductase (KR), dehydratase (DH), and enoyl reductase (ER) domains execute a cryptic biosynthetic program to selectively reduce the nascent β-keto moieties after each chain extension step into a β-alcohol, an alkene, or an alkane.5–7 Partially reducing PKSs lack ER domains and afford relatively simple cyclic structures.8 Finally, nonreducing PKSs (nrPKSs) lack all reductive domains, but incorporate a starter unit : ACP transacylase (SAT) domain for priming unit selection,9,10 and a product template (PT) domain for regiospecific closure of the first aromatic ring.10,11 In addition, nrPKSs typically feature a C-terminal product release domain, such as a C-C bond-forming thioesterase/Claisen cyclase that yields terminal benzene moieties;12 a Type I O-C bond-forming thioesterase (TE) that affords macrolactones, pyrones, complex esters or simple carboxylates;13 or a reductive release domain that produces aromatic aldehydes or acylresorcinols.12,14
The biosynthesis of the majority of fungal polyketides requires a single iterative PKS each. However, some polyketides, including the congeners of the benzenediol lactones (BDLs), polylactones, sorbicillinoids, tricholignans and some azaphilones are assembled by a collaborating hrPKS - nrPKS pair acting in sequence.13–15 In these quasi-modular systems the hrPKS produces a linear polyketide chain with an idiosyncratic reduction pattern and configuration (Fig. 1A). This priming unit is transferred by the SAT domain onto the downstream nrPKS for further chain elongation without reduction, and first ring aldol cyclization.16 In the case of BDL congeners, chain release typically involves macrolactone formation.17 Further tailoring reactions may then afford the mature bioactive molecules.18–20 BDLs featuring a C2-C7 connectivity at the benzene ring (F-type cyclization) are the resorcylic acid lactones (RALs), while those having a C8-C3 connectivity (S-type cyclization) are known as dihydroxyphenylacetic acid lactones (DALs).16,21 Natural RALs contain 12- or 14-membered macrocycles (RAL12 or RAL14). These compounds are rich pharmacophores with wide-ranging biological activities, such as resorcylide, a RAL12 mineralocorticoid receptor antagonist; lasiodiplodin, a RAL12 prostaglandin biosynthesis inhibitor; radicicol, a RAL14 heat-shock protein 90 inhibitor; and hypothemycin, a RAL14 selective inhibitor of mitogen-associated protein kinases (SI Fig. S1). As opposed to RALs, Nature synthesizes a much more confined library of DALs, exemplified by sporostatin, a DAL10 kinase inhibitor, and 10,11-dehydrocurvularin, a DAL12 p97 inhibitor.22,23 One hallmark of fungal iterative PKSs is their enigmatic programming rules that control starter unit selection; the extent of chain extension; the degree and configuration of β-keto reduction; and the chemo- and regio-specificity of ring closure. For BDL congeners, an additional level of programming complexity arises from the interactions of the BDL synthase subunits (i.e. the hrPKS and the nrPKS) as a result of the inter-protein handover of the priming unit.24 Insights into the intrinsic biosynthetic programs of fungal iterative PKS domains (i.e. their catalytic competence towards a given intermediate), their extrinsic programming (i.e. domain interactions and the kinetic competition for a given intermediate) and the emergent metaprogram of the PKS (i.e. the overall observable program of the whole system) may accelerate the discovery of novel polyketide scaffolds, and guide the rational engineering of iterative PKSs for the production of “unnatural” natural products (uNPs).
Figure 1. Biosynthesis of DALs.

A. Scheme for the biosynthesis of the DAL12 10,11-dehydrocurvularin (4a) and its spontaneous hydration products 11-hydroxycurvularins (5a/b) by the collaborating hrPKS – nrPKS pair AtCurS1 – AtCurS2.20 The tetraketide 4Pa is transferred from AtCurS1 to AtCurS2 by the SAT domain, and used as a priming unit to initiate four more cycles of chain extension (“4+4”), followed by PT-catalyzed S-type (C8-C3) aldol cyclization and aromatization, and TE-catalyzed macrolactone formation. B. Schematic representation of the orphan rrdal biosynthetic gene cluster of Rhytidhysteron rufulum CBS 306.38.25 C. Comparison of the domain architectures of the RrDalS1 – RrDalS2 and the AtCurS1 – AtCurS2 hrPKS – nrPKS pairs (percent amino acid sequence identities of the domains are shown), and the product profile (reversed-phase LC-MS trace recorded at 300 nm with a photodiode array detector) of a representative culture of S. cerevisiae BJ5464-NpgA26,27 expressing RrDalS1 and RrDalS2. Peaks labeled with a star are unidentified host metabolites. See Fig. 3 for product structures.
We have been investigating the programming rules and exploring the promiscuity and plasticity of collaborating hrPKS – nrPKS pairs in fungal BDL synthases using in vivo combinatorial synthetic biology. We generated novel BDL scaffolds in the heterologous host Saccharomyces cerevisiae by BDL synthase subunit shuffling, combinatorial domain exchanges and rational engineering of the active site cavities of domains.16,17,24,28 We also explored combinatorial BDL scaffold decoration with non-cognate tailoring enzymes.29,30 Although product structures may be affected in artificial contexts such as some heterologous hosts or during in vitro reconstitution of biosynthetic enzymes31, BDL synthases and similar fungal iterative PKSs show remarkably high programmatic fidelity upon heterologous expression in yeast or Aspergillus chassis.3,14 Here we report the resuscitation of an unproductive orphan BDL synthase system, discovered by genome mining, by exploiting its combinatorial biosynthetic potential. In addition to generating unexpected non-macrocyclic DAL congeners, our combinatorial synthetic biology approaches also reveal novel layers of programming exerted by the SAT and TE domains on the BDL synthase system, and demonstrate that evolutionary divergence in the intrinsic programs of iterative PKS programming subroutines may create conflicts in the metaprogram of the system and thus incapacitate these semi-modular synthases.
RESULTS
Functional Characterization of an Orphan DAL Biosynthetic Gene Cluster from Rhytidhysteron rufulum
During the course of our genome mining efforts, we encountered a BDL synthase biosynthetic gene cluster in the published genome sequence25 of the plant pathogenic fungus Rhytidhysteron rufulum CBS 306.38 (Dothideomycetes, Hysteriales). This cluster harbors a pair of genes (rrdalS1 and rrdalS2) that encode an orphan hrPKS – nrPKS pair (RrDalS1 – RrDalS2, Figure 1B and C).25 These iterative PKS genes are bracketed by rrE1 and rrE2 encoding two major facilitator superfamily (MFS) exporters (Supporting Information, SI Table S1). However, no genes encoding potential polyketide decoration enzymes are present in this cluster, suggesting that the scaffold generated by RrDalS1 – RrDalS2 does not undergo post-PKS tailoring. Global sequence comparisons showed that RrDalS1 and RrDalS2 are orthologous to characterized BDL-producing hrPKSs and nrPKSs, respectively, with identities in the 55% to 66% range (SI Table S2). Detailed sequence comparisons indicated that the domains of RrDalS1 and RrDalS2 share the highest sequence identities with those of DAL-type BDL synthases, and predicted that PTRrDalS2 catalyzes an S-type (C8-C3) aldol condensation to yield a DAL scaffold (SI Table S2).16 Since DALs are relatively rare in Nature,22 we set out to functionally characterize this BDL synthase using the heterologous host Saccharomyces cerevisiae BJ5464-NpgA.26,27 Characterization of the native product of the system in R. rufulum was not attempted due to difficulties in obtaining the strain, the absence of information on the expression conditions of the cluster, and the lack of developed methods for the genetic manipulation of the fungus. Co-expressing the intron-free rrdalS1 and rrdalS2 genes synthesized with codon optimization for expression in yeast led to the production of several polyketide products in minute quantities insufficient for isolation (Entry #1 in Fig. 2 and SI Fig. S3). Thus, we opted for a combinatorial synthetic biology approach where RrDalS1 and separately RrDalS2 were paired with non-cognate BDL synthase partners from four model systems: AtCurS1 – AtCurS2 for 10,11-dehydrocurvularin biosynthesis,20 AzResS1 – AzResS2 for trans-resorcylide production,19 LtLasS1 – LtLasS2 for desmethyl-lasiodiplodin assembly,19 and CcRadS1 – CcRadS2 for monocillin II synthesis.18 These voucher systems offer pre-defined biosynthons that assemble to a variety of products with different scaffold types (DAL and RAL), macrolactone ring sizes (12- and 14-membered rings), macrolactone redox patterns (ketone, alkene, and alkane) and configurations (R or S configuration of the alcohol at the penultimate carbon).
Figure 2. Combinatorial biosynthesis with BDL synthase heterocombinations.

The deduced structures of the priming unit thioesters are presented below the cartoons of the appropriate hrPKSs. nrPKS cartoons show the SAT domains (chevron), the KS-AT-PT-ACP chassis (rectangle), and the Type I TE domains (block arrow), color-coordinated with the hrPKSs. Blue, RrDalS; red, AtCurS; green, AzResS; yellow, LtLasS; maroon, CcRadS. Products are listed in the rightmost column in the order of decreasing abundance; compound numbers in red indicate macrocycles. See Fig. 3 for product structures. First heatmap block, distribution (in percentages) of pentaketide (5P), tetraketide (4P), and triketide (3P)-primed polyketides among all detected products of the strain expressing the hrPKS – nrPKS combination. Second heatmap block, relative quantification (in percentage) of all macrocyclic (MCL) products among all detected products of the strain. Third heatmap block, relative overall polyketide productivity (in percentage) of the strain, compared to the overall polyketide productivity of the strain expressing RrDalS1 – RrDalS2(TEAtCurS2) (the most productive BDL synthase combination in these experiments). The heatmap color code is given at the bottom of the figure, crosshatch indicates no production of the given product type. Fermentations were conducted in triplicates and repeated a minimum of three times (n=9, means are shown), as described in SI Materials and Methods.
First, the hrPKS RrDalS1 was co-expressed with the DAL-making nrPKS AtCurS2 in S. cerevisiae. The resulting heterocombination produced the same polyketides as those obtained with the native R. rufulum PKS pair, but with an improved titer for compound 11 (Entry #3 in Fig. 2 and SI Fig. S3). Structure elucidation of 11 (Fig. 3) using HRESIMS, NMR, and other spectroscopic techniques showed that this compound is an acyl dihydroxyphenylacetic acid (ADA) ethyl ester featuring an (R,2E,4E)-7-hydroxyocta-2,4-dienoyl moiety, derived from the tetraketide priming unit 4Pc (Fig. 2). The minor product 1b and its hydrated analogues 2c/d turned out to be ADA ethyl esters derived from the triketide 3Pa, a priming unit that results from the incorporation of one fewer ketide units.15,28,32 Thus, the increased productivity of 11 at the expense of 1b and 2c/d with AtCurS2 effectively reversed the proportion of triketide-derived vs. tetraketide-derived products (3:1 with the native pair, 1:3 with the heterocombination, Entries #1 vs. #3 in Fig. 2 and SI Fig. S3). To reveal the structural basis for this shift, we replaced the KS-AT-PT-ACP chassis of RrDalS2 with that of AtCurS2. The resulting chimera produced the same array of polyketides as the native pair when challenged with RrDalS1, but with an even lower productivity (Entry #2 in Fig. 2 and SI Fig. S3). This indicated that the RrDalS2 polyketide homologation machinery (KS, AT and ACP) and first ring cyclase (PT) are not the primary determinants for the observed low productivity of the R. rufulum BDL synthase. Moreover, the ratio of triketide vs. tetraketide-derived products was the same as that with the native RrDalS pair (3:1), indicating that the SAT and/or the TE domains are the decisive factor(s) that determine the length distribution of the RrDalS1-produced priming units.
Figure 3. Chemical structures of detected products.

Compounds in dashed boxes were isolated and identified on the basis of their spectroscopic data. Other products were identified by their LC-MS spectra and comparisons with isolated standards.
To further probe the program of RrDalS1, we created heterocombinations with the RAL-producing nrPKSs AzResS2, LtLasS2, and CcRadS2 (Entries #4, #5 and #6 in Fig. 2 and SI Fig. S3). These RAL nrPKSs accepted tetraketide 4Pc as the priming unit, with no 3Pa-derived product evident, and carried out their characteristic biosynthetic program for chain elongation and formation of the benzene ring in the C2-C7 register. Off-program products were also obtained, however these all originated from anomalies in the chain elongation programs of the RAL-type nrPKSs and not that of RrDalS1. Thus, ARA ethyl ester 22 formed when AzResS2 conducted only three extension cycles instead of the on-program four, while isocoumarin 21 derived from the incorporation of four ketide units instead of the on-program three by LtLasS2 (Entries #4 and #5 in Fig. 2 and SI Fig. S3). The products were released by transesterification to ethanol produced by the yeast host (1b, 2c/d, 11, 20 and 22), or by formation of an α-pyrone (21) as a result of an intramolecular nucleophilic attack of the C-9 enol on the C-1 carbonyl. The absence of DAL or RAL products in these experiments may reflect the difficulty for the RrDalS1-derived short (3Pa) or very rigid (4Pc) acyl priming units to adopt a conformation conducive for macrolactone formation. Taken together, these heterocombination experiments show that RrDalS1 is a competent hrPKS that synthesizes tetraketide 4Pc with an R-configured secondary alcohol and an α,β,γ,δ-unsaturated ketone functionality as its typical product, but may also afford the shorter homologue 3Pa.
Next, we turned to clarify the biosynthetic program of RrDalS2 using heterocombinations with voucher hrPKSs. All four resulting noncognate pairs produced a panel of novel polyketides in low to fair amounts (Entries #11, #18, #25 and #31 in Fig. 2 and SI Fig. S4). RrDalS2 accepted the enantiomeric tetraketide priming units 4Pa (from AtCurS1) and 4Pb (from AzResS1), readily executed four rounds of chain elongation, faithfully carried out the predicted S-type (C8-C3) aldol condensation, and exclusively released the final products as ADA alkyl esters with no macrolactones detectable (Entries #11 and #18 in Fig. 2 and SI Fig. S4). The expected α,β-enone functionality from 4Pa or 4Pb is transformed into β-hydroxyl or α-tetrahydropyranyl ketone moieties via spontaneous inter- or intramolecular oxy-Michael addition in the matured products. These compounds (Fig. 3, 7a/b, 8a/b, 9a and 10a from AtCurS1 – RrDalS2 vs. 7c/d, 8c/d, 9b, and 10b from AzResS1 – RrDalS2) are enantiomers in a pairwise manner due to the enantiomeric nature of 4Pa and 4Pb. Replacement of the KS-AT-PT-ACP chassis of RrDalS2 with that of AtCurS2 led to no change in the product profiles (Entries #12 and #19 in Fig. 2 and SI Fig. S4), although significantly improved overall productivity.
The LtLasS1 – RrDalS2 noncognate pair showed low productivity (Entry #25 in Fig. 2 and SI Fig. S4), but afforded the expected DAL lasilarin (14) and its ADA ester analogue (16), both derived from the on-program pentaketide priming unit of LtLasS1 (5Pa). Nevertheless, the main product was epi-curvularin (6, Fig. 3), a DAL derived from the off-program, shorter tetraketide priming unit 4Pd. Replacing the KS-AT-PT-ACP chassis of RrDalS2 with that of AtCurS2 again increased productivity (Entry #26 in Fig. 2 and SI Fig. S4), revealing additional minor pentaketide or tetraketide-derived ADA ethyl esters such as 19 (from priming unit 5Pc), 12 and 13 (from 4Pd) and 7c/d and 8d (from 4Pb). While these minor products increased the share of ADA esters at the expense of macrocyclic products, priming unit chain length preferences remained largely unchanged with the chimera. Curiously, this heterocombination also produced (R)-9-hydroxydecanoic acid (23, Fig. 3), the free on-program pentaketide priming unit 5Pa of LtLasS1. Release of hrPKS products by hydrolysis was observed earlier28 and may reflect a proofreading process (presumably by the TE) that removes unproductive intermediates from the nrPKS.
The CcRadS1 – RrDalS2 noncognate pair was unproductive, but could be revived by enforcing subunit coupling via SATRrDalS2 replacement with SATCcRadS2 (Entry #31 in Fig. 2 and SI Fig. S4). While overall productivity remained very low, this domain replacement nevertheless allowed product formation from 5Pb, the on-program pentaketide primer from CcRadS1. However, ADA ethyl ester 17 and its hydrated analogues (18a/b) were the dominant products, with the expected DAL14 radilarin (15) barely detectable. Like compounds 7a/b and 7c/d, the isomeric mixture 18a/b was generated by spontaneous hydration of the α,β-unsaturated bond of the enone. The diastereomers 18a and 18b were separated on a chiral column and their absolute configurations were assigned by ECD/TDDFT calculations as 11S,17R and 11R,17R, respectively (Fig. 3, SI Fig. S8.4). Replacing the KS-AT-PT-ACP chassis of RrDalS2 with that of AtCurS2 led to the production of the same pentaketide priming unit-derived products in trace amounts (Entry #32 in Fig. 2 and SI Fig. S4).
Taken together, these heterocombination experiments show that RrDalS2 is a competent nrPKS that is programmed to append an S-type (C8-C3) tetraketide biosynthon to a tetraketide priming unit, and release the product predominantly as an ADA ester. Pentaketide priming units are disfavored, but may allow the synthase to afford minute amounts of macrocyclic (DAL14) products. DAL12 products are also possible when using a completely saturated, highly flexible tetraketide priming unit such as 4Pd. The KS-AT-PT-ACP chassis seems largely irrelevant in determining product distribution between macrocycles and linear products, or amongst compounds derived from priming units of different lengths, emphasizing that the chassis does not influence priming unit selection or chain termination chemistry.
SAT Domains as Proactive Logic Gates
Our subunit shuffling approach revealed that the individual RrDalS subunits are catalytically competent enzymes; uncovered their likely biosynthetic programs; and yielded a panel of unprecedented polyketides. Importantly, these experiments showed that AtCurS2 and RrDalS2 are orthologous DAL synthases that extend various priming units by incorporating four more malonate equivalents; however, these enzymes also show idiosyncratic differences in their chain initiation and termination chemistries. Considering the well-established role of SAT domains in priming unit loading,9,10 we created hybrid RrDalS2 / AtCurS2 enzymes by swapping their SAT domains, and interrogated these chimeric enzymes by offering them the priming units produced by RrDalS1 or the voucher hrPKSs.
Grafting the SATRrDalS2 domain onto AtCurS2 led to surprisingly large shifts in the product distributions of all noncognate pairs, clearly favoring shorter, and if possible, triketide priming units (Entries #7, #14, #21, #28 and #34 in Fig. 2 and SI Fig. S5). Thus, with RrDalS1 as the partner, the formerly not isolable triketide-primed compounds 1b and 2c/d became the exclusive products, delivered at fair titers (Entry #7 vs. Entry #3 in Fig. 2 and SI Fig. S5). Compounds 2c and 2d are C-11 epimeric ADA ethyl esters that feature a 1,3-diol moiety in the acyl sidechain (Fig. 3). The configuration of C-11 was determined as 11S for 2c and 11R for 2d by ECD/TDDFT calculations (SI Fig. S8.1), considering that the secondary alcohol at C-13 should adopt an R configuration as was seen in compound 11. Purified 1b was rapidly converted to a mixture of 1b and 2c/d in aqueous solution, suggesting that it has an α,β-enone functionality that is prone to spontaneous oxy-Michael addition.
When pairing with AtCurS1 or AzResS1, the chimeric AtCurS2(SATRrDalS2) accepted the enantiomeric tetraketide starter units 4Pa or 4Pb, and predominantly released the products as DALs (4a/b and 5a–d) (Entries #14 and #21 in Fig. 2 and SI Fig. S5). However, triketide-primed polyketides 1a, 2a/b and 3 (for AtCurS1), and 1b and 2c/d (for AzResS1) were also detected. Compounds 2b and 2a were elucidated to have 11R,13S and 11S,13S configurations, respectively. Thus, these are the enantiomers of 2c and 2d in a pairwise manner (Fig. 3, SI Fig S8.1 and Table S5.2). Similar to 2c and 2d, the OH-11 of 2b and 2a also result from spontaneous hydration of the E-double bond in 1a. Compound 3 is an ADA ethyl ester with a 5-hydroxyhexanoyl side chain (Fig. 3). We reckon that the corresponding highly saturated triketide priming unit 3Pc is another off-program product of AtCurS1 rather than originating from a fortuitous modification by the host, considering that we have never observed analogous reductions by endogenous yeast enzymes.
The product profile of LtLasS1 – AtCurS2(SATRrDalS2) also changed appreciably compared to LtLasS1 – AtCurS2 (Entries #28 vs. #27 in Fig. 2 and SI Fig. S5). Instead of the on-program pentaketide-primed lasilarin (14), the tetraketide 4Pd-derived DAL12 epi-curvularin (6) became the major product. Triketide 3Pa-derived 1b and 2c/d appeared as minor products, in amounts far exceeding that of the on-program pentaketide-derived compound. Surprisingly, CcRadS1 – AtCurS2(SATRrDalS2) successfully generated the on-program pentaketide-derived compounds 15 and 18a/b (Entry #34 in Fig. 2 and SI Fig. S5). However, tetraketide 4Pc and triketide 3Pa were also apparently downloaded by the nrPKS, as evidenced by the appearance of 11 and 2c/d in substantial amounts. Taken together, these SAT swap experiments show that SATRrDalS2 is selecting shorter priming units when those are readily offered by the hrPKS partner, and further, it forces the handover of derailed, off-program priming units in a proactive manner.
The complementary experiment (replacing SATRrDalS2 with SATAtCurS2) was unproductive despite our success with the SATCcRadS2 donor using an analogous switchover site. Varying the switchover site could not rescue productivity either (four additional, plausible sites were investigated, located in front of, within, or after the SAT/KS linker region, as described in the SI Supplementary Methods), regardless of the hrPKS partner used for the resulting chimeras. Considering that our previous experiments clearly show that both constituent parts of these chimeras (i.e. the SATAtCurS2 domain and the KS-AT-PT-ACP-TE segment of RrDalS2) are catalytically competent with the hrPKS-derived priming units, we hypothesize that unfavorable inter-domain structural interactions prevented product turnover with RrDalS2(SATAtCurS2).
TE Domains Are Logic Gates for Product Conformation
In addition to their differences in priming unit selection, AtCurS2 and RrDalS2 also diverge in their product release modes. Thus, AtCurS2 overwhelmingly produces DALs (Entries #13, #27 and #33 in Fig. 2 and SI Fig. S5), unless the priming unit is too rigid for macrolactone formation (Entry #3 in Fig. 2 and SI Fig. S5). In contrast, RrDalS2 typically delivers the final products as ADA alkyl esters (Entries #11, #18 and #31 in Fig. 2 and SI Fig. S4), unless encountering an especially flexible priming unit whose ω−1 alcohol moiety is able to redirect product release towards intramolecular cyclization (Entry #25 in Fig. 2 and SI Fig. S4). Considering the established gatekeeping roles of TE domains in product release,17,33 we reasoned that these discrepancies may reflect differences in the intrinsic programs of the TE domains in the two nrPKSs. Thus, we created RrDalS2 / AtCurS2 chimeras by swapping their TE domains.
As expected, chain extension and S-type first ring formation remained unaffected in AtCurS2(TERrDalS2). When challenged with voucher hrPKSs, priming unit selection also remained conservative, matching the expected, on-program tetraketides or pentaketides routinely offered by the partner enzymes (Entries #15, #22, #29, and #35 in Fig. 2 and SI Fig. S6). Importantly, heterocombination of the chimera with RrDalS1 failed to afford any triketide-derived products (Entry #8 in Fig. 2 and SI Fig. S6), in spite of the reasonable productivity of this noncognate BDL synthase, and the compatibility of SATAtCurS2 with such short priming units (Entry #3 in Fig. 2 and SI Fig. S5). The most important consequence of the TE replacement was the dramatic alteration of the mode of product release with all hrPKS partners, with predominant production of ADA alkyl esters at the expense of DAL macrocycles (Entries #8, #15, #22, #29, and #35 in Fig. 2 and SI Fig. S6).
Replacement of the TE domain of RrDalS2 with TEAtCurS2 in the complementary experiment markedly shifted the product distribution towards macrocyclic DALs at the expense of ADA analogues (Entries #16, #23, #30, #36 and #37 in Fig. 2 and SI Fig. S6). As expected, macrocycle formation remained out of reach with triketide priming units (Entry #9 in Fig. 2 and SI Fig. S6). However, the combination with the cognate hrPKS partner RrDalS1 was remarkably productive. This pair not only produced 70 times the amount of polyketides obtained from the admittedly dismal RrDalS1 – RrDalS2 native pair, but was also by far the most productive BDL synthase, outperforming even the native AtCurS1 – AtCurS2 pair by well over 30%. In contrast, polyketide productivity was strongly diminished when the chimera was coupled to AtCurS1 (Entry #16 in Fig. 2 and SI Fig. S6). Surprisingly, a low level of heterocoupling with CcRadS1 was also observed even without SAT domain replacement (Entry #36 in Fig. 2 and SI Fig. S6). Nevertheless, the CcRadS1 – RrDalS2(TEAtCurS2) pair remained a marginal producer, affording only trace amounts of the expected DAL14 product, radilarin (15). Instead, the chain length fidelity of CcRadS1 broke down, and the product profile became dominated by polyketides derived from off-program, shortened priming units, such as the ADA ethyl ester (11) from tetraketide primer 4Pc, and ADA ethyl esters (2c/d) from triketide 3Pa. In fact, hrPKS fidelity is compromised in most pairings with RrDalS2(TEAtCurS2). Thus, RrDalS1 appears to be an efficient triketide (and not a tetraketide) synthase that affords 3Pa exclusively; AtCurS1 and AzResS1 offer 4:1 mixtures of tetraketides and triketides instead of tetraketides only; and LtLasS1 generates mostly a tetraketide and even a triketide instead of its native pentaketide (Entries #9, #16, #23 and #30 in Fig. 2 and SI Fig. S6).
The striking productivity improvement upon introducing TEAtCurS2 into RrDalS2 led us to consider that TERrDalS2 may be mismatched with the nrPKS chassis.34 Thus, this TE may have “expected” an F-type (C2-C7) resorcylate intermediate instead of the S-type (C8-C3) phenylacetate offered by the rest of RrDalS2. Thus, we grafted TERrDalS2 onto the AzResS2 chassis, and paired this chimera with RrDalS1, AtCurS1 and AzResS1 (Entries #10, #17 and #24 in Fig. 2 and SI Fig. S6). The resulting noncognate BDL synthases were all barely functional, producing minute amounts of the isocoumarins 21, 24a, 24b or 25 (Fig. 3) derived from the on-program priming units offered by the hrPKSs, and feature an F-type resorcylic acid motif afforded by PTAzResS2. These isocoumarins may result from the spontaneous release of the enthalpically and entropically favored pyrone from the ACP-bound, stalled resorcylate intermediates that are not accepted by the grafted TERrDalS2.17,34 The complementary experiment (introducing TEAzResS2 into RrDalS2) provided only unproductive BDL synthase heterocombinations. This may indicate that TEAzResS2 is unable to release a phenylacetate product, while spontaneous pyrone formation is not possible for S-type intermediates.17,34
Taken together, these TE swaps indicate that the intrinsic program of TERrDalS2 favors ADA ester formation regardless of product chain length, but it is not amenable to release F-type (resorcylate) intermediates. In contrast, TEAtCurS2 is programmed to generate macrolactones, and is permissive towards products from shortened priming units transferred by the short chain-preferring SATRrDalS2.
DISCUSSION
Understanding the points of failure for engineered biocatalysts such as iterative PKSs originating from domain replacements or subunit shuffling is pivotal to advance the biosynthetic production of unnatural natural products (uNPs). To improve our design of custom biocatalytic machines for combinatorial synthetic uNP production, structural enzymology studies would need to predict folding deficiencies and protein-level domain interaction failures in engineered systems.35–37 At the same time, it is necessary to understand the intrinsic programming (the catalytic competencies of domains for an intermediate) and the extrinsic programming (domain-level interactions and the kinetic competition of various catalytically competent domains for the same intermediate) of PKSs, and the “metaprogram” emerging from all these subroutines. A novel avenue for developing such an understanding became evident when we encountered a seemingly derelict, largely unproductive natural BDL synthase system from Rhytidhysteron rufulum. To our surprise, subunit shuffling and domain exchanges in a combinatorial context revealed that all constituent parts of the RrDalS system were functional and highly efficient in certain contexts. Nevertheless, the natural combination of these parts was almost completely unproductive in the heterologous host. In effect, Nature provided us with a natural example of an iterative PKS system with a faulty overall (meta)program.
To decipher the programming of the RrDalS system, we first paired the two subunits with voucher BDL synthase partners. Next, we built nrPKS chimeras by exchanging domains between RrDalS2 and AtCurS2, a highly productive DAL-producing nrPKS orthologue of RrDalS2 with subtle but consistent programming differences in priming unit selection and product release mechanisms. Based on previous studies,14 we presumed that these differences would largely be determined by the SAT and the TE domains, with the nrPKS KS-AT-ACP polyketone homologation chassis and the PT domain exercising only marginal influence over the programmed outcomes. The resulting RrDalS2/AtCurS2 hybrid enzymes were challenged with voucher hrPKSs providing priming units with different lengths, reduction levels, and ω−1 alcohol configurations. Priming units of different length and functionalization have been seen to provoke (compensatory) changes in product length by the acceptor PKSs in bacterial iterative modules,38 Type II PKSs39 and fungal iterative PKSs.32,40 However, in our extended set of experiments the nrPKS chain length program remained remarkably constant, regardless of the priming unit utilized (from triketides to pentaketides with different functional groups). Thus, the observed product structures could be easily ascribed to the engineered programming alterations upon SAT and TE exchanges. These experiments revealed several novel programming constraints that determine the overall metaprogram of BDL synthases, and by extension, those of similar PKS-containing natural or engineered biosynthetic systems.
Product Release Modes Are Determined by the Intrinsic Program of the TE Domain
In fungal nrPKSs, O-C bond-forming TE domains dictate product release modes that generally correlate with the phylogenetic grouping of the nrPKS.14 Thus, these TEs may release products by hydrolysis (such as with orsellinic acid synthases) or 6-membered pyrone ring formation (such as with the cercosporin synthase).41 An intriguing example is a group of TEs that catalyze cross-coupling (transesterification) to a different, complex biosynthon.42 However, TE-catalyzed product release for BDLs is generally thought to favor macrocyclization, with occasional hydrolysis or pyrone formation taken as an indication of the bypass of a TE that is unable to process a particular substrate.
In our experiments, TEAtCurS2 efficiently facilitated the intramolecular marocyclization of octa- or nonaketide scaffolds derived from priming units 4Pa, 4Pb, 4Pd, 5Pa, and 5Pb, showing permissiveness for the length, redox pattern and ω−1 alcohol configuration of the acyl chain. In contrast, the preferred mode of release for TERrDalS2 is intermolecular transesterification to a short-chain alcohol from primary metabolism to afford ADA esters, even when TEAtCurS2 releases the very same carbon skeleton overwhelmingly as a macrocycle (cf. Entries #15 and #13, #11 and #16 and so on, Fig. 2). TERrDalS2 is still able to catalyze macrolactonization with a few scaffolds derived from pentaketide primers, or those originating from the very flexible, saturated tetraketide 4Pd. However, macrocycles remain minor products even in these cases (Entries #29, #31, #32, and #35), and/or the overall product yield drops considerably (cf. Entries #25 and #26). Thus, preference for macrocycle vs. linear product formation in BDL synthases is a part of the intrinsic program of competent TE domains, and may be determined by the shape, charge distribution and volume of the TE cyclization chamber. Products derived from triketide primers, and those from the rigid tetraketide 4Pc could not be released as macrolactones by either TEAtCurS2 or TERrDalS2. Inside the active site cavity, the acyl tails of these polyketides may be unable to fold back to use their distal alcohol for a nucleophilic attack on the Ser oxoester. Modulation of the TE catalytic chamber may then allow different BDL synthase systems to occupy varying positions on the macrocycle – linear product spectrum. Meanwhile, the active site of TERrDalS2 must still be accessible to simple alcohols (but not to water) for product release as ethyl-, isobutyl-, or phenethyl esters.
SAT Domains Are Proactive Selectors Contributing to the hrPKS Metaprogram
In vitro reconstitution of the dissected aflatoxin biosynthetic nrPKS has indicated that the SAT domain and the polyketide homologation machinery were remarkably tolerant towards a range of artificial priming units.40 Similar results from the chaetoviridin or the asperfuranone collaborating iterative PKSs32,43 and the hypothemycin BDL synthase pair,13 together with the in vivo permissiveness of SAT domains during subunit shuffling24 were interpreted to mean that the intrinsic program of the SAT domains in collaborating, quasi-modular systems (such as FAS – nrPKS or hrPKS – nrPKS pairs) involves little discrimination towards the priming unit presented by the hrPKS.40 Thus, SAT domains have been described to resemble “passive docking domains that enable vectorial substrate transfer in modular PKSs”.36
Our current results show that the SAT of AtCurS2 (and that of AzResS2) was indeed behaving as a promiscuous, permissive catalyst. Thus, SATAtCurS2 faithfully transferred the on-program tetraketide or pentaketide priming units offered by the hrPKS noncognate partners, with the exception of 5Pb, the priming unit from CcRadS1. On the contrary, SATRrDalS2 displayed a clear intrinsic selectivity, consistently choosing shorter, and if possible triketide priming units at the expense of longer, especially pentaketide ones (Entries #7, #9, #14, #16, #21, #23, #28, #30, #34 and #36, Fig. 2). Although SATRrDalS2 could also accept tetraketides or even pentaketides to a lesser extent (including small amounts of 5Pb from CcRadS1), it behaved in a proactive way and forced the hrPKS partner to offer derailment products. Thus, SATRrDalS2 downloaded large amounts of premature priming units from all hrPKS partners that were one or even two ketide units shorter, and less reduced, than the on-program products of the hrPKSs. Such versatility of the voucher hrPKSs to assemble polyketides of irregular lengths and redox patterns has not been observed before, and indicates that their biosynthetic output is strongly influenced by the off-loading preferences of the SAT domain. Thus, SAT domains are not “passive docking domains”,36 but follow an intrinsic program to discriminate priming units according to their lengths. What’s more, this intrinsic program then becomes a part of the extrinsic metaprogram of the hrPKSs in collaborating systems.
SATRrDalS2 typically intercepted the programmed elongation and reduction sequence of the hrPKS before the ER-catalyzed reduction step one or two extension cycles before product maturation (priming units 3Pa, 3Pb, 4Pb, 4Pc). Interception before the ER recreated the α,β-enone functionality of the full-length, RrDalS1-generated tetraketide priming unit 4Pc, indicating a strong preference for this motif by SATRrDalS2. Moreover, the same priming unit (3Pa) was downloaded by SATRrDalS2 from three different hrPKSs with distinct product types (tetraketide synthase AzResS1 and two pentaketide synthases, LtLasS1 and CcRadS1). Forced handover of intermediates before chain extension but after full reduction (4Pd in Entries #25, #26, #28 and #30; 3Pc in Entries #14 and #16) or before β-keto processing (5Pc, Entry #26) were also observed, although less frequently. Parallel work with dissected modules from Type I modular PKSs showed that heterologous TE domains may similarly intercept polyketide intermediates before the KR-catalyzed β-keto reduction.44
The role of downstream, off-loading domains in the chain length control of hrPKSs has been extensively investigated in hrPKS-NRPS (nonribosomal peptide synthetase) hybrid enzymes for acyltetramic acid or lovastatin biosynthesis. While the NRPS condensation (C) domain shows considerable selectivity during acyltetramic acid biosynthesis,45 it only acts when the hrPKS module exhausted all its synthetic possibilities.46 A closer parallel to the proactive off-loading activity of SATRrDalS2 is found in soppiline B biosynthesis where a downstream chalcone synthase actively intercepts and offloads a shorter, premature polyketide from the partner hrPKS.47
Taken together, our results support and extend a model in which the hrPKS metaprogram (the overall program of the whole enzyme) emerges from the kinetic competition of domains that are catalytically competent for the ACP-bound intermediates.36,46 In BDL synthases and similar collaborating systems, kinetic competition between the hrPKS cis-acting domains KS, KR, DH and ER is supplemented in trans by that of the SAT domain from the nrPKS subunit. This multipartite competition is responsible for setting the biosynthetic trajectories of the nascent β-keto intermediates on the hrPKS, channeling them towards β-keto processing, further chain extension, or transacylation to the nrPKS partner.
SAT and TE Domain Interactions Influence the BDL Synthase Metaprogram
The TE domain has emerged as one of the most important decision gates in polyketide biosynthesis.17,27,44,48,49 In a combinatorial context, an appropriate BDL synthase TE domain may confer high productivity, but an incompatible TE may render the same hrPKS – nrPKS unproductive.17 Apart from proofreading during chain extension, TE domains monitor the formation50 and the register17,34 of the first aromatic ring as stringent logic gates. However, macrocycle-forming BDL synthase TEs are permissive towards the configuration of the nucleophilic alcohol, and the oxidation state of the acyl chain.17,34 TE domains are also permissive for the length of the product (typically within one ketide unit), with the KS as the primary determinant for nrPKSs product size.14 Correspondingly, the chain length of the nrPKS biosynthon did not vary with AtCurS2 or RrDalS2, irrespective of the SAT or TE domains or the hrPKS subunit partners used. In contrast, the TE domains substantially modulated the distribution of products derived from different priming units, and thus moderated or intensified the effects of SAT domain exchanges. Thus, TEAtCurS2 is a permissive gatekeeper that successfully releases heptaketide, octaketide and nonaketide products derived from triketide, tetraketide or pentaketide priming units, respectively. In particular, TEAtCurS2 facilitates the efficient production of triketide-primed polyketides when this domain and SATRrDalS2 are appended to an AtCurS2 or RrDalS2 chassis (Entries #7, #9, #14, #16, #21, #23, #28, #30, #34 and #36, Fig. 2). In contrast, TERrDalS2 is much less efficient in releasing triketide-primed products when present in the same enzyme assemblies as SATRrDalS2, triggering large shifts towards longer polyketide homologues, and depressing the overall product yield (e.g. Entries #2 vs. #7, #1 vs. #9, #12 vs. #14, Fig. 2). On the other hand, appropriate combinations of SAT and TE domains significantly enhance overall product yields. Thus, when SATRrDalS2 is matched with TEAtCurS2 in the RrDalS2 chassis, the production of 1b and 2c/d with RrDalS1 as the partner increases seventyfold as compared to the native RrDalS1 – RrDalS2 pair (Entries #9 vs. #1, Fig. 2). These observations indicate that TE domains work as size-selective filters in BDL synthases. Thus, the intrinsic TE program modulates the observable metaprogram of the hrPKS subunits by facilitating or obstructing product formation from certain priming units. For example, RrDalS1 would be perceived as a pure triketide synthase when combined with an nrPKS chassis featuring SATRrDalS2 and TEAtCurS2. However, the same hrPKS would be categorized as a mixed triketide/tetraketide synthase when paired with an analogous nrPKS equipped with TERrDalS2 (cf. Entries #7 and #9 with #1 and #2, Fig. 2). This apparent control of the hrPKS chain length manifests at a remote level, since the TE (as opposed to the SAT) is not in a kinetic competition for hrPKS-bound intermediates, but monitors advanced intermediates on the nrPKS ACP after chain extension and first ring cyclization. Since the metaprogram of the hrPKS, and in turn that of the whole BDL synthase is an emergent property that derives from the intrinsic selectivities of the constituent domains, their extrinsic interactions and kinetic competition for the same ACP-bound intermediates, the result of an engineered change affecting only a few factors is rarely clear-cut. Instead, multiple products are biosynthesized, and complex patterns emerge depending on the overall biosynthetic context (such as in chimeric PKSs, or when an engineered nrPKS is paired with different hrPKS partners).
Evolutionary Perspectives on BDL Synthase Metaprogram Emergence
To obtain a perspective on the evolution of BDL synthase programming, we inferred the evolutionary history of O-C bond-forming Type I nrPKS TE domains using the C-C bond-forming thioesterase/Claisen cyclase domains as the outgroup, and cross-referenced the resulting phylogenetic tree with the products of the synthases, if known. BDL-type TEs form a large superfamily (Fig. 4), accompanied by basal clades with TE domains for the hydrolytic release of orsellinic acid (OA); OA cross-coupling to other scaffolds; TEs affording polylactones; and two other clades with no known metabolic products. The BDL TE superfamily neatly splits into two sister clades according to the register of the first aromatic ring. This dichotomy is in agreement with a previously observed functional correlation between PTs and TEs from F-type (C2-C7) vs. S-type (C8-C3) systems, whereby neither TE group can macrocyclize intermediates with the opposite type of aromatic ring.17,34 Accordingly, a PT phylogenetic tree constructed from the same nrPKSs shows a very similar clading pattern, with the F-type vs. S-type register as its main organizing principle (SI Fig. S7). Further, F-type PTs and TEs both branch to RAL macrocycles where the nrPKS extends the priming unit through three additional extension cycles (X+3 split); RAL macrocycles with an X+4 split; pyrones with a C2-C7 first aromatic ring; and a group with unknown products where F-type cyclization is clearly indicated by diagnostic PT residues.16 For S-type PTs and TEs, clades for DAL macrocycles and unknown C8-C3 products are observable. The PT and TE domains of RrDalS2 are ensconced in a clade with no other functionally characterized synthases. This clade is sister to those of macrocyclic DALs, and may include other cross-coupling, ADA ester-synthesizing enzymes. Thus, PT and TE domains in BDL synthase nrPKSs follow similar evolutionary trajectories, reflecting the close collaboration of these domains in product formation.
Figure 4. Phylogenetic trees for nrPKS TE and SAT domains.

nrPKSs with O-C bond-forming Type I TEs were collected from NCBI GenBank in a comprehensive search (>40% amino acid identity, >80% coverage to “bait” sequences), aligned using MEGA 7, and their evolutionary histories were inferred with the neighbor joining method. nrPKSs with thioesterase/Claisen cyclase domains were used as the outgroup (dark teal triangle). The percentage (if >50%) of replicate trees where the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. Branches basal to BDL synthases were collapsed and shown as colored wedges (brown, collaborating hrPKS – nrPKS systems with a discrete TE protein; purple, TEs from orsellinic acid hybrid metabolite synthases; yellow, polylactone TEs; green, TEs from orsellinic acid synthases; black, TEs from synthases with unknown products). Colored dots represent TEs from voucher BDL synthase systems (maroon, zearalenone,51 lasiodiplodin19 and hypothemycin52 synthases; red, resorcylide19 and monocillin II18,52 synthases; pink, cladosporin53 synthase; blue, RrDalS2, navy, dehydrocurvularin20,54 synthases). Blue arrows indicate the relocation of the ADA ester and the unknown S-type clade into the RAL X+4 clade.
The inferred evolutionary history of nrPKS SAT domains shows surprising differences compared to the TE and PT reconstructions (Fig. 4). Although the BDL superfamily is still distinct and accompanied by similar basal clades, there is no clean dichotomy for F- vs. S-type product skeletons. The BDL SAT domain superfamily is not organized according to the chain length or the redox/stereochemical characteristics of the priming units either. Instead, the RrDalS2 ADA ester clade, and another S-type (C8-C3) clade with unknown products “invades” the RAL X+4 group (Fig. 4), a pattern also apparent in a phylogenetic tree of entire hrPKS subunits (SI Fig. S7). This indicates that in BDL synthase systems, SAT domains and hrPKSs follow similar evolutionary trends, consistent with the role of the SAT domains in creating the hrPKS metaprogram. However, considering the divergence of the TE (PT) and the SAT (hrPKS) trees, these programming subroutines may travel different evolutionary trajectories. This may lead to structural innovations in BDL biosynthesis. However, this disjointed evolution also poses the danger of developing a programmatic conflict among BDL synthase subroutines and even a breakdown of the overall metaprogram, as is often seen with chimeric synthases inadvertently constructed from ill-matched parts.46 We believe the native RrDalS system is a victim of such a fate: the RrDalS2 SAT (and its hrPKS partner) developing a strong preference for triketide priming units, while the TE domain drifting towards a preference for tetraketide-initiated products. The emerging metaprogram of the BDL synthase thus becomes self-contradictory, leading to an unproductive tug-of-war between the SAT and the TE, nearly incapacitating the RrDalS system in the heterologous host.
CONCLUSIONS
A combination of rational domain swaps with subunit shuffling in a heterologous host allowed the revitalization of a seemingly derelict BDL synthase system identified by genome mining in R. rufulum, and generated a library of structurally diversified DALs and ADA esters in isolable amounts. Although the structure of the native product of the BDL synthase system in R. rufulum remains uncharacterized, our synthetic biology experiments in S. cerevisiae further expanded the chemical diversity of the relatively rare S-type fungal BDL congeners by 23 compounds which are new to Nature. Even more importantly, our results revealed constraints and programming complexities that genetic engineers would need to take into account when designing iterative PKS genetic machines for combinatorial synthetic microbiology.
Combinatorial biosynthetic production of uNPs in native or heterologous hosts relies on the promiscuity and plasticity of natural product biosynthetic machineries such as iterative PKSs. Structure-guided active site cavity engineering, and swaps of sub-domains, domains, or even subunits presupposes that protein-protein interactions among engineered and native units will remain operational, and the altered product intermediates will be acceptable to all functional units. Previous experiments with fungal dual PKS systems suggested that the constituent hrPKS and nrPKS enzymes are autonomous, and assemble their own biosynthons in a largely independent manner. Successful subunit collaboration depends on an appropriate SAT domain functionally coupling these synthases into a BDL synthase system. Our current findings demonstrate a complementary, but more dynamic scenario where the overall BDL synthase metaprogram emerges from the intrinsic substrate specificities of the individual domains, and from the extrinsic interactions and kinetic competition among catalytically competent domains for the growing polyketide intermediates. Thus, the growing hrPKS biosynthon can be intercepted in trans by a downstream SAT domain with a strict intrinsic substrate preference, resulting in the facile transfer of a shortened priming unit, often with an altered reduction pattern, to the nrPKS. Successful product formation is gated by the terminal TE domain that not only monitors the formation of the first aromatic ring, but also works as a size selectivity filter. Thus, the size of the priming unit (the hrPKS chain length metaprogram) and that of the overall product (the BDL synthase chain length metaprogram) involves the complex interplay of cis-acting and trans-acting subroutines from both the hrPKS and the nrPKS. In addition to its contribution to chain length determination, the intrinsic program of the TE domain also sets the overall product shape by adopting a download mode from a repertoire that includes macrocycle formation, hydrolysis, transesterification, and pyrone formation. It is instructive that PT and TE domains on one hand, and SAT domains and hrPKS subunits on the other travel related evolutionary paths in native BDL synthase systems. However, it is also instructive that divergence of these evolutionary paths may lead to unproductive combinations even in nature, as exemplified by the BDL synthase system of R. rufulum. To avoid such a fate, combinatorial synthetic microbiology must carefully characterize and standardize biosynthetic parts, and devise iterative PKS metaprograms from mutually compatible subroutines.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Key Research and Development Program of China (2018YFA0901800 to Y. X.); the National Natural Science Foundation of China (21807110 to C. W. and 31870076 to Y. X.); the China Postdoctoral Science Foundation (2019T120162 to C.W.); the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP to Y. X. and L. Z.); the Joint Genomics Institute of the U.S. Department of Energy (WIP ID 1349 to I. M.); the USDA National Institute of Food and Agriculture (Hatch project ARZT-1361640-H12-224 to I. M.); the Higher Education Institutional Excellence Program of the Ministry of Human Capacities in Hungary (NKFIH-1150-6/2019 to I. M.); and the U.S. National Institutes of Health (NIGMS 5R01GM114418 to I. M.). We thank Dr. Mei Zhang (the Beijing Center for Physical and Chemical Analysis) for providing access to the NMR device and Dr. Lida Han (the Research Facility Center of Biotechnology Research Institute) for the HR-ESIMS measurement. I. M. has disclosed financial interests in TEVA Pharmaceuticals Hungary and the University of Debrecen, Hungary, which are unrelated to the subject of the research presented here. All other authors declare no competing financial interests.
Footnotes
SUPPORTING INFORMATION
Supporting Information. Experimental and bioinformatic procedures, descriptions on the isolation and structure elucidation of compounds, spectroscopic data, supplementary figures including additional structures, product distributions during combinatorial biosynthesis, and phylogenetic trees.
REFERENCES
- 1.Hertweck C The biosynthetic logic of polyketide diversity. Angew. Chem., Int. Ed. 2009, 48, 4688–4716. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ; Cragg GM Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- 3.Chooi YH; Tang Y Navigating the fungal polyketide chemical space: from genes to molecules. J. Org. Chem. 2012, 77, 9933–9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keatinge-Clay AT The structures of type I polyketide synthases. Nat. Prod. Rep. 2012, 29, 1050–1073. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H; Gao Z; Qiao K; Wang J; Vederas JC; Tang Y A fungal ketoreductase domain that displays substrate-dependent stereospecificity. Nat. Chem. Biol. 2012, 8, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisch KM; Bakeer W; Yakasai AA; Song Z; Pedrick J; Wasil Z; Bailey AM; Lazarus CM; Simpson TJ; Cox RJ Rational domain swaps decipher programming in fungal highly reducing polyketide synthases and resurrect an extinct metabolite. J. Am. Chem. Soc. 2011, 133, 16635–16641. [DOI] [PubMed] [Google Scholar]
- 7.Yakasai AA; Davison J; Wasil Z; Halo LM; Butts CP; Lazarus CM; Bailey AM; Simpson TJ; Cox RJ Nongenetic reprogramming of a fungal highly reducing polyketide synthase. J. Am. Chem. Soc. 2011, 133, 10990–10998. [DOI] [PubMed] [Google Scholar]
- 8.Sun H; Ho CL; Ding F; Soehano I; Liu X-W; Liang Z-X Synthesis of (R)-mellein by a partially reducing iterative polyketide synthase. J. Am. Chem. Soc. 2012, 134, 11924–11927. [DOI] [PubMed] [Google Scholar]
- 9.Crawford JM; Dancy BCR; Hill EA; Udwary DW; Townsend CA Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 16728–16733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford JM; Thomas PM; Scheerer JR; Vagstad AL; Kelleher NL; Townsend CA Deconstruction of iterative multidomain polyketide synthase function. Science 2008, 320, 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawford JM; Korman TP; Labonte JW; Vagstad AL; Hill EA; Kamari-Bidkorpeh O; Tsai SC; Townsend CA Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization. Nature 2009, 461, 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du L; Lou L PKS and NRPS release mechanisms. Nat. Prod. Rep. 2010, 27, 255–278. [DOI] [PubMed] [Google Scholar]
- 13.Zhou H; Qiao K; Gao Z; Meehan MJ; Li JW-H; Zhao X; Dorrestein PC; Vederas JC; Tang Y Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J. Am. Chem. Soc. 2010, 132, 4530–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox RJ; Skellam EJ, Fungal non-reducing polyketide synthases In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier: 2019. [Google Scholar]
- 15.Zhou H; Zhan J; Watanabe K; Xie X; Tang Y A polyketide macrolactone synthase from the filamentous fungus Gibberella zeae. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6249–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Y; Zhou T; Zhou Z; Su S; Roberts SA; Montfort WR; Zeng J; Chen M; Zhang W; Lin M; Zhan J; Molnár I Rational reprogramming of fungal polyketide first-ring cyclization. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5398–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y; Zhou T; Zhang S; Xuan L-J; Zhan J; Molnár I Thioesterase domains of fungal nonreducing polyketide synthases act as decision gates during combinatorial biosynthesis. J. Am. Chem. Soc. 2013, 135, 10783–10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S; Xu Y; Maine EA; Wijeratne EMK; Espinosa-Artiles P; Gunatilaka AAL; Molnár I Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem. Biol. 2008, 15, 1328–1338. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y; Zhou T; Espinosa-Artiles P; Tang Y; Zhan J; Molnár I Insights into the biosynthesis of 12-membered resorcylic acid lactones from heterologous production in Saccharomyces cerevisiae. ACS Chem. Biol. 2014, 9, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y; Espinosa-Artiles P; Schubert V; Xu Y-M; Zhang W; Lin M; Gunatilaka AAL; Süssmuth R; Molnár I Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl. Environ. Microbiol. 2013, 79, 2038–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas R A biosynthetic classification of fungal and Streptomycete fused-ring aromatic polyketides. ChemBioChem 2001, 2, 612–627. [DOI] [PubMed] [Google Scholar]
- 22.Shen W; Mao H; Huang Q; Dong J Benzenediol lactones: a class of fungal metabolites with diverse structural features and biological activities. Eur. J. Med. Chem. 2015, 97, 747–777. [DOI] [PubMed] [Google Scholar]
- 23.Tillotson J; Bashyal BP; Kang M; Shi T; De La Cruz F; Gunatilaka AAL; Chapman E Selective inhibition of p97 by chlorinated analogues of dehydrocurvularin. Org. Biomol. Chem. 2016, 14, 5918–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y; Zhou T; Zhang S; Espinosa-Artiles P; Wang L; Zhang W; Lin M; Gunatilaka AAL; Zhan J; Molnár I Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12354–12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohm RA; Feau N; Henrissat B; Schoch CL; Horwitz BA; Barry KW; Condon BJ; Copeland AC; Dhillon B; Glaser F; Hesse CN; Kosti I; LaButti K; Lindquist EA; Lucas S; Salamov AA; Bradshaw RE; Ciuffetti L; Hamelin RC; Kema GH; Lawrence C; Scott JA; Spatafora JW; Turgeon BG; de Wit PJ; Zhong S; Goodwin SB; Grigoriev IV Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8, e1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma SM; Li JW-H; Choi JW; Zhou H; Lee KKM; Moorthie VA; Xie X; Kealey JT; Da Silva NA; Vederas JC; Tang Y Complete reconstitution of a highly reducing iterative polyketide synthase. Science 2009, 326, 589–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H; Qiao K; Gao Z; Vederas JC; Tang Y Insights into radicicol biosynthesis via heterologous synthesis of intermediates and analogs. J. Biol. Chem. 2010, 285, 41412–41421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai J; Lu Y; Xu Y-M; Zhang W; Chen M; Lin M; Gunatilaka AAL; Xu Y; Molnár I Diversity-oriented combinatorial biosynthesis of hybrid polyketide scaffolds from azaphilone and benzenediol lactone biosynthons. Org. Lett. 2016, 18, 1262–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie L; Zhang L; Wang C; Wang X; Xu Y-M; Yu H; Wu P; Li S; Han L; Gunatilaka AAL; Wei X; Lin M; Molnár I; Xu Y Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E4980–E4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X; Wang C; Duan L; Zhang L; Liu H; Xu Y-M; Liu Q; Mao T; Zhang W; Chen M; Lin M; Gunatilaka AAL; Xu Y; Molnár I Rational reprogramming of O-methylation regioselectivity for combinatorial biosynthetic tailoring of benzenediol lactone scaffolds. J. Am. Chem. Soc. 2019, 141, 4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfeifer BA; Khosla C Biosynthesis of polyketides in heterologous hosts. Microbiol. Mol. Biol. Rev. 2001, 65, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T; Sanchez JF; Chiang Y-M; Oakley BR; Wang CCC Rational domain swaps reveal insights about chain length control by ketosynthase domains in fungal nonreducing polyketide synthases. Org. Lett. 2014, 16, 1676–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korman TP; Crawford JM; Labonte JW; Newman AG; Wong J; Townsend CA; Tsai S-C Structure and function of an iterative polyketide synthase thioesterase domain catalyzing Claisen cyclization in aflatoxin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6246–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horsman ME; Hari TPA; Boddy CN Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat. Prod. Rep. 2016, 33, 183–202. [DOI] [PubMed] [Google Scholar]
- 35.Herbst DA; Huitt-Roehl CR; Jakob RP; Kravetz JM; Storm PA; Alley JR; Townsend CA; Maier T The structural organization of substrate loading in iterative polyketide synthases. Nat. Chem. Biol. 2018, 14, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbst DA; Townsend CA; Maier T The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai S-C The structural enzymology of iterative aromatic polyketide synthases: a critical comparison with fatty acid synthases. Annu. Rev. Biochem. 2018, 87, 503–531. [DOI] [PubMed] [Google Scholar]
- 38.Curran SC; Hagen A; Poust S; Chan LJG; Garabedian BM; de Rond T; Baluyot M-J; Vu JT; Lau AK; Yuzawa S; Petzold CJ; Katz L; Keasling JD Probing the flexibility of an iterative modular polyketide synthase with non-native substrates in vitro. ACS Chem. Biol. 2018, 13, 2261–2268. [DOI] [PubMed] [Google Scholar]
- 39.Nicholson TP; Winfield C; Westcott J; Crosby J; Simpson TJ; Cox RJ First in vitro directed biosynthesis of new compounds by a minimal type II polyketide synthase: evidence for the mechanism of chain length determination. Chem. Commun. 2003, 686–687. [DOI] [PubMed] [Google Scholar]
- 40.Huitt-Roehl CR; Hill EA; Adams MM; Vagstad AL; Li JW; Townsend CA Starter unit flexibility for engineered product synthesis by the nonreducing polyketide synthase PksA. ACS Chem. Biol. 2015, 10, 1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman AG; Vagstad AL; Belecki K; Scheerer JR; Townsend CA Analysis of the cercosporin polyketide synthase CTB1 reveals a new fungal thioesterase function. Chem. Commun. 2012, 48, 11772–11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lackner G; Bohnert M; Wick J; Hoffmeister D Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 2013, 20, 1101–1106. [DOI] [PubMed] [Google Scholar]
- 43.Winter JM; Cascio D; Dietrich D; Sato M; Watanabe K; Sawaya MR; Vederas JC; Tang Y Biochemical and structural basis for controlling chemical modularity in fungal polyketide biosynthesis. J. Am. Chem. Soc. 2015, 137, 9885–9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch AA; Schmidt JJ; Lowell AN; Hansen DA; Coburn KM; Chemler JA; Sherman DH Probing selectivity and creating structural diversity through hybrid polyketide synthases. Angew. Chem., Int. Ed. 2020, doi: 10.1002/anie.202004991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kakule TB; Lin Z; Schmidt EW Combinatorialization of fungal polyketide synthase-peptide synthetase hybrid proteins. J. Am. Chem. Soc. 2014, 136, 17882–17890. [DOI] [PubMed] [Google Scholar]
- 46.Yang X-L; Friedrich S; Yin S; Piech O; Williams K; Simpson TJ; Cox RJ Molecular basis of methylation and chain-length programming in a fungal iterative highly reducing polyketide synthase. Chem. Sci. 2019, 10, 8478–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaneko A; Morishita Y; Tsukada K; Taniguchi T; Asai T Post-genomic approach based discovery of alkylresorcinols from a cricket-associated fungus, Penicillium soppi. Org. Biomol. Chem. 2019, 17, 5239–5243. [DOI] [PubMed] [Google Scholar]
- 48.Heberlig GW; Brown JTC; Simard RD; Wirz M; Zhang W; Wang M; Susser LI; Horsman ME; Boddy CN Chemoenzymatic macrocycle synthesis using resorcylic acid lactone thioesterase domains. Org. Biomol. Chem. 2018, 16, 5771–5779. [DOI] [PubMed] [Google Scholar]
- 49.Chen X-P; Shi T; Wang X-L; Wang J; Chen Q; Bai L; Zhao Y-L Theoretical studies on the mechanism of thioesterase-catalyzed macrocyclization in erythromycin biosynthesis. ACS Catal. 2016, 6, 4369–4378. [Google Scholar]
- 50.Wang M; Zhou H; Wirz M; Tang Y; Boddy CN A Thioesterase from an iterative fungal polyketide synthase shows macrocyclization and cross coupling activity and may play a role in controlling iterative cycling through product offloading. Biochemistry 2009, 48, 6288–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim Y-T; Lee Y-R; Jin J; Han K-H; Kim H; Kim J-C; Lee T; Yun S-H; Lee Y-W Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. Mol. Microbiol. 2005, 58, 1102–1113. [DOI] [PubMed] [Google Scholar]
- 52.Reeves CD; Hu Z; Reid R; Kealey JT Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol. 2008, 74, 5121–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cochrane RVK; Sanichar R; Lambkin GR; Reiz B; Xu W; Tang Y; Vederas JC Production of new cladosporin analogues by reconstitution of the polyketide synthases responsible for the biosynthesis of this antimalarial agent. Angew. Chem., Int. Ed. 2016, 55, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cochrane RVK; Gao Z; Lambkin GR; Xu W; Winter JM; Marcus SL; Tang Y; Vederas JC Comparison of 10,11-dehydrocurvularin polyketide synthases from Alternaria cinerariae and Aspergillus terreus highlights key structural motifs. ChemBioChem 2015, 16, 2479–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.