

Abstract
Background
Body mass index (BMI)-associated loci are used to explore the effects of obesity using Mendelian randomization (MR), but the contribution of individual tissues to risks remains unknown. We aimed to identify tissue-grouped pathways of BMI-associated loci and relate these to cardiometabolic disease using MR analyses.
Methods
Using Genotype-Tissue Expression (GTEx) data, we performed overrepresentation tests to identify tissue-grouped gene sets based on mRNA-expression profiles from 634 previously published BMI-associated loci. We conducted two-sample MR with inverse-variance-weighted methods, to examine associations between tissue-grouped BMI-associated genetic instruments and type 2 diabetes mellitus (T2DM) and coronary artery disease (CAD), with use of summary-level data from published genome-wide association studies (T2DM: 74 124 cases, 824 006 controls; CAD: 60 801 cases, 123 504 controls). Additionally, we performed MR analyses on T2DM and CAD using randomly sampled sets of 100 or 200 BMI-associated genetic variants.
Results
We identified 17 partly overlapping tissue-grouped gene sets, of which 12 were brain areas, where BMI-associated genes were differentially expressed. In tissue-grouped MR analyses, all gene sets were similarly associated with increased risks of T2DM and CAD. MR analyses with randomly sampled genetic variants on T2DM and CAD resulted in a distribution of effect estimates similar to tissue-grouped gene sets.
Conclusions
Overrepresentation tests revealed differential expression of BMI-associated genes in 17 different tissues. However, with our biology-based approach using tissue-grouped MR analyses, we did not identify different risks of T2DM or CAD for the BMI-associated gene sets, which was reflected by similar effect estimates obtained by randomly sampled gene sets.
Keywords: Mendelian randomization analysis, body mass index, type 2 diabetes mellitus, coronary artery disease, waist circumference, anthropometry
Key Messages
Body mass index (BMI)-associated genes are differentially expressed in the brain, digestive system, spleen and kidney cortex.
The tissue-grouped gene sets did not show different effects on risks of type 2 diabetes mellitus and coronary artery disease.
BMI-associated tissue-grouped gene sets are all associated with a higher waist circumference and more total body fat.
Introduction
The prevalence of obesity and obesity-related diseases is increasing worldwide.1 Previous studies identified many genetic variants associated with an increased risk of developing obesity and a high body mass index (BMI).2–4 A Genome-Wide Association Study (GWAS) by Yengo et al. (2018) identified 656 independent genetic variants associated with BMI, which collectively explained up to 6% of the total variation in BMI.4 High BMI is a well-known risk factor for cardiometabolic disease, such as type 2 diabetes mellitus and cardiovascular disease,5–8 which has been confirmed in Mendelian randomization (MR) analyses, in which genetic variants associated with an exposure are used as instrumental variables.9
Characterization of genetic variants that are associated with BMI, and the genes corresponding to these single nucleotide polymorphisms (SNPs), can provide insight into the biological causes of a high BMI. In an earlier MR study on the association between BMI and anxiety,10 SNPs were categorized based on three mechanistic domains through which they were likely to influence BMI: appetite, adipogenesis and cardiopulmonary function, derived from an earlier GWAS on BMI.3 As an alternative and complementary approach to gene categorization based on mechanistic domains, tissue-grouped gene sets of BMI-associated genes can be identified by analysing the differential expression of BMI-associated genes in tissues: genes whose expression is significantly up- or downregulated in a given tissue compared with other tissues.11
Enrichment of BMI-associated genes has been observed in tissues of the central nervous system, notably in the hypothalamus, pituitary gland, hippocampus and limbic system,2 which are brain areas involved in appetite regulation, cognition and emotion, and thereby collectively highlighted the potential biological processes involved in the regulation of body composition. Additionally, a recent study combined genes for BMI, waist–hip ratio (WHR) and WHR adjusted for BMI (WHRadjBMI), which are all proxies of body composition, to identify enrichment of these genes on a tissue level.12 In this study, differential expression of these body-composition-related genes was observed in the central nervous system, adipocyte-related tissues and the digestive and urogenital systems.12 Additionally, BMI-associated genes that show differential expression in specific tissues, thereby reflecting potential tissue-grouped causes of a high BMI, may have distinct effects on disease risk as well.13–16 A more detailed investigation of these biological causes will allow a comprehensive overview of potential targets for interventions.
For this reason, we aimed to group BMI-associated genetic instruments based on tissue-expression profiles. With these newly identified combinations of BMI-associated genetic instruments, we aimed to conduct two-sample MR analyses on cardiometabolic diseases and measures of abdominal adiposity, in order to unravel the underlying pathways of tissue-grouped causes of a high BMI leading to cardiometabolic-disease risk.
Methods
Selection of genetic variants and gene annotation for BMI
We selected SNPs for all genetic loci independently associated with BMI, which were identified in the GWAS meta-analysis by Yengo et al.4
For the meta-analysis, results of the GWAS on BMI in the UK Biobank were combined with publicly available summary statistics of an earlier GWAS on BMI in the Genetic Investigation of ANthropometric Traits (GIANT) consortium, comprising around 700 000 individuals of mainly European ancestry in total. After meta-analysis, 656 independent SNPs associated with BMI were identified in their main genome-wide significance association (p-value <5 × 10–8) (see Supplementary Table 1, available as Supplementary data at IJE online).
The 656 SNPs were annotated and mapped to genes based on several lines of evidence using the SNP2GENE function within the Functional Mapping and Annotation (FUMA) v1.3.5e web application (date of accession: 12 November 2019).17 First, we uploaded a file with the GWAS summary statistics, including the chromosome and position, reference SNP (rs) ID, p-value, effect allele, non-effect allele, beta and standard deviation for each SNP, and the sample size of the GWAS.
We did not predefine lead SNPs of a genomic region, but instead used the following default parameters: minimum p-value of lead SNPs <5e–8, maximum p-value cut-off <0.05, r2 threshold to define independent significant SNPs ≥0.6, second r2 threshold to define lead SNPs ≥0.1, reference panel population 1000 G Phase3 EUR, minimum Minor Allele Frequency (MAF) ≥0 and maximum distance between linkage disequilibrium (LD) blocks to merge into a locus <250 kb. We included variants in the reference 1000 G reference panel (non-GWAS tagged SNPs in LD with an independent significant SNP).18 Gene annotation was based on positional mapping obtained from Annotate Variation (ANNOVAR) with a maximum distance to genes of 10 kilobases.19 For the gene types, we used Ensembl v92 and selected protein-coding gene types. A set of prioritized genes is derived from the combined mapping strategies, based on the provided GWAS summary statistics and filter settings. The output from FUMA comprised genome-wide plots, a summary of the results and result tables, from which we selected the table with independently significant lead SNPs and the table with mapped genes, and merged these to obtain a list with lead SNPs and their mapped genes. Due to the use of a filter to identify independent SNPs, 22 lead SNPs were omitted, resulting in 634 mapped genes of interest that were used to identify the tissue-grouped gene sets. All exact parameters used for gene annotation and mapping can be found in the Supplementary Methods, available as Supplementary data at IJE online.
Tissue-grouped gene sets
Normalized gene expressions (reads per kilo base per million, RPKM) of 54 non-diseased tissue types were available in GTEx version 8,11 of which we excluded five tissue types because no eQTL analysis was available (bladder, cervix-ectocervix and -endocervix, fallopian tube and kidney medulla). Tissue types included different brain areas, adipose tissue depots, skin, blood, spleen, kidney cortex and tissues involved in digestion (e.g. oesophagus, stomach, small intestine, pancreas and colon) or reproduction (e.g. ovaries, uterus, vagina and testis). In the GENE2FUNC in FUMA, the average of the normalized expression [zero mean of log2(RPKM +1)] per tissue per gene was shown as expression value, to allow comparison of expression across tissue types. By this means, we identified differentially expressed gene sets (genes of which the expression is significantly up- or downregulated in a given tissue compared with other tissues) for each of the 54 tissue types available in GTEx v8. Two-sided Student’s t-tests with Bonferroni correction were performed per gene per tissue against all other tissues to identify differentially expressed genes in a given tissue. In GENE2FUNC in FUMA, genes are tested against those differentially expressed gene sets by hypergeometric tests to evaluate whether the prioritized genes (genes of interest) are overrepresented in differentially expressed gene sets in the 54 specific tissue types. We entered the 634 BMI-associated genes (our mapped genes of interest based on the independent lead SNPs) in the GENE2FUNC function and selected ‘All’ background genes, Ensembl version v92 and the GTEx v8 54 tissue type (date of accession: 12 November 2019). We selected the tissues that showed significant differential gene expression in GTEx v8 after adjusting for multiple testing using a false-discovery-rate threshold of 5%. Using the summary data retrieved from FUMA, we identified the specific genes that were differentially expressed in each tissue to establish the gene sets used in the tissue-grouped MR analyses. All tissue-grouped gene sets were considered strong instruments for BMI based on their F-statistics. All of the exact parameters used to generate the tissue-grouped gene sets can be found in the Supplementary Methods, available as Supplementary data at IJE online.
Outcome data on type 2 diabetes and coronary artery disease
As study outcomes for the MR analyses, we used publicly available meta-analysis summary statistics of GWAS on type 2 diabetes mellitus and coronary artery disease by the DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) and the Coronary Artery Disease Genome wide Replication and Meta-analysis plus the Coronary Artery Disease Genetics (CARDIoGRAMplusC4D) consortia, respectively. These datasets, based on the largest number of participants to date, contain the per-allele beta-estimates of all investigated SNPs on the outcomes, accompanying standard errors and the effect alleles.
The report of the DIAGRAM consortium was a meta-analysis of 32 different cohort studies of European ancestry (including the UK Biobank), consisting of 74 124 cases of T2DM and 824 006 controls.20 T2DM was defined as a fasting glucose concentration >6.9 mmol/L or 2-hour plasma glucose of ≥11.1 mmol/L, treatment with glucose-lowering agents, diagnosis by a general practitioner or medical specialist, or self-report.
Within the CARDIoGRAMC4D consortium, GWAS data of 48 studies were meta-analysed, with 60 801 cases and 123 504 controls.21 CAD was defined by a history of myocardial infarction, acute coronary syndrome, chronic stable angina or >50% coronary stenosis.
Outcome data on anthropometric traits
The underlying mechanism of the different effects of the tissue-grouped gene sets on disease risk could be via the effect of the tissue-grouped gene sets on body-fat distribution measured by waist circumference or total body fat. By this means, we aimed to give insight into the underlying mechanisms of tissue-grouped causes of a high BMI and their effect on disease risk. We used publicly available meta-analysis summary statistics of anthropometric traits conducted by the GIANT consortium and by the Neale Lab. The data of the GIANT consortium were reported as a meta-analysis of 57 multi-ancestry cohorts, with oversampling of cohorts of European ancestry, genotyped by genome-wide SNP arrays and of 44 cohorts genotyped with the Metabochip. In the present analysis, we used waist circumference (in centimetres, unadjusted for BMI) in up to 224 459 individuals.22
The GWAS on total body fat (as a percentage) was based on data from the UK Biobank, in up to 331 117 individuals of White British genetic ancestry.23 Closely related individuals, individuals with sex chromosome aneuploidies and individuals who had withdrawn consent from the UK Biobank study had been removed from the analysis.
MR analysis
Methods for MR analyses of summary-level data based on two study samples have been described in detail previously.24,25 We estimated the associations between all genetic instruments for BMI, the tissue-grouped gene sets and the outcome measures (CAD, type 2 diabetes mellitus and anthropometric traits). We allowed the use of LD proxies (R2 > 0.8) and we allowed palindromic SNPs with a MAF threshold of 0.3. Using inverse-variance-weighted (IVW) analyses, we combined the effects of the individual genetic instruments to obtain a genetically determined association between exposure and outcome under the assumption of the absence of horizontal pleiotropy. The IVW analyses resulted in a weighted mean estimate of a genetically determined increase of 1 standard deviation (SD) of BMI (kg/m2) on the odds ratio (OR) of type 2 diabetes mellitus and CAD, or on waist circumference (SD, in centimetres), and on total body fat (SD, percentage). In the present study, an increase of 1 SD of BMI (kg/m2) corresponded to an increase in BMI of 4.8 kg/m2.
MR analyses could suffer from bias due to pleiotropic effects, when a genetic variant also affects other phenotypes and thereby influences the outcome via alternative pathways other than through the exposure. Therefore, as sensitivity analyses, we performed MR-Egger regression26 and weighted median-estimator (WME) analyses.27 MR-Egger accounts for potential pleiotropy and tests for the presence of directional pleiotropy, whereas WME estimates a weighted median effect instead of a weighted mean effect. Similarity of the IVW and MR-Egger and WME effect estimates indicates that the results of the MR analysis are robust. In addition to MR-Egger and WME, we used the recently described Weighted Mode to detect causal effects—a method that is consistent when the largest number of similar individual causal-effect estimates comes from valid instruments, even if the majority of instruments are invalid.28
For MR analyses, we removed the genetic instrument mapped to TCF7L2 (rs7903146) from the gene sets (and thereby all MR analyses) given its previously described complex and pleiotropic effect.29 Discrepancies between the total numbers of genetic variants in the tissue-grouped gene sets and the numbers of genetic variants in the tissue-grouped gene sets that were used in the MR analyses are due to the removal of palindromic genetic instruments with intermediate allele frequencies.
The combined effects of the genetic variants in the overall and sensitivity analyses were estimated using the R-based package ‘TwoSampleMR’.30 The R script for the MR analyses can be found in the Supplementary Methods, available as Supplementary data at IJE online.
MR analyses using random samples of genetic variants
To gain insight into whether GTEx-based subsets of genetic instruments are the result of chance, we additionally determined the effect estimates for CAD and T2DM by randomly selecting genetic variants. We randomly sampled 1000 gene sets, consisting of 100 or 200 genetic variants from the 633 BMI-associated genetic variants, and performed MR analyses (IVW) on CAD and T2DM with these random gene sets. The R script for the MR analyses using random samples of genetic variants can be found in the Supplementary Methods, available as Supplementary data at IJE online.
Results
Identification of tissue-grouped expression gene sets
Figure 1 shows the analysis procedure we followed to obtain the tissue-grouped BMI-associated gene sets from 656 BMI-associated SNPs. Based on the analysis with GTEx data, we identified 17 tissues in which genes associated with BMI were differentially expressed (Figure 2A), of which 12 were derived from brain areas. Other tissues were derived from the arteries, digestive system, spleen or kidney cortex. Differentially expressed genes in each tissue were identified to establish the gene sets (see Figure 2A). There was substantial overlap in the genes that were differentially expressed in the brain tissues, but none had a completely overlapping expression profile. Gene symbols and their expression in the different tissue-grouped gene sets are provided in Supplementary Table 1, available as Supplementary data at IJE online. Clustering of the BMI-associated genes in the tissue-grouped gene sets is shown in Figure 2B. It should be noted that a substantial number of genetic instruments was not differentially expressed in any of the identified tissues.
Figure 1.

The analysis procedure. (i) We selected 656 single nucleotide polymorphisms (SNPs) independently associated with BMI.4 (ii) Using the SNP2GENE function in the FUMA web application, gene annotation was performed for the selected SNPs based on positional mapping obtained from Annotate Variation (ANNOVAR).19 In this step, 22 SNPs were omitted based on a r2 threshold of ≥0.6. (iii) The 634 BMI-associated genes were grouped into 17 tissue-grouped gene sets using the GENE2FUNC function in Functional Mapping and Annotation (FUMA). These gene sets were based on differential gene expression in different tissues with the use of GTEx version 8. (iv) The 17 tissue-grouped gene sets were used as exposures in Mendelian randomization (MR) analyses, with the outcomes type 2 diabetes mellitus (T2DM), coronary artery disease (CAD), waist circumference and total body fat. The genetic instrument rs7903146 was removed from the gene sets (and thereby all MR analyses) given its pleiotropic effect.
Figure 2.

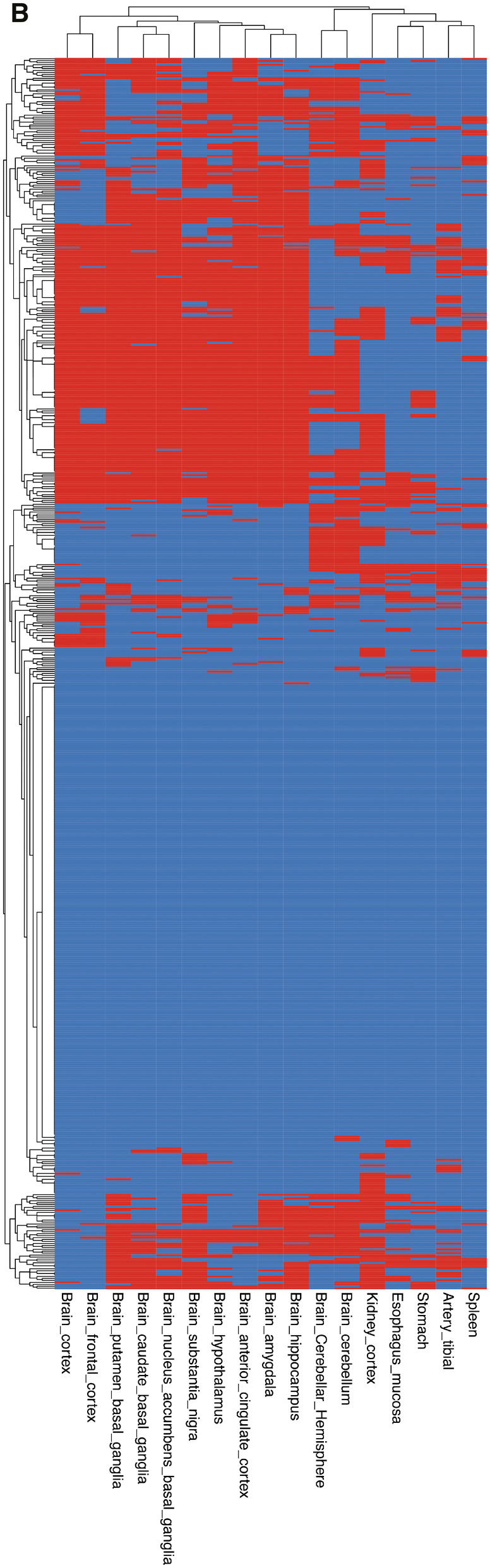
Based on the analyses using GTEx v8 54 tissue-types data, we identified 17 tissues in which body mass index (BMI)-associated genes were differentially expressed, indicated by black/red bars. (A) Was directly taken from the online tool Functional Mapping and Annotation (FUMA).17 (B) Clustering of the BMI-associated genes in the tissue-grouped gene sets. Differentially expressed genes in a given tissue are shown in black/red; grey/blue indicates no differential expression in a given tissue.
Tissue-grouped MR analyses
Type 2 diabetes mellitus and CAD
We performed MR with the tissue-grouped gene sets on T2DM and CAD (Table 1). Considering all BMI genetic instruments, an increase in BMI was associated with an increased risk of both T2DM [per SD BMI OR 2.71, 95% confidence interval (CI) 2.49; 2.94] and CAD (per SD BMI, OR 1.48, 1.37; 1.59).
Table 1.
Association between the BMI-overall and BMI-associated tissue-grouped gene sets and risk of type 2 diabetes and coronary artery disease using (tissue-grouped) inverse-variance-weighted analyses
| Type 2 diabetes |
Coronary artery disease |
|||||
|---|---|---|---|---|---|---|
| n genes | Beta (SE) | Odds ratio (95% CI) | n genes | Beta (SE) | Odds ratio (95% CI) | |
| BMI—all genetic variants | 616 | 1.00 (0.04) | 2.71 (2.49; 2.94) | 613 | 0.39 (0.04) | 1.48 (1.37; 1.59) |
| Artery tibial | 86 | 0.93 (0.13) | 2.54 (1.99; 3.50) | 86 | 0.43 (0.10) | 1.53 (1.26; 1.87) |
| Brain amygdala | 231 | 0.90 (0.08) | 2.45 (2.08; 2.89) | 230 | 0.41 (0.07) | 1.51 (1.32; 1.72) |
| Brain anterior cingulate cortex | 232 | 0.93 (0.08) | 2.54 (2.19; 2.94) | 231 | 0.43 (0.06) | 1.54 (1.37; 1.73) |
| Brain caudate basal ganglia | 222 | 0.89 (0.09) | 2.42 (2.04; 2.89) | 221 | 0.40 (0.07) | 1.49 (1.30; 1.69) |
| Brain cerebellar hemisphere | 160 | 1.08 (0.09) | 2.94 (2.49; 3.47) | 159 | 0.34 (0.08) | 1.41 (1.22; 1.63) |
| Brain cerebellum | 214 | 1.00 (0.08) | 2.72 (2.33; 3.19) | 213 | 0.37 (0.07) | 1.45 (1.28; 1.65) |
| Brain cortex | 213 | 0.95 (0.08) | 2.59 (2.22; 3.03) | 212 | 0.43 (0.06) | 1.53 (1.36; 1.73) |
| Brain frontal cortex | 207 | 0.97 (0.08) | 2.63 (2.25; 3.07) | 206 | 0.43 (0.06) | 1.54 (1.37; 1.73) |
| Brain hippocampus | 237 | 0.96 (0.08) | 2.60 (2.22; 3.05) | 237 | 0.44 (0.06) | 1.55 (1.37; 1.75) |
| Brain hypothalamus | 214 | 0.97 (0.08) | 2.65 (2.27; 3.09) | 213 | 0.40 (0.06) | 1.49 (1.33; 1.68) |
| Brain nucleus accumbens basal ganglia | 224 | 0.99 (0.08) | 2.70 (2.31; 3.16) | 223 | 0.40 (0.06) | 1.48 (1.31; 1.68) |
| Brain putamen basal ganglia | 237 | 0.92 (0.08) | 2.52 (2.16; 2.95) | 237 | 0.46 (0.06) | 1.58 (1.40; 1.78) |
| Brain substantia nigra | 226 | 0.95 (0.08) | 2.58 (2.20; 3.03) | 225 | 0.44 (0.06) | 1.55 (1.37; 1.75) |
| Oesophagus mucosa | 99 | 1.00 (0.13) | 2.72 (2.12; 3.50) | 99 | 0.33 (0.09) | 1.39 (1.17; 1.65) |
| Kidney cortex | 191 | 1.00 (0.09) | 2.72 (2.29; 3,22) | 190 | 0.36 (0.07) | 1.44 (1.25; 1.65) |
| Spleen | 63 | 1.00 (0.12) | 2.72 (2.17; 3,42) | 63 | 0.28 (0.13) | 1.32 (1.02; 1.70) |
| Stomach | 81 | 0.94 (0.12) | 2.55 (2.01; 3.24) | 81 | 0.41 (0.09) | 1.51 (1.25; 1.82) |
Betas and odds ratios are obtained using Mendelian randomization analyses. The odds ratio can be interpreted per increase of 1 standard deviation of BMI (kg/m2), which is equivalent to an increase of 4.8 kg/m2. ngenes indicates the number of genes in each gene set.
BMI, body mass index; SE, standard error; CI, confidence interval.
In the tissue-grouped MR analyses, all BMI-associated tissue-grouped gene sets were associated with an increased risk of T2DM (Table 1), with ORs ranging from 2.42 (2.04; 2.89) for the brain caudate basal ganglia to 2.94 (2.49; 3.47) for the brain cerebellar hemisphere. For CAD, the ORs were ranging from 1.32 (1.02; 1.70) for the spleen to 1.58 (1.40; 1.78) for the brain putamen basal ganglia. The sensitivity analyses (MR-Egger, WME and Weighted Mode) provided similar effects of the tissue-grouped gene sets on T2DM and CAD, although the effect estimates were slightly larger compared with the IVW results (see Supplementary Table 2, available as Supplementary data at IJE online).
Waist circumference and total body fat
We examined the associations of the tissue-grouped gene sets with the anthropometric traits waist circumference and total body fat as outcomes to explore potential underlying mechanisms of cardiometabolic disease (see Table 2). Considering all BMI genetic instruments, an increase of 1 SD of BMI was associated with an increase in waist circumference of 0.83 cm (95% CI 0.80; 0.85) and total body fat of 0.61% (95% CI 0.60; 0.63). All BMI-associated tissue-grouped gene sets had a positive effect on waist circumference and total body fat. Overall, the alternative MR methods MR-Egger, WME and Weighted Mode analyses indicated a positive effect of the tissue-grouped gene sets on waist circumference and total body fat (see Supplementary Table 3, available as Supplementary data at IJE online).
Table 2.
Association between the BMI-overall and BMI-associated tissue-grouped gene sets and waist circumference and total body fat using (tissue-grouped) inverse-variance-weighted analyses
| Waist circumference (cm) |
Total body fat (%) |
|||
|---|---|---|---|---|
| n genes | Beta (95% CI) | n genes | Beta (95% CI) | |
| BMI—all genetic variants | 610 | 0.83 (0.80; 0.85) | 615 | 0.61 (0.60; 0.63) |
| Artery tibial | 84 | 0.84 (0.78; 0.90) | 86 | 0.61 (0.57; 0.65) |
| Brain amygdala | 229 | 0.84 (0.80; 0.88) | 231 | 0.64 (0.61; 0.67) |
| Brain anterior cingulate cortex | 230 | 0.85 (0.82; 0.89) | 232 | 0.65 (0.63; 0.68) |
| Brain caudate basal ganglia | 219 | 0.86 (0.82; 0.90) | 221 | 0.64 (0.61; 0.67) |
| Brain cerebellar hemisphere | 156 | 0.85 (0.81; 0.90) | 160 | 0.61 (0.58; 0.64) |
| Brain cerebellum | 210 | 0.85 (0.81; 0.89) | 214 | 0.64 (0.61; 0.67) |
| Brain cortex | 212 | 0.86 (0.82; 0.90) | 213 | 0.65 (0.62; 0.68) |
| Brain frontal cortex | 205 | 0.85 (0.82; 0.89) | 206 | 0.66 (0.63; 0.69) |
| Brain hippocampus | 236 | 0.84 (0.80; 0.88) | 237 | 0.64 (0.61; 0.67) |
| Brain hypothalamus | 212 | 0.84 (0.81; 0.88) | 214 | 0.64 (0.61; 0.66) |
| Brain nucleus accumbens basal ganglia | 222 | 0.86 (0.82; 0.90) | 224 | 0.65 (0.62; 0.68) |
| Brain putamen basal ganglia | 236 | 0.83 (0.79; 0.86) | 237 | 0.65 (0.62; 0.67) |
| Brain substantia nigra | 224 | 0.84 (0.80; 0.87) | 226 | 0.63 (0.60; 0.66) |
| Oesophagus mucosa | 97 | 0.84 (0.78; 0.89) | 99 | 0.61 (0.56; 0.65) |
| Kidney cortex | 188 | 0.82 (0.78; 0.86) | 191 | 0.63 (0.60; 0.66) |
| Spleen | 62 | 0.85 (0.78; 0.91) | 63 | 0.59 (0.54; 0.64) |
| Stomach | 80 | 0.82 (0.75; 0.88) | 81 | 0.61 (0.56; 0.66) |
Betas are obtained using Mendelian randomization analyses. The betas can be interpreted per increase of 1 standard deviation of BMI (kg/m2). ngenes indicates the number of genes in each gene set.
BMI, body mass index; CI, confidence interval.
MR analyses using random samples of genetic variants
The distribution of ORs obtained by random sampling of 100 or 200 genetic variants (from the 633 BMI-associated genetic variants) was similar to the results of the tissue-grouped MR (IVW method), as presented in the histograms in Figure 3. For T2DM, the ORs ranged from 1.9 to 3.8 for the sampled gene sets of 100 variants and from 1.7 to 3.4 for the sampled gene sets of 200 variants, with the most frequent OR around 2.7, which was similar to the estimates that were observed in the tissue-grouped MR analyses. Similar to the tissue-grouped results, the ORs of the randomly sampled gene sets for the outcome CAD were smaller than for T2DM, ranging from 1.2 to 2.1, with the most frequent OR around 1.4.
Figure 3.

Histograms displaying the distribution of odds ratios [from inverse-variance weighted (IVW) analyses] for the association between randomly sampled sets of 100 or 200 genetic variants and type 2 diabetes mellitus (T2DM) and coronary artery disease (CAD). (A) Random sample of 100 genetic variants—T2DM. (B) Random sample of 200 genetic variants—T2DM. (C) Random sample of 100 genetic variants—CAD. (D) Random sample of 200 genetic variants—CAD.
Discussion
The aims of the present study were to identify tissue-grouped pathways of BMI-associated loci and their relation to cardiometabolic disease and anthropometric measures using MR analyses. The results of our study imply that genetic variants associated with BMI show enrichment in different tissues. In earlier studies, enrichment of BMI-associated genes was observed in the central nervous system,2 in accordance with the results of the present study. Expression of genetic variants that were associated with a decreased WHR was enriched in the digestive and urogenital systems.12 We observed enrichment of BMI-grouped genes in the oesophagus mucosa, stomach, spleen and kidney cortex.
Instead of performing MR using all genetic instruments for a given exposure, we examined the contribution of associated tissues to the risk of cardiometabolic disease, in order to unravel the role of specific tissues in the genetic risk of cardiometabolic disease due to overweight or obesity. Using MR, the associations between BMI and T2DM and CAD have been well established.5–8 Although the use of tissue-grouped gene sets of genetic instruments would mean a reduction in statistical power to identify effects of BMI, we were still able to identify effect estimates similar to the overall MR analysis comprising all genetic instruments, which was also reflected in the F-statistics of the tissue-grouped gene sets.31 Therefore, the increased availability of large GWAS datasets allows examination of the individual contributions of grouped genetic variants in more detail.
The results of the present study imply that genetic variants associated with BMI have similar effects on type 2 diabetes and CAD risk, regardless of the tissue in which the genes show differential expression, although there was some variation in the observed effect estimates. All BMI-associated gene sets were shown to have an effect on waist circumference and total body fat, which are both known to be associated with increased risk of T2DM and CAD. These results are supported by findings of additional analyses, in which we randomly selected 100 or 200 genetic instruments from the 633 BMI-associated genetic variants. After we repeatedly performed Mendelian randomization analyses on T2DM and CAD with randomly sampled BMI-associated gene sets, the distribution of the effect estimates was similar to the results of the tissue-grouped MR analyses. This suggests that the effect estimates of the BMI-associated gene sets based on tissue-expression profiles were similar to the BMI-associated gene sets obtained by random sampling of genetic instruments.
The identification of groups of genetic variants that have differential effects on an outcome measure, representing different mechanisms of disease, can be approached from various perspectives. In the present study, we explored the added value of clustering genetic instruments based on a biological perspective. Importantly, in previous research, a statistical approach has been proposed.32 The authors were able to group high-density lipoprotein cholesterol variants according to their effect on the risk of coronary heart disease and thus to identify groups of genetic variants showing different effects on disease. We therefore hypothesize that grouping the BMI-associated genetic variants by a statistical approach might identify groups of BMI-associated genetic variants that have different effects on disease risk.
A potential limitation of our study is the methodology of the clustering of the genetic instruments in the tissue-grouped gene sets. Currently, the GTEx analyses are performed using a standard overrepresentation test that does not take into account the effect size of the genetic instrument on the exposure. Genetic variants with large effect sizes therefore have the same weight in the clustering as genetic variants with much smaller effect sizes. As the GWAS by Yengo et al. identified many genetic variants for BMI with small effect sizes, this likely will have affected the clustering. Additionally, tissue-grouped gene sets identified by the enrichment analyses by GTEx are influenced by the number of tissue samples available for the different tissues. Subsequently, this has implications for the results of the MR analyses.
Another limitation of our study is the overlap between the genes within the tissue-grouped gene sets, which made it more difficult to mutually compare the tissue-grouped gene sets. Due to the overlap, the gene sets might be less specific to distinguish between the different tissues, although they are specific for investigating the relation with cardiometabolic disease.
To conclude, this study explored whether we can add insight in causal disease biology when analysing tissue-grouped gene sets and by examining the effects of these gene sets on different outcome measures using MR analyses. In the context of BMI-associated genetic variants, our novel approach does not provide additional insight into the role of specific tissues in the genetic risk for specific cardiometabolic disease due to overweight or obesity.
Author contributions
I.V., R.d.M., R.S., S.T., F.R., D.v.H., K.W.v.D and R.N.: Conceptualization; I.V., R.d.M., R.S., S.T., K.W.v.D. and R.N.: Methodology; I.V. and R.N.: Formal analysis; I.V.: Writing – Original draft; I.V., R.d.M., R.S., S.T., F.R., D.v.H., K.W.v.D. and R.N.: Writing – Review & Editing; I.V. and R.N.: Visualization.
Funding
This work was supported by an internal PhD fellowship of the board of directors of the Leiden University Medical Center (IV) and by the European Commission funded project HUMAN (Health-2013-INNOVATION-1-602757 to D.v.H. and R.N.).
Supplementary Material
Acknowledgements
We thank the DIAGRAM and CARDIoGRAM consortia for making available their GWAS meta-analyses. We thank Kim Luijken for her help with the R script for generating random samples of genetic variants.
Conflict of interest
None declared.
References
- 1.World Health Organization (WHO). Obesity and overweight. Fact sheet No 311. 2020. https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (27 March 2020, date last accessed).
- 2. Locke AE, Kahali B, Berndt SI et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Speliotes EK, Willer CJ, Berndt SI et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yengo L, Sidorenko J, Kemper KE et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 2018;27:3641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dale CE, Fatemifar G, Palmer TM et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a Mendelian randomization analysis. Circulation 2017;135:2373–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lyall DM, Celis-Morales C, Ward J et al. Association of body mass index with cardiometabolic disease in the UK Biobank: a Mendelian randomization study. JAMA Cardiol 2017;2:882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hagg S, Fall T, Ploner A et al. Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int J Epidemiol 2015;44:578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wade KH, Chiesa ST, Hughes AD et al. Assessing the causal role of body mass index on cardiovascular health in young adults: Mendelian randomization and recall-by-genotype analyses. Circulation 2018;138:2187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–63. [DOI] [PubMed] [Google Scholar]
- 10. Walter S, Glymour MM, Koenen K et al. Do genetic risk scores for body mass index predict risk of phobic anxiety? Evidence for a shared genetic risk factor. Psychol Med 2015;45:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.GTEx Project. Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat Genet 2017;49:1664–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Winkler TW, Gunther F, Hollerer S et al. A joint view on genetic variants for adiposity differentiates subtypes with distinct metabolic implications. Nat Commun 2018;9:1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell 2015;161:119–32. [DOI] [PubMed] [Google Scholar]
- 14. Yeo GS, Heisler LK. Unraveling the brain regulation of appetite: lessons from genetics. Nat Neurosci 2012;15:1343–9. [DOI] [PubMed] [Google Scholar]
- 15. Timper K, Bruning JC. Hypothalamic circuits regulating appetite and energy homeostasis: pathways to obesity. Dis Model Mech 2017;10:679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lenard NR, Berthoud HR. Central and peripheral regulation of food intake and physical activity: pathways and genes. Obesity (Silver Spring) 2008;16: S11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Auton A, Brooks LD, Durbin RM et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mahajan A, Taliun D, Thurner M et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikpay M, Goel A, Won HH et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shungin D, Winkler TW, Croteau-Chonka DC et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015;518:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Neale Lab. Rapid GWAS of thousands of phenotypes for 337,000 samples in the UK Biobank 2017. 2017. http://www.nealelab.is/blog/2017/7/19/rapid-gwas-of-thousands-of-phenotypes-for-337000-samples-in-the-uk-biobank (27 March 2020, date last accessed).
- 24. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. EPIC-InterAct Consortium. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 2015;30:543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noordam R, Smit RA, Postmus I, Trompet S, van Heemst D. Assessment of causality between serum gamma-glutamyltransferase and type 2 diabetes mellitus using publicly available data: a Mendelian randomization study. Int J Epidemiol 2016;45:1953–60. [DOI] [PubMed] [Google Scholar]
- 26. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 2017;46:1985–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corbin LJ, Richmond RC, Wade KH et al. BMI as a modifiable risk factor for type 2 diabetes: refining and understanding causal estimates using Mendelian randomization. Diabetes 2016;65:3002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hemani G, Zheng J, Elsworth B et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol 2011;40:755–64. [DOI] [PubMed] [Google Scholar]
- 32. Burgess S, Foley CN, Allara E, Staley JR, Howson J. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun 2020;11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.