

Abstract
Molecules containing trifluoromethoxyaryl groups are of interest in pharmaceutical, agrochemical, and materials science research, due to their unique physical and electronic properties. Many of the known methods to synthesize aryl trifluoromethyl ethers require harsh reagents and highly controlled reaction conditions and rarely occur when heteroaromatic units are present. The two-step O-trifluoromethylation of phenols via aryl xanthates is one such method that suffers from these drawbacks. Herein, we report a method for the synthesis of aryl trifluoromethyl ethers from phenols by the facile conversion of the phenol to the corresponding aryl and heteroaryl xanthates with newly synthesized imidazolium methylthiocarbonothioyl salts and conversion of these xanthates to the trifluoromethyl ethers under mild reaction conditions.
Graphical Abstract
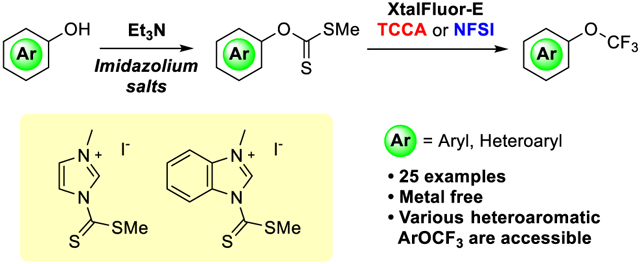
INTRODUCTION
Organic molecules containing fluorine are common in pharmaceutical, agrochemical, and materials science, due to their unique physical properties and bioactivities.1 Therefore, new synthetic methodologies to introduce fluorine into organic molecules have been studied extensively in recent years.2 The OCF3 group is of particular interest to pharmaceutical research, owing to its high electronegativity (χ = 3.7), unique orthogonal conformation relative to the aromatic ring,3 high lipophilicity,4 and high metabolic stability. Although the OCF3 group possesses an intriguing combination of properties, few pharmaceuticals exist that contain the moiety. The absence of such structures is due, in part, to the relatively harsh and toxic reaction conditions needed for their synthesis and lack of existing methods that tolerate heterocyclic diversity, both of which are important considerations in pharmaceutical research.
Known methods to form aryl trifluoromethyl ethers include silver-mediated direct trifluoromethylation of phenols with the Ruppert-Prakash reagent,5 silver-mediated trifluoromethoxylation of arylstannanes6a and aryldiazonium salts,6b C–H trifluoromethoxylation via redox-active catalysis or trifluoromethoxy migration,7 and fluorodecarboxylation of aryloxydi-fluoroacetic acids.8 Other useful methods9–11 have been reported, but many of them rely on harsh fluorinating reagents or are of narrow substrate scope.
In this work, we focused our efforts on utilizing readily available aromatic and heteroaromatic alcohols as precursors for the synthesis of aryl trifluoromethyl ethers via aryl and heteroaryl xanthates (Scheme 1). In their pioneering work on the subject, Hiyama and co-workers reported the conversion of phenols to trifluoromethoxy arenes via methylxanthates.9c,e The trifluoromethylation reactions, however, only proceeded with an exceptionally large excess of highly toxic HF-pyridine. Leroux and co-workers9f investigated this transformation further and determined that heterocyclic functionality, with the exception of 2-halo-azines, was not tolerated. (See the Supporting Information.) Some operational improvements to this procedure were made by Umemoto et al.,9g using FluoLead in combination with antimony trichloride, but the reported substrate scope for aryl xanthates was limited to a single unisolated example. Finally, to the best of our knowledge, there have been few advancements on the synthesis of aryl xanthates. Most preparations use a two-step protocol consisting of alcohol deprotonation with a strong base followed by condensation and alkylation with CS2 and MeI, respectively. In our experience, this reaction is highly sensitive to the steric and electronic properties of the phenol. (See the Supporting Information.)
Scheme 1.
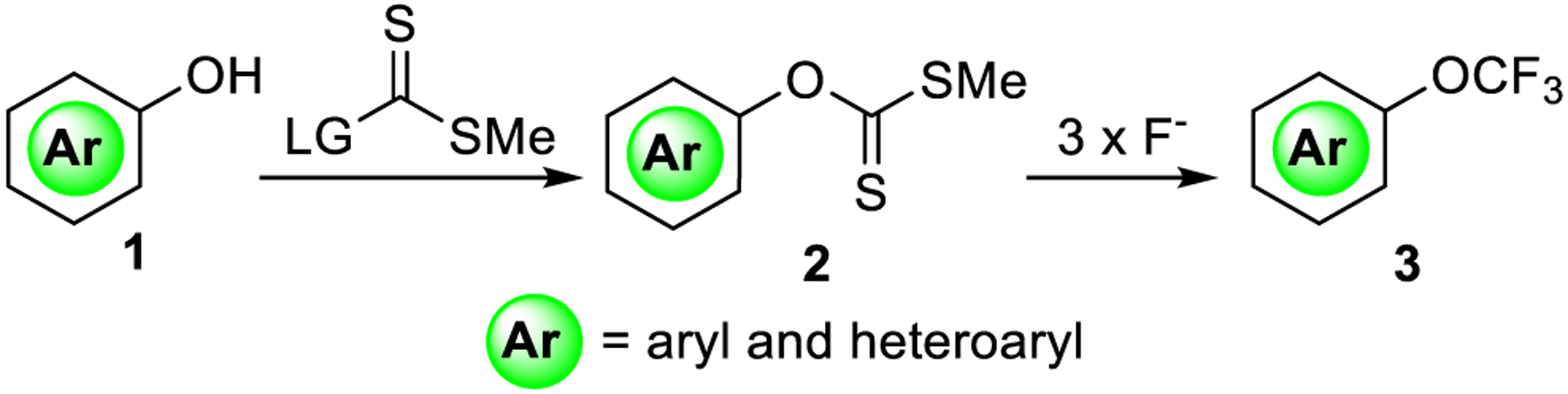
Two-Step Trifluoromethoxylation of Phenol
RESULTS AND DISCUSSION
We began our investigation by first developing a robust method to form xanthates from aromatic and heteroaromatic alcohols. Under standard conditions with NaH, CS2, and MeI, 4-dimethylaminocarboxy phenol 1a (Table 1) reacted to low conversion, likely due to the low nucleophilicity of 1a. To improve this transformation, we evaluated several potential reagents that could form xanthates. The reaction of 1a with commercially available imidazole carbodithioate 4,12 which is reported to be more reactive than CS2, did not proceed in appreciable yield (entry 1). The same reaction with benzotriazole analog 5 was much more effective and afforded 2a in 70% yield, along with unreacted starting material (entry 2). Therefore, we designed imidazolium and benzimidazolium analogs 6 and 7,13 which we expected to be more electrophilic than 5.14 After investigating the effects of added base, we found that the combination of imidazolium 6 and triethylamine formed xanthate 2a in 99% yield (entry 3). The analogous reaction with benzimidazolium salt 7 afforded 2a in lower yield (entry 4, 2a: 79%), due to a side reaction, which was later determined to be thiocarbonylation of the aromatic ring, owing to the stronger electrophilicity of 7 than of 6. The conversion of phenol 1a to the corresponding xanthate with 6 in MeCN was as effective as that in DMF (entry 5, 2a: 98%). The analogous reaction of the 4-alkoxycarbonyl-substituted phenol 1b with 6 and 7 also formed the xanthate in a high yield (91% and 99%, entries 6 and 7). The reactions with these reagents to form aromatic xanthates are operationally simple and convenient.
Table 1.
Reactions of Phenols with a Series of Xanthalating Reagentsa
 | |||||
|---|---|---|---|---|---|
| entryb | phenol | reagent | base | solvent | yieldc |
| 1 | 1a | 4 | NaH | DMF | 2a: 5%d |
| 2 | 1a | 5 | NaH | DMF | 2a: 70% |
| 3 | 1a | 6 | Et3N | DMF | 2a: 99% |
| 4 | 1a | 7 | Et3N | DMF | 2a: 79% |
| 5 | 1a | 6 | Et3N | MeCN | 2a: 98% |
| 6 | 1b | 6 | Et3N | MeCN | 2b: 91% |
| 7 | 1b | 7 | Et3N | MeCN | 2b: 99% |
LG: leaving group.
Standard reaction conditions: 1 (0.5 mmol), xanthate-forming reagent (0.5 mmol), base (0.6 mmol), and solvent (5 mL).
Isolated yields.
Yield was determined by 1H NMR using Me2SO2 as an internal standard instead of isolated yield.
Having established reliable conditions for the formation of aromatic and heteroaromatic xanthates from phenols, we investigated the conversion of 4-dimethylaminocarbonyl phenoxyxanthate 2a to the corresponding aryl trifluoromethyl ether (Table 2). XtalFluor-E, a commercially available, free-flowing solid, was chosen as a fluoride source due to its good stability under air and low proclivity for thermal degradation.15 Control investigations with other potential fluoride sources, including PyFluor, HF/KF, HF/Pyr, DeoxoFluor, DAST, XtalFluor-M, and XtalFluor-E with TCCA showed that the yield of the trifluoromethyl ether from xanthate 2a was the highest with XtalFluor-E as the source of fluoride. (See the Supporting Information.)
Table 2.
Development of Conditions for the Conversion of Aryl Xanthates to Aryl Trifluoromethyl Ether
 | ||||
|---|---|---|---|---|
| entrya | xanthate | X+ source (equiv) | additive (equiv) | yieldb |
| 1 | 2a | I2 (3.0) | none | 3a: 0% |
| 2 | 2a | NBS (3.0) | none | 3a: 4% |
| 3 | 2a | DBH (1.5) | none | 3a: 6% |
| 4 | 2a | DBCA (1.5) | none | 3a: 13% |
| 5 | 2a | NFSI (3.0) | none | 3a: 9% |
| 6 | 2ad | TCCA (1.0) | none | 3a: 78% |
| 7c | 2ad | TCCA (1.0) | none | 3a: 32% |
| 8 | 2a | TCCA (1.0) | H2O (1.0) | 3a: 76% |
| 9 | 2bd | TCCA (1.0) | none | 3b: 56% |
| 10 | 2bd,e | NFSI (3.0) | none | 3b: 75% |
| 11 | 2bd,e | NFSI (3.0) | H2O (1.0) | 3b: 70% |
| 12 | 2cd | TCCA (1.0) | none | 3c: 56% |
| 13 | 2cd,e | NFSI (3.0) | none | 3c: 80% |
| 14 | 2cd,e | NFSI (3.0) | H2O (1.0) | 3c: 75% |
Standard conditions: 2 (50 mmol, 1.0 equiv), XtalFluor-E (250 mmol, 5.0 equiv), X+ source (50–150 mmol, 1–3 equiv), additives, (CH2Cl)2 (1 mL), 80 °C, 3 h.
Yields were determined by 19F NMR using PhCF3 as an internal standard.
The reaction was conducted in a glovebox.
0.5 mmol of 2 was used.
3.0 equiv of XtalFluor-E was used.
To optimize the formation of trifluoromethyl ether 3a from xanthate 2a with XtalFluor-E, we tested a series of reactions with various halonium oxidants as soft Lewis acids. Previous computational studies by Kepp16 showed that halogen atoms have a relatively high thiophilicity and are particularly useful Lewis acids toward thiocarboyls. Hiyama and co-workers9c,e demonstrated this principal when they utilized various sources of electrophilic bromides (i.e., NBS or DBH) to accelerate the fluorination of xanthates in the presence of HF-pyridine. Under our conditions, the yields of the reactions with fluorinating, brominating, and iodinating reagents as Lewis acid were low (entries 1–6); in contrast, the yield of the aryl trifluoromethyl ether from the reaction with trichloroisocyanuric acid (TCCA) was high (entry 6, 3a: 78%). The same reaction conducted in a glovebox gave 2a in a significantly lower 32% yield (entry 7). On the basis of the hypothesis that water in the air facilitates the reaction, we found that reactions with added water (1.0 equiv) were reproducible and scalable (entry 8, 3a: 76%).17
The reaction of the more electron-deficient 4-ethoxycarbonyl phenoxyxanthate 2b occurred in high yield under slightly different conditions. We found that the reaction of 4-ethoxycarbonyl phenoxyxanthate 2b with XtalFluor-E and TCCA afforded a difluorochloromethyl ether (ArOCF2Cl; 18%) side product, in addition to the desired trifluoromethyl ether (entry 9, 2b: 56%). In light of this result, fluoronium reagents, such as N-fluorobenzenesulfoneiminde (NFSI) and Selectfluor, were investigated as alternative halonium Lewis acid sources. Although reactions were slower (24 h), the treatment of 2b with 3 equiv of NFSI and XtalFluor-E afforded trifluoromethyl ether 3b in a high 75% yield (entry 10). Under these conditions with XtalFluor-E, the yield of the reaction was the same with or without 1 equiv of added water (entry 11, 2b: 70%). The reaction of the more electron-rich 4-adamantyl phenoxyxanthate 2c was similar to that of 2b. The reaction of 2c with XtalFluor-E and TCCA formed both the difluorochloromethyl ether and the trifluoromethyl ether (Table 2; entry 12, 2c: 56%), whereas the reaction of 2c with NFSI led to the desired 3c without the chlorinated side product (Table 2; entry 13, 2c: 80%).
The scope of the conversion of aromatic and heteroaromatic alcohols to the corresponding aryl trifluoromethyl ethers under the conditions discovered for the two-step trifluoromethoxylation of phenols is shown in Table 3. This operationally simple method allowed for the facile formation of a broad range of xanthates from the corresponding phenols, using a minimal amount of reagent 6 or 7 (1 equiv) and mild base (1.1 equiv) in MeCN. Phenols 1a–1w were converted to the corresponding xanthates 2a–2w in over 90% yield with reagent 6, with a few exceptions. The formation of xanthates from electron-poor phenols, such as ethylbenzoate 2c, chromenone 2h, and hydroxypyrazole 2w, proceeded in higher yield with the more electrophilic benzimidazole-derived reagent 7. The formation of the xanthate from 2,4-di-t-butylphenol 1f with either reagent 6 or 7 resulted in poor yield, because decomposition of the xanthalating reagents is faster than the desired xanthate formation. (See the Supporting Information.) Instead, formation of 2f occurred quantitatively with the less electrophilic benzotriazole 5.
Table 3.
Substrate Scope of Two-Step Trifluoromethylation of Phenol

|
Standard condition of xanthate formation: 1 (2.0 mmol), 6 (2.0 mmol), Et3N (2.0 mmol), MeCN (10 mL), 0 °C, 1 h.
Condition A: 2 (0.5 mmol), XtalFluor-E (2.5 mmol), TCCA (0.5 mmol), H2O (0.5 mmol), and (CH2Cl)2 (10 mL), 80 °C.
Condition B: 2 (0.5 mmol), XtalFluor-E (1.5 mmol), NFSI (1.5 mmol), and (CH2Cl)2 (10 mL), 80 °C.
NMR yields determined by 19F NMR using PhCF3 as an internal standard were reported in the parentheses.
Reagent 7 was used instead of 6.
Reagent 5 (1.5 equiv), Cs2CO3 (2.0 equiv), and DMF (0.1 M) were used instead of the standard conditions.
Selectfluor (0.5 mmol, 1.0 equiv) was added.
Selectfluor (1.0 mmol, 2.0 equiv) was added.
The conversion of the xanthates produced by this method to the corresponding aryl trifluoromethyl ethers was performed under one of two conditions: condition A, with XtalFluor-E (5 equiv), TCCA (1 equiv), and H2O (1 equiv) in 1,2-dichloroethane at 80 °C for 3–12 h, or condition B, with XtalFluor-E (3 equiv) and NFSI (3 equiv) in 1,2-dichloroethane at 80 °C for 12–48 h. All the reactions were performed under atmospheric conditions with no added controls. Generally, the reactions with substrates containing Lewis-basic functional groups, such as amides, nitriles, or nitrogen-containing heteroaromatic rings, occurred in higher yield under condition A than under condition B. Reactions of substrates lacking Lewis basic functionality occurred in higher yield under condition B than under condition A.
Using condition A or B, we synthesized a variety of aryl trifluoromethyl ethers from the corresponding xanthates in modest to excellent yields. In several cases, the isolated yields were lower than the yields determined by NMR spectroscopy because of the low boiling point of the product or the necessity for preparative SFC or both. Both electron-poor and electron-rich xanthates readily reacted to yield the desired aryl trifluoromethyl ethers. For certain substrates containing electron-rich aromatic rings, electrophilic chlorination reactions were observed as side processes from the reaction with TCCA. This side reaction could be avoided by utilizing condition B, with NFSI in place of TCCA (for example: 3f, 56%; 3g: 65%). Trifluoromethoxylation of more electron-neutral 4-bromophenol and 4-iodophenol also reacted smoothly to afford useful building blocks 3k and 3l in 66% and 67% yield, respectively.
A few examples of the conversion of aromatic alcohols containing heterocyclic substituents or heteroaromatic alcohols to the corresponding trifluoromethyl ethers have been reported.5,11 We found that the conversion of xanthates derived from these classes of alcohols to the corresponding trifluoromethyl ether occurred under both conditions A and B. The reactions of 2m–2q, in which the aryl xanthate is substituted with a heteroaromatic substituent, afforded the corresponding trifluoromethyl ethers. Likewise, xanthates derived from phenols substituted with six-membered heteroarenes, such as 2o, 2p, and 2q, formed the corresponding heteroaryl trifluoromethyl ethers in good yield.
Heteroaromatic xanthates, in which the xanthyl group is connected directly to a heteroaromatic ring, such as a pyridine or quinoline, also formed the corresponding trifluoromethyl ethers. Although yields were modest in most cases, these reactions enabled the conversions of heteroaromatic alcohols to several previously unknown trifluoromethoxy heteroarenes (3r, 3u–3w). In certain instances, such as compound 3s, the volatility prevented complete isolation, even though the product was formed in substantial yield, as determined by 1H and 19F NMR spectroscopy. Perhaps most notable product from reaction of a heteroaromatic xanthate is example 3w. To the best of our knowledge, this example represents the first known preparation of a five-membered, nitrogen-containing heterocycle containing an OCF3 group derived from the corresponding alcohol.
Observations made during the fluorination reactions of xanthates provided insight into a potential reaction mechanism. For example, reactions under condition A gave Ar-OCF2Cl as a byproduct in certain cases.18 In addition, under condition A, the reactions with one equivalent of water occurred in higher yields than those lacking added water. These data suggest that the reactions conducted under conditions A and B proceed through slightly different pathways (Scheme 2).
Scheme 2.

Likely Mechanism for Conversion of Aryl Xanthate to Aryl Trifluoromethyl Ether
The first step of the mechanism is likely coordination of the thiocarbonyl group of the xanthates to TCCA or NFSI to form activated species 9. Nucleophilic addition of fluoride then would yield monofluoro intermediate 10. Two additional substitutions of fluoride would ultimately afford the desired trifluoromethyl ether 3. It is possible in the last step involving difluoro intermediate 11 that homolytic cleavage of the C–S bond and trapping with a source of Cl· occurs in competition with nucleophilic substitution by fluoride. Consistent with the possibility of these two parallel pathways for the final step, no byproduct 8 was observed in the presence of BHT.
Further analysis of the reaction mixture with xanthate 2a revealed the presence of carbamothioate byproduct 12a (Scheme 3). The formation of 12a can be attributed to the liberation of diethylamine from the degradation of XtalFluor-E, followed by a Newman-Kwart-type rearrangement.19 Indeed, a control experiment conducted with 2a, Et2NH, and TCCA produced the byproduct 12a, along with the hydrolyzed product 1a. From the water present in condition A, a small amount of HF is likely generated, and this HF would protonate the diethylamine to suppress this side reaction. It is uncertain why added water does not have the same effect on the reaction by path B, but it is possible that the smaller amount of XtalFluor-E and lower Lewis acidity of NFSI causes the competing Newman-Kwart-type rearrangement to be slower.
Scheme 3.

Control Experiment with Diethylamine
In summary, we have described an improved, two-step trifluoromethoxylation of phenols via xanthate intermediates by simple procedures and easily-handled reagents. In the first step, phenols or heteroaryl alcohols react with imidazolium salt 6 or benzimidazolium salt 7 to form xanthates in high yield. In the second step, XtalFluor-E reacts with xanthate in the presence of trichloroisocyanuric acid (TCCA) or N-fluorosulfonimide (NFSI) to form a range of aryl and heteroaryl trifluoromethyl ethers from common phenols and heteroaromatic alcohols. All reactions were performed under atmospheric conditions with no added controls. This methodology should be useful for pharmaceutical, agrochemical, and materials science research in which aromatic and heteroaromatic trifluoromethyl ethers are highly desired.
EXPERIMENTAL SECTION
General Remarks.
All xanthate formations were conducted with N2-flushed round-bottom flasks. The trifluoromethylation was conducted with 20 mL screw cap vials in air. All the solvents for the reactions were purchased from Sigma-Aldrich (anhydrous grade equipped sure seal) and used as received. Unless otherwise stated, the reaction temperatures above 23 °C refer to the temperatures of an aluminum heating block, which were controlled by an electronic temperature modulator. NMR spectra were recorded on Bruker AVQ-400 and AV-600 instruments at UC Berkeley and a 500 MHz Bruker Avance III with a CryoProbe at Kyushu University. Chemical shifts (δ) are reported in ppm, relative to the residual solvent signal. Data from 1H NMR spectra are reported as follows: chemical shift (multiplicity, coupling constants, number of hydrogens). Abbreviations are as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and bs (broadening singlet). All 13C NMR spectra were proton-decoupled. GC-MS data were obtained on an Agilent 6890-N GC system containing an Alltech EC-1 capillary column and an Agilent 5973 mass-selective detector. High-resolution electron impact (EI) mass spectral data were obtained from the University of California, Berkeley Mass Spectrometry Laboratory. High-resolution electron spray ionization (ESI) TOF-MS and atmospheric pressure chemical ionization (APCI) TOF-MS were obtained from the Lawrence-Berkeley National Laboratory Catalysis Center.
Synthesis of Phenols.
1a, 1b, 1c, 1d, 1e, 1f, 1g, 1h, 1i, 1j, 1k, 1l, 1m, 1n, 1q, 1s, 1t, 1u, and 1v were commercially available. The syntheses of 1o, 1p, 1r, and 1w are described below.
3-Hydroxy-6-dimethylaminocarbonylpyridine (1r).
Ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI, 2.11 g, 11.0 mmol, 1.1 equiv) was added to a solution of 5-hydroxypicolinic acid (1.39 g, 10.0 mmol, 1.0 equiv), dimethylamine hydrochloride (897 mg, 11.0 mmol, 1.1 equiv), 4-dimethylaminopyridine (DMAP, 1.22 g, 10.0 mmol, 1.0 equiv), and diisopropylethylamine (3.8 mL, 22 mmol, 2.2 equiv) in CH2Cl2 (20 mL) at room temperature. After stirring at room temperature for 12 h, the solution was directly poured onto a silica gel column and purified (EtOAc/MeOH 100:0 to 80:20) to give 1.00 g of 1r (61%); white crystal; 1H NMR (600 MHz, CDCl3) δ 10.38 (bs, 1H), 8.09 (d, J = 2.9 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.23 (dd, J = 8.4, 2.9 Hz, 1H), 3.00 (bs, 3H), 2.96 (bs, 3H); 13C NMR (151 MHz, CDCl3) δ 167.9, 154.3, 145.1, 135.9, 124.7, 122.6, 38.6, 35.2; HRMS (ESI), calcd for C8H11N2O2+ (M + H)+ 167.0815, found 167.0818.
4-(6-Chloropyridin-3-yl)phenol (1o).
Procedure A: To a 100 mL single-neck round-bottom flask, 4-hydroxyphenylboronic acid (690 mg, 6.00 mmol, 1.2 equiv), 2-chloro-5-bromopyridine (962 mg, 5.00 mmol, 1.0 equiv), sodium carbonate (1.59 g, 15.0 mmol, 3.0 equiv), 1,4-dioxane (19 mL), and H2O (6 mL) were added. The mixture was degassed by bubbling with nitrogen for 30 min. [1,1′-Bis-(diphenylphosphino)ferrocene]dichloropalladium (PdCl2(dppf), 73.2 mg, 2 mol %) was added to the mixture at room temperature. The mixture was warmed at 80 °C, stirred for 24 h, and then cooled to room temperature. The resulting mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (EtOAc/hexane 50:50 to 100:0) to give 905 mg of phenol 1o (88%); white crystal; 1H NMR (500 MHz, (CD3)2SO) δ 9.76 (s, 1H), 8.64 (d, J = 2.5 Hz, 1H), 8.05 (dd, J = 8.2, 2.5 Hz, 1H), 7.58–7.54 (m, 2H), 7.52 (d, J = 8.2 Hz, 1H), 6.90–6.86 (m, 2H); 13C NMR (126 MHz, (CD3)2SO) δ 158.1, 148.1, 147.0, 136.9, 135.0, 128.1, 126.2, 124.2, 116.0; HRMS (ESI), calcd for C11H9ClNO+ (M + H)+ 206.0367, found 206.0368.
4-(Pyridin-3-yl)phenol (1p).
Procedure A was followed for the reaction of 3-bromopyridine (790 mg, 5.00 mmol). The title compound 1p was obtained in 94% yield; white crystal; 1H NMR (500 MHz, (CD3)2SO) δ 9.69 (s, 1H), 8.81 (d, J = 1.9 Hz, 1H), 6.98 (dd, J = 4.7, 1.3 Hz, 1H), 7.97 (ddd, J = 7.9, 1.9, 1.3 Hz, 1H), 7.57–7.53 (m, 2H), 7.42 (dd, J = 7.9, 4.7 Hz, 1H), 6.90–6.86 (m, 2H); 13C NMR (126 MHz, (CD3)2SO) δ 157.6, 147.4, 147.0, 135.6, 133.1, 128.0, 127.7, 123.8, 115.9; HRMS (ESI), calcd for C11H10NO+ (M + H)+ 172.0757, found 172.0756.
1-(4-Chlorophenyl)-1H-pyrazol-4-ol (1w).
Ethyl-4-chloroacetoacetate (2.7 m L, 20 mmol) was added to aqueous conc. HCl (5 mL) at room temperature. After stirring at room temperature for 24 h, the solution was poured into ice and extracted with EtOAc (3 × 20 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated under high vacuum at 0 °C to give 4-chloroacetoacetic acid, which was diluted with 6 mL of water (solution A).
Sodium nitrite (1.38 g, 20.0 mmol, 1.0 equiv) was added dropwise to a solution of p-chloroaniline (2.52 g, 20.0 mmol, 1.0 equiv), conc. HCl (5 mL), and H2O (20 mL) at 0 °C (solution B). After maintaining solution B for 30 min at 0 °C, this solution was transferred into solution A by pipet and then stirred for 10 min. An aqueous 6 M solution of sodium acetate (10 mL) was added to the resulting solution dropwise. (CAUTION: CO2 bubbles violently.) After stirring for 1 h, the precipitate in the mixture was collected by filtration, washed with H2O, and dried under high vacuum to give 2.02 g of (E)-1-chloro-3-(2-(4-chlorophenyl)hydrazineylidene)-propan-2-one (13) (58%); yellow powder; 1H NMR (600 MHz, CDCl3) δ 11.67 (bs, 1H), 7.38–7.35 (m, 2H), 7.31 (d, J = 1.3 Hz, 1H), 7.25–7.22 (m, 2H), 4.87 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 189.2, 141.8, 132.6, 129.3, 125.7, 115.4, 45.2; HRMS (ESI), calcd for C9H8Cl2N2ONa+ (M + Na)+ 252.9906, found 252.9900.
Sodium hydroxide (1.00 g, 25.0 mmol, 3.2 equiv) was added to a solution of 13 (1.82 g, 7.88 mmol, 1.0 equiv) and MeOH (50 mL) at room temperature, causing the temperature rise to approximately 40 °C. After stirring for 1 h at room temperature, the mixture was concentrated and diluted with water (10 mL). Insoluble material was removed by filtration. The solution then was neutralized with conc. HCl. The resulting precipitate was filtered, washed with water, and dried under high vacuum to give 1.51 g of 4-hydroxylpyrrazole 1w (99%); pale-yellow powder; 1H NMR (600 MHz, CDCl3) δ 7.53 (s, 1H), 7.38–7.35 (m, 2H), 7.53–7.48 (m, 2H), 7.41 (s, 2H), 7.40–7.36 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 143.1, 138.8, 131.9, 131.6, 129.7, 119.9, 114.0; HRMS (ESI), calcd for C9H8ClN2O+ (M + H)+ 195.0320, found 195.0319.
Synthesis of Xanthate Forming Reagents.
Reagent 4 is commercially available. N-(Methylthiothiocarbonyl)benzotriazole 5 was synthesized following a reported procedure.20 The methods to synthesize imidazolium reagent 6 and benzimidazolium reagent 7 are described below.
Imidazolium Salt (6).
A 250 mL single-neck-shield flask equipped with a stir bar and septum was purged with nitrogen. Methylthio-(thiocarbonyl)imidazole 4 (7.09 g, 44.9 mmol, 1.0 equiv)21 in benzene (120 mL) was transferred to the flask via cannula. Iodomethane (28 mL, 450 mmol, 10 equiv) was added to the mixture. The septum was replaced with a screw cap quickly. The resulting mixture was warmed to 80 °C with an oil bath. While stirring for 3 h, an orange precipitate formed. The precipitate was filtered, washed with anhydrous benzene (100 mL), and dried under high vacuum to give 6.44 g of imidazolium reagent 6 (48%); a bright orange solid; mp. 89 °C; 1H NMR (600 MHz, (CD3)2SO), δ 10.14 (bs, 1H), 8.50 (dd, J = 2.2, 1.5 Hz, 1H), 7.97 (dd, J = 2.2, 1.5 Hz, 1H), 3.94 (s, 3H), 2.95 (s, 3H); 13C NMR (151 MHz, (CD3)2SO) δ 197.6, 136.3, 125.1, 119.5, 36.7, 21.2; HRMS (ESI), calcd for C6H9N2S2+ (M + H)+ 173.0202, found 173.0208.
Benzimidazolium Salt (7).
A 250 mL single-neck-shield flask equipped with a stir bar and septum was purged with nitrogen. N-Methylthio(thiocarbonyl)-benzimidazole (9.00 g, 43.2 mmol, 1.0 equiv)22 in MeCN (90 mL) was transferred to the flask via cannula. Iodomethane (22 mL, 350 mmol, 8.0 equiv) was added to the mixture. The septum was replaced with a screw cap quickly. The resulting mixture was warmed to 80 °C with an oil bath. While stirring for 4.5 h, a bright orange precipitate was generated. The precipitate was filtered, washed with EtOAc/hexane 3:1 (100 mL), and dried under high vacuum to give 10.9 g of benzimidazolium reagent 7 (72%) as a yellowish orange solid; mp. 140 °C; 1H NMR (500 MHz, CDCl3, rotamer was observed) δ 10.629 (bs, 1/2H), 10.627 (bs, 1/2H), 8.59–8.54 (m, 1H), 8.17–8.12 (m, 1H), 7.84–7.79 (m, 2H), 4.182 (s, 3/2H), 4.181 (s, 3/2H), 3.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.0, 143.0, 132.7, 129.4, 128.5, 127.8, 116.0, 114.5, 34.0, 21.5; HRMS (ESI), calcd for C10H11N2S2+ (M + H)+ 223.0358, found 223.0355.
General Procedure B for the Formation of Xanthates.
Reagent 6 or 7 (2.00 mmol, 1.0 equiv) was added to a solution of the phenol 1a–1w (2.00 mmol, 1.0 equiv), triethylamine (330 μL, 2.2 mmol, 1.1 equiv), and MeCN (10 mL) at 0 °C. After stirring at 0 °C for 1 h, the mixture was quenched with saturated aqueous NaHCO3 (20 mL) and extracted with EtOAc (2 × 20 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography or recrystallization to give xanthates 2a–2w.
4-Dimethylaminocarbonylphenoxy Xanthate (2a).
General Procedure B was followed for the reaction of 4-dimethylaminocarbonyl phenol 1a (330 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:1) to give the product as a white solid (510 mg, >99%). 2a: 1H NMR (600 MHz, CDCl3) δ 7.48 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 3.10 (bs, 3H), 3.01 (bs, 3H); 13C NMR (151 MHz, CDCl3) δ 215.4, 170.7, 155.3, 134.6, 128.8, 122.3, 39.8, 35.6, 20.2; HRMS (ESI), calcd for C11H14NO2S2+ (M + H)+ 256.0460, found 256.0464.
4-Ethoxycarbonylphenoxy Xanthate (2b).
General Procedure B was followed for the reaction of 4-ethoxycarbonylphenol 1b (332 mg, 2.00 mmol) and benzimidazolium reagent 7 (700 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:9) to give the product as a white solid (510 mg, 99%). 2b: 1H NMR (600 MHz, CDCl3) δ 8.14–8.10 (m, 2H), 7.19–7.15 (m, 2H), 4.39 (q, J = 6.9, 2H), 2.68 (s, 3H), 1.39 (t, J = 6.9, 3H); 13C NMR (151 MHz, CDCl3) δ 215.3, 165.8, 158.0, 131.3, 128.9, 122.4, 61.3, 20.2, 14.5; HRMS (ESI), calcd for C11H12O3S2Na+ (M + Na)+ 279.0120, found 279.0115.
4-Adamantylphenoxy Xanthate (2c).
General Procedure B was followed for the reaction of 4-adamantylphenol 1c (456 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 0:1 to 1:100) to give the product as a white solid (598 mg, 94%). 2c: 1H NMR (600 MHz, CDCl3) δ 7.41–7.38 (m, 2H), 7.05–7.02 (m, 2H), 2.67 (s, 3H), 2.12–2.08 (m, 3H), 1.92 (d, J = 2.2 Hz, 6H), 1.82–1.72 (m, 6H); 13C NMR (151 MHz, CDCl3) δ 216.0, 152.5, 149.8, 126.2, 121.4, 43.4, 36.9, 36.3, 29.1, 20.1; HRMS (ESI), calcd for C18H23OS2+ (M + H)+ 319.1185, found 319.1186.
4-Cyanophenoxy Xanthate (2d).
General Procedure B was followed for the reaction of 4-cyanophenol 1d (238 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:1) to give the product as a white solid (351 mg, 84%). 2d: 1H NMR (600 MHz, CDCl3) δ 7.75–7.71 (m, 2H), 7.25–7.22 (m, 2H), 2.68 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 214.9, 157.6, 133.9, 123.7, 118.2, 110.7, 20.3; HRMS (ESI), calcd for C9H7NOS2Na+ (M + Na)+ 231.9861, found 231.9854.
Phenoxy Xanthate (2e).
General Procedure B was followed for the reaction of phenol 1e (376 mg, 4.00 mmol) and imidazolium reagent 6 (1.20 g, 4.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a pale-yellow oil (676 mg, 92%). 2e: 1H NMR (600 MHz, CDCl3) δ 7.45–7.41 (m, 2H), 7.30 (tt, J = 7.7, 1.1 Hz, 1H), 7.13–7.09 (m, 2H), 2.68 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.9, 154.8, 129.7, 126.7, 122.2, 20.1; HRMS (EI), calcd for C8H8OS2+ (M)+ 184.0017, found 184.0015.
2,4-Di-t-butylphenoxy Xanthate (2f).
Cesium carbonate (1.30 g, 4.00 mmol, 2.0 equiv) was added to a single-neck flask, dried by heat gun under vacuum, cooled, and filled back with nitrogen. 2,4-Di-t-butyl-phenol 1f (413 mg, 2.00 mmol, 1.0 equiv) and DMF (20 mL) were added to the flask. The resulting mixture was cooled to 0 °C, and benzotriazole reagent 5 (628 mg, 3.00 mmol, 1.5 equiv) was added. After stirring for 3 h at 0 °C, the mixture was treated with saturated aqueous NaHCO3 and extracted with hexane (20 mL × 3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (EtOAc/hexane 0:100) to give the product as a white solid (587 mg, 99%). 2f: 1H NMR (600 MHz, CDCl3) δ 7.46–7.44 (m, 1H), 7.26–7.23 (m, 1H), 7.01–6.98 (m, 1H), 2.70 (s, 3H), 1.38 (s, 9H), 1.35 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 215.5, 151.0, 149.0, 140.4, 124.6, 123.69, 123.68, 34.91, 34.88, 31.6, 30.6, 20.1; HRMS (ESI), calcd for C16H24OS2Na+ (M + Na)+ 319.1161, found 319.1170.
4-(Methoxycarbonylethyl)phenoxy Xanthate (2g).
General Procedure B was followed for the reaction of methyl 3-(4-hydroxyphenyl)propanoate 1g (360 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:9) to give the product as a white solid (524 mg, 97%). 2g: 1H NMR (600 MHz, CDCl3) δ 7.26–7.23 (m, 2H), 7.04–7.00 (m, 2H), 3.67 (s, 3H), 2.97 (t, J = 7.7 Hz, 2H), 2.66 (s, 3H), 2.65 (t, J = 7.7 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 216.0, 173.2, 153.2, 139.0, 129.5, 122.1, 51.8, 35.6, 34.88, 31.6, 30.6, 20.1; HRMS (ESI), calcd for C12H14O3S2Na+ (M + Na)+ 293.0277, found 293.0278.
O-Methylthiocarbonyl-7-hydroxycoumarin (2h).
General Procedure B was followed for the reaction of 7-hydroxycoumarin (umbelliferone) 1h (324 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:9 to 1:3) to give the product as a white solid (490 mg, 97%). 2h: 1H NMR (600 MHz, CDCl3) δ 7.71 (d, J = 9.5 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.11 (d, J = 1.8 Hz, 1H), 7.06 (dd, J = 8.4, 1.8 Hz, 1H), 6.43 (d, J = 9.5 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 215.1, 160.3, 156.8, 154.9, 142.9, 128.8, 119.2, 117.4, 116.6, 111.4, 20.3; HRMS (ESI), calcd for C11H9O3S2+ (M + H)+ 252.9988, found 252.9982.
4-Benzoylphenoxy Xanthate (2i).
General Procedure B was followed for the reaction of 4-benzoylphenol 1i (396 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:3 to 1:2) to give the product as a white solid (538 mg, 93%). 2i: 1H NMR (600 MHz, CDCl3) δ 7.91–7.88 (m, 2H), 7.81 (dd, J = 8.4, 1.4 Hz, 2H), 7.60 (tt, J = 7.4, 1.4 Hz, 1H), 7.49 (dd, J = 8.4, 7.4 Hz, 1H), 7.24–7.21 (m, 2H), 2.69 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.2, 195.5, 157.7, 137.5, 135.7, 132.6, 131.8, 130.0, 128.5, 122.3, 20.2; HRMS (ESI), calcd for C15H12O2S2Na+ (M + Na)+ 311.0171, found 311.0168.
4-Trifluoromethylphenoxy Xanthate (2j).
General Procedure B was followed for the reaction of 4-trifluoromethylphenol 1j (324 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a pale-yellow oil (475 mg, 94%). 2j: 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 2.69 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 215.3, 156.9, 128.9 (q, J = 33 Hz, C), 127.1 (q, J = 3.6 Hz, CH), 123.9 (q, J = 272 Hz, CF3), 123.0 20.2; 19F NMR (470 MHz, CDCl3) δ 62.3 (s); HRMS (EI), calcd for C9H7F3OS2 251.9890, found 251.9894.
4-Bromophenoxy Xanthate (2k).
General Procedure B was followed for the reaction of 4-bromophenol 1k (346 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a pale-yellow oil (525 mg, >99%). 2k: 1H NMR (600 MHz, CDCl3) δ 7.55–7.52 (m, 2H), 7.01–6.97 (m, 2H), 2.67 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.6, 153.7, 132.8, 124.1, 120.0, 20.2; HRMS (EI), calcd for C8H7BrOS2 261.9122, found 261.9124.
4-Iodophenoxy Xanthate (2l).
General Procedure B was followed for the reaction of 4-iodophenol 1l (440 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a white solid (602 mg, 97%). 2l: 1H NMR (600 MHz, CDCl3) δ 7.75–7.71 (m, 2H), 6.89–6.85 (m, 2H), 2.67 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.5, 154.5, 138.8, 124.5, 91.0, 20.2; HRMS (EI), calcd for C8H7IOS2 309.8983, found 309.8988.
4-(1H-Imidazol-1-yl)phenoxy Xanthate (2m).
General Procedure B was followed for the reaction of 4-(1H-imidazol-1-yl)phenol 1m (320 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:2 to 1:1) to give the product as a white solid (500 mg, >99%). 2m: 1H NMR (600 MHz, CDCl3), δ 7.86 (dd, J = 1.2, 1.2 Hz, 1H), 7.47–7.42 (m, 2H), 7.28 (dd, J = 1.2, 1.2 Hz, 1H), 7.25–7.21 (m, 3H), 2.69 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.8, 153.4, 135.7, 135.6, 130.7, 123.8, 122.7, 118.4, 20.2; HRMS (ESI), calcd for C11H11N2OS2+ (M + H)+ 251.0307, found 251.0304.
4-(Benzothiazole-2-yl)phenoxy Xanthate (2n).
General Procedure B was followed for the reaction of 4-(benzothiazole-2-yl)phenol 1n (422 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:20 to 1:10) to give the product as a white solid (549 mg, 91%). 2n: 1H NMR (600 MHz, CDCl3) δ 8.18–8.15 (m, 2H), 8.08 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.51 (dd, J = 8.1, 7.3 Hz, 1H), 7.40 (dd, J = 8.1, 7.3 Hz, 1H), 7.27–7.23 (m, 2H), 2.70 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.5, 166.8, 156.6, 154.2, 135.3, 129.0, 126.6, 125.5, 123.5, 123.1, 121.8, 20.2; HRMS (ESI), calcd for C15H12NOS3+ (M + H)+ 318.0076, found 318.0081.
4-(6-Chloropyridine-3-yl)phenoxy Xanthate (2o).
General Procedure B was followed for the reaction of 4-(6-chloropyridine-3-yl)phenol 1o (411 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:1 to 1:2) to give the product as a white solid (575 mg, 97%). 2o: 1H NMR (600 MHz, CDCl3) δ 8.61 (dd, J = 2.6, 0.7 Hz, 1H), 7.84 (dd, J = 8.4, 2.6 Hz, 1H), 7.61–7.58 (m, 2H), 7.41 (dd, J = 8.4, 0.7 Hz, 1H), 7.24–7.21 (m, 2H), 2.70 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.9, 155.0, 150.8, 148.1, 137.3, 135.0, 134.8, 128.4, 124.4, 123.2, 20.2; HRMS (ESI), calcd for C13H11ClNOS2+ (M + H)+ 295.9965, found 295.9961.
4-(Pyridine-3-yl)phenoxy Xanthate (2p).
General Procedure B was followed for the reaction of 4-(pyridine-3-yl)phenol 1p (342 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:1 to 1:2) to give the product as a yellow oil (499 mg, 95%). 2p: 1H NMR (600 MHz, CDCl3) δ 8.85 (d, J = 2.2 Hz, 1H), 8.61 (dd, J = 4.8, 1.5 Hz, 1H), 7.88 (ddd, J = 7.7, 2.2, 1.8 Hz, 1H), 7.65–7.61 (m, 2H), 7.38 (dd, J = 7.7, 4.8 Hz, 1H), 7.24–7.21 (m, 2H), 2.69 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.9, 154.8, 148.7, 148.3, 136.3, 135.8, 134.6, 128.4, 123.8, 123.0, 20.2; HRMS (ESI), calcd for C13H12NOS2+ (M + H)+ 262.0355, found 262.0361.
4-(Pyrimidine-5-yl)phenoxy Xanthate (2q).
General Procedure B was followed for the reaction of 4-(pyrimidine-5-yl)phenol 1q (344 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:2 to 1:4) to give the product as a white solid (445 mg, 85%). 2q: 1H NMR (600 MHz, CDCl3) δ 9.22 (s, 1H), 8.97 (s, 2H), 7.66–7.62 (m, 2H), 7.29–7.25 (m, 2H), 2.70 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.8, 157.7, 155.4, 155.0, 133.6, 132.7, 128.4, 123.5, 20.2; HRMS (ESI), calcd for C12H11N2OS2+ (M + H)+ 263.0307, found 263.0308.
O-(6-(Dimethylcarbamoyl)pyridin-3-yl) S-Methyl Carbonodithioate (2r).
General Procedure B was followed for the reaction of 6-carboxyamide-3-hydroxypyridine 1r (332 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:2 to 1:4) to give the product as a white solid (445 mg, 87%). 2r: 1H NMR (600 MHz, CDCl3) δ 8.37 (d, J = 2.6 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.55 (dd, J = 8.4, 2.6 Hz, 1H), 3.14 (s, 3H), 3.12 (s, 3H), 2.70 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.2, 168.1, 152.2, 151.4, 142.4, 131.1, 124.9, 39.3, 36.1, 20.2; HRMS (ESI), calcd for C10H13N2O2S2+ (M + H)+ 257.0413, found 257.0410.
O-(6-Chloropyridin-3-yl) S-Methyl Carbonodithioate (2s).
General Procedure B was followed for the reaction of 6-chloro-3-hydroxypyridine 1s (220 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:3) to give the product as a pale-yellow solid (352 mg, 94%). 2s: 1H NMR (600 MHz, CDCl3) δ 8.20 (d, J = 2.9 Hz, 1H), 7.44 (dd, J = 8.4, 2.9 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 215.4, 150.2, 148.7, 143.8, 133.2, 124.9, 20.4; HRMS (ESI), calcd for C7H7ClNOS2+ (M + H)+ 219.9652, found 219.9648.
O-(Quinoline-3-yl) S-Methyl Carbonodithioate (2t).
General Procedure B was followed for the reaction of 3-hydroxyquinoline 1t (290 mg, 2.00 mmol) and imidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 0:1 to 1:9) to give the product as a brown oil (395 mg, 84%). 2t: 1H NMR (600 MHz, CDCl3) δ 8.74 (d, J = 1.8 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.72 (dd, J = 8.4, 7.3 Hz, 1H), 7.57 (dd, J = 8.1, 7.3 Hz, 1H), 2.70 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.8, 148.0, 146.3, 146.1, 129.6, 129.5, 128.2, 127.8, 127.5, 127.1, 20.3; HRMS (ESI), calcd for C11H10NOS2+ (M + H)+ 236.0198, found 236.0196.
O-(Quinoline-7-yl) S-Methyl Carbonodithioate (2u).
General Procedure B was followed for the reaction of 3-hydroxyquinoline 1u (290 mg, 2.00 mmol) and benzimidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:9) to give the product as a white solid (459 mg, 98%). 2u: 1H NMR (600 MHz, CDCl3) δ 8.93 (dd, J = 4.4, 1.5 Hz, 1H), 8.16 (d, J = 9.2 Hz, 1H), 8.15 (dd, J = 8.4, 1.5 Hz, 1H), 7.54 (d, J = 2.6 Hz, 1H), 7.50 (dd, J = 9.2, 2.6 Hz, 1H), 7.43 (dd, J = 8.4, 4.4 Hz, 1H), 2.71 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.6, 152.4, 150.7, 146.7, 136.1, 131.3, 128.7, 125.2, 121.8, 119.2, 20.4; HRMS (ESI), calcd for C11H10NOS2+ (M + H)+ 236.0198, found 236.0195.
O-(Pyridine-3-yl) S-Methyl Carbonodithioate (2v).
General Procedure B was followed for the reaction of 4-ethoxycarbonyl phenol 1v (185 mg, 2.00 mmol) and benzimidazolium reagent 6 (600 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:1 to 3:1) to give the product as a yellow oil (353 mg, 98%). 2v: 1H NMR (600 MHz, CDCl3) δ 8.53 (dd, J = 4.8, 1.1 Hz, 1H), 8.42 (d, J = 2.6 Hz, 1H), 7.46 (ddd, J = 8.4, 2.6, 1.1 Hz, 1H), 7.37 (dd, J = 8.4, 4.8 Hz, 1H), 2.68 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 215.6, 151.3, 147.6, 144.1, 136.1, 131.3, 128.7, 125.2, 121.8, 119.2, 20.4; HRMS (ESI), calcd for C11H10NOS2+ (M + H)+ 236.0198, found 236.0195.
O-(1-(4-Chlorophenyl)-1H-pyrazol-4-yl) S-Methyl Carbonodithioate (2w).
General Procedure B was followed for the reaction of 4-hydroxypyrazole 1w (389 mg, 2.00 mmol) and benzimidazolium reagent 7 (700 mg, 2.00 mmol). The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:9) to give the product as a white solid (563 mg, 99%). 2w: 1H NMR (600 MHz, CDCl3) δ 8.03 (s, 1H), 7.68 (s, 1H), 7.62–7.59 (m, 2H), 7.44–7.41 (m, 2H), 2.68 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 214.5, 140.1, 138.7 134.2, 132.5, 129.7, 120.2, 118.9, 20.4; HRMS (ESI), calcd for C11H9ClN2OS2+ (M + Na)+ 306.9737, found 306.9740.
General Procedures for the Synthesis of Aryltrifluoromethyl Ethers (ArOCF3) 3a–3w.
General Procedure C (Reaction Conditions with TCCA).
A 20 mL vial was charged with xanthate 2 (0.5 mmol, 1.0 equiv), TCCA (116 mg, 0.5 mmol, 1.0 equiv), and XtalFluor-E (573 mg, 2.5 mmol, 5.0 equiv) in air. Then, anhydrous dichloroethane (5 or 10 mL) and water (9.0 mg, 0.5 mmol, 1.0 equiv) were added to the vial. (*One equivalent of water was weighed into a pipet, and the water was transferred to the flask by forced air.) The resulting mixture was stirred at 80 °C for the reported reaction time. After cooling to room temperature, the mixture was quenched with saturated aqueous NaHCO3 (5 mL), stirred for 30 min, and filtered through a pad of Celite. The filtrate was extracted with EtOAc (2 × 10 mL). Activated charcoal powder (ca. 1 g) and silica gel (ca. 1 g) were added to the combined organic layer. (Note: Charcoal helps to remove brown byproducts derived from XtalFluor-E.) After stirring for 1 min, the mixture was filtered with Celite and washed with EtOAc. The filtrate was concentrated and purified by silica gel column chromatography to give ArOCF3 (3).
General Procedure D (Reaction Conditions with NFSI).
A 20 mL vial was charged with xanthates 2 (0.5 mmol, 1.0 equiv), NFSI (473 mg, 1.5 mmol, 3.0 equiv), and XtalFluor-E (343 mg, 1.5 mmol, 3.0 equiv) under air. Then, anhydrous dichloroethane (2 or 5 mL) was added to the vial. The resulting mixture was stirred at 80 °C for desired reaction time. After cooling to room temperature, the mixture was quenched with saturated aqueous NaHCO3 (5 mL), stirred for 30 min, and filtered through a pad of Celite. The filtrate was extracted with EtOAc (2 × 10 mL). Activated carbon powder (ca. 1 g) and silica gel (ca. 1 g) were added to the combined organic layer. After stirring for 1 min, the mixture was filtered with Celite and washed with EtOAc. The filtrate was concentrated and purified by silica gel column chromatography to give ArOCF3 (3).
4-(Dimethylaminocarbonyl)trifluoromethoxybenzene (3a).
General Procedure C was followed for the reaction of xanthate 2a (128 mg, 0.50 mmol). The reaction was conducted with dichloroethane (10 mL) for 3 h. The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 1:3 to 1:1) to give a mixture of 3a (65.0 mg, 56%) and ArOCF2Cl byproduct (1.9 mg, 1.5%). 3a: a pale-yellow solid; 1H NMR (600 MHz, CDCl3) δ 7.48–7.44 (m, 2H), 7.26–7.22 (m, 2H), 3.11 (s, 3H), 2.98 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.4, 149.9, 134.9, 129.0, 120.8, 120.4 (q, J = 258.7, CF3), 39.6, 35.5; 19F (376 MHz, CDCl3) δ −59.0 (s); HRMS (ESI), calcd for C10H11F3NO2+ (M + H)+ 234.0736, found 234.0742.
4-(Ethoxycarbonyl)trifluoromethoxybenzene (3b).
General Procedure D was followed for the reaction of xanthate 2b (128 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a pale-yellow oil (56.0 mg, 48%). 3b: 1H NMR (600 MHz, CDCl3) δ 7.48–7.44 (m, 2H), 7.26–7.22 (m, 2H), 3.11 (s, 3H), 2.98 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 170.4, 149.9, 134.9, 129.0, 120.8, 120.4 (q, J = 258.7, CF3), 39.6, 35.5; 19F (376 MHz, CDCl3) δ −59.0 (s); HRMS (EI), calcd for C10H9F3O3+ (M)+ 234.0504, found 234.0507.
4-(Adamantyl)trifluoromethoxybenzene (3c).
General Procedure D was followed for the reaction of xanthate 2c (159 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a white solid (118 mg, 80%). 3c: 1H NMR (600 MHz, CDCl3) δ 7.39–7.35 (m, 2H), 7.17–7.13 (m, 2H), 2.13–2.09 (m, 3H), 1.90 (d, J = 2.2, 6H), 1.82–1.72 (m, 6H); 13C NMR (151 MHz, CDCl3) δ 150.1, 147.2, 126.4, 120.7 (q, J = 257, CF3), 120.6, 43.4, 36.8, 36.2, 29.1; 19F NMR (376 MHz, CDCl3) δ −59.1 (s), HRMS (EI), calcd for C17H19F3O+ (M)+ 296.1388, found 296.1392.
2,4-(Di-t-butyl)trifluoromethoxybenzene (3f).
General Procedure D was followed for the reaction of xanthate 2f (148 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a colorless oil (77.1 mg, 56%). 3f: 1H NMR (600 MHz, CDCl3) δ 7.42 (d, J = 2.6 Hz, 1H), 7.22 (dd, J = 8.4, 2.6 Hz, 2H), 7.18–7.14 (m, 1H), 1.41 (s, 9H), 1.33 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 148.5, 146.6, 139.9, 124.9, 124.1, 120.9 (q, J = 258, CF3), 118.4, 35.1, 34.8, 31.6, 30.4; 19F NMR (376 MHz, CDCl3) δ −55.8 (s), HRMS (EI), calcd for C15H21F3O+ (M)+ 274.1544, found 274.1549.
4-(Methoxycarbonylethyl)trifluoromethoxybenzene (3g).
General Procedure D was followed for the reaction of xanthate 2g (135 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a pale-yellow oil (81.1 mg, 65%). 3g: 1H NMR (600 MHz, CDCl3) δ 7.23–7.20 (m, 2H), 7.15–7.11 (m, 2H), 3.67 (s, 3H), 2.95 (t, J = 7.7 Hz, 2H), 2.95 (t, J = 7.7 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 173.1, 147.8, 139.4, 129.7, 121.2, 120.6 (q, J = 258, CF3), 51.8, 35.6, 30.3; 19F NMR (376 MHz, CDCl3) δ −57.9 (s), HRMS (EI), calcd for C11H11F3O3+ (M)+ 248.0660, found 248.0662.
7-Trifluoromethoxy Coumarin (3h).
General Procedure D was followed for the reaction of xanthate 2g (126 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 6 h. The crude reaction mixture was filtered through a pad of silica gel (EtOAc/hexane 3:1) and then purified by preparative SFC to remove (PhSO2)2NH as a main contaminant (95% CO2/40% MeOH; flow: 3.0 mL/min; Princeton PPU 250 mm × 10.0 mm 5 mm) to give the product as a white solid (25.0 mg, 22%). 3h: 1H NMR (CDCl3, 400 MHz) δ 7.71 (d, J = 9.8 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.22 (s, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.45 (d, J = 9.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 159.9, 154.8, 151.3, 142.4, 129.1, 120.3 (q, J = 256, CF3), 117.3, 116.9, 116.9, 109.3; 19F NMR (376 MHz, CDCl3) δ −57.9 (s); HRMS (APCI) Calculated for C10H6F3O3+ (M + H)+ 231.0260, found 231.0261.
4-(Benzoyl)trifluoromethoxybenzene (3i).
General Procedure D was followed for the reaction of xanthate 2i (144 mg, 0.50 mmol). The reaction was conducted with dichloroethane (2 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography (hexane) to give the product as a colorless oil (68.3 mg, 51%). 3i: 1H NMR (600 MHz, CDCl3) δ 7.89–7.85 (m, 2H), 7.79 (d, J = 7.7 Hz, 2H), 7.61 (t, J = 7.7 Hz, 1H), 7.50 (t, J = 7.7 Hz, 2H), 7.34–7.30 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 195.2, 152.2, 137.2, 136.0, 132.8, 132.1, 130.0, 128.5, 120.5 (q, J = 259, CF), 120.3; 193 F NMR (376 MHz, CDCl3) δ −58.8 (s); HRMS (EI), calcd for C14H9O2F3+ (M)+ 266.0555, found 266.0558.
4-(1H-Imidazol-1-yl)trifluoromethoxybenzene (3m).
General Procedure C was followed for the reaction of xanthate 2m (125 mg, 0.50 mmol). The reaction was conducted with dichloroethane (10 mL) for 12 h. The crude reaction mixture was filtered through a pad of silica gel (EtOAc) and then purified by preparative SFC to remove byproducts derived from XtalFluor-E (85% CO2/15% MeOH (0.2% 7 M NH3 in MeOH); flow: 3.0 mL/min; Princeton PPU 250 mm × 10.0 mm 5 mm) to give the product as a white solid (41.0 mg, 36%). 3q: 1H NMR (CDCl3, 400 MHz) δ 7.69 (s, 1H), 7.30–7.26 (m, 2H), 7.22–7.18 (m, 2H), 7.11–7.07 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 157.8, 154.9, 149.9, 133.9, 133.2, 128.6, 121.9, 120.5 (q, J = 258, CF3); 19F NMR (376 MHz, CDCl3) δ −58.0 (s); HRMS (APCI) calcd for C11H8F3N2O+ (M + H)+ 241.0583, found 241.0586.
4-(Benzothiazole-2-yl)trifluoromethoxybenzene (3n).
General Procedure C was followed for the reaction of xanthate 2n (151 mg, 0.50 mmol). The reaction was conducted with dichloroethane (10 mL) for 3 h. The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 0:1 to 1:100) to give a mixture of 3n (53.6 mg, 38%), ArOCF2Cl byproduct (5.9 mg, 3.8%), and ArOCFCl2 byproduct (3.6 mg, 2.2%).23 3n: pale-yellow solid; 1H NMR (600 MHz, CDCl3) δ 8.16–8.12 (m, 2H), 8.08 (d, J = 8.1 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.52 (dd, J = 8.1, 7.3 Hz, 1H), 7.41 (dd, J = 8.1, 7.3 Hz, 1H), 7.34 (d, J = 8.4 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 166.3, 154.2, 151.2, 135.3, 132.3, 129.2, 126.7, 125.6, 123.5, 121.8, 121.3, 120.5 (q, J = 259, CF3); 19F NMR (376 MHz, CDCl3) δ −58.1 (s); HRMS (ESI), calcd for C14H9F3NOS+ (M + H)+ 296.0351, found 296.0347.
4-(6-Chloropyridine-3-yl)trifluoromethoxybenzene (3o).
General Procedure C was followed for the reaction of xanthate 2o (148 mg, 0.50 mmol). The reaction was conducted with dichloroethane (5 mL) for 12 h. The crude reaction mixture was purified by silica gel column chromatography (EtOAc/hexane 9:1 to 3:1) to give a mixture of 3o (68.5 mg, 50%), ArOCF2Cl byproduct (2.7 mg, 1.9%), and ArOCFCl2 byproduct (2.5 mg, 1.6%).23 3o: pale-yellow solid; 1H NMR (CDCl3, 600 MHz) δ 8.58 (d, J = 2.6 Hz, 1H), 7.81 (dd, J = 8.4, 2.6 Hz, 1H), 7.58–7.55 (m, 2H), 7.41 (d, J = 8.4 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 150.9, 149.6, 147.9, 137.2, 135.3, 134.4, 128.6, 124.5, 121.7, 120.5 (q, J = 257, CF3); 19F NMR (376 MHz, CDCl3) δ −59.0 (s); HRMS (APCI) calcd for C12H8ClF3NO+ (M + H)+ 274.0241, found 274.0240.
4-(Pyridine-3-yl)trifluoromethoxybenzene (3p).
General Procedure D was followed for the reaction of xanthate 2p (131 mg, 0.50 mmol) with the exception adding Selectfluor (117 mg, 0.50 mmol, 1.0 equiv). The reaction was conducted with dichloroethane (2 mL) for 12 h. The crude reaction mixture was filtered through a pad of silica gel (EtOAc/hexane 3:1) and then purified by preparative SFC to remove (PhSO2)2NH as a main contaminant (95% CO2/5% MeOH (0.2% 7 M NH3 in MeOH); flow: 3.0 mL/min; Princeton PPU 250 mm × 10.0 mm 5 mm) to give the product as a colorless oil (55.0 mg, 46%). 3p: 1H NMR (CDCl3, 400 MHz) δ 8.90–8.79 (m, 1H), 8.63 (d, J = 4.8 Hz, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.66–7.57 (m, 2H), 7.43–7.32 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 149.3, 148.8, 148.1, 136.5, 136.0, 134.5, 128.6, 123.7, 121.6, 120.5 (q, J = 258, CF3); 19F NMR (376 MHz, CDCl3) δ −57.8 (s); HRMS (APCI) calcd for C12H9F3NO+ (M + H)+ 240.0631, found 240.0624.
4-(Pyrimidine-3-yl)trifluoromethoxybenzene (3q).
General Procedure D was followed for the reaction of xanthate 2q (131 mg, 0.50 mmol) with the exception adding Selectfluor (354 mg, 1.00 mmol, 2.0 equiv). The reaction was conducted with dichloroethane (2 mL) for 12 h. The crude reaction mixture was filtered through a pad of silica gel (EtOAc/hexane 1:1) and then purified by preparative SFC to remove (PhSO2)2NH as a main contaminant (95% CO2/5% MeOH (0.2% 7 M NH3 in MeOH); flow: 3.0 mL/min; Princeton PPU 250 mm × 10.0 mm 5 mm) to give the product as a white solid (35.0 mg, 29%). 3q: 1H NMR (CDCl3, 400 MHz) δ 9.25 (s, 1H), 8.95 (s, 2H), 7.63 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 157.8, 154.9, 149.9, 133.9, 133.2, 128.6, 121.9, 120.5 (q, J = 258, CF); 19F NMR (376 MHz, CDCl3) δ −58.0 (s); HRMS (APCI) calcd for C11H8F3N2O+ (M + H)+ 241.0583, found 241.0586.
5-Dimethylaminocarbonyl-3-trifluoromethoxypyridine (3r).
General Procedure C was followed for the reaction of xanthate 2r (128 mg, 0.50 mmol). The reaction was conducted with dichloroethane (5 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography to give a mixture of 3q (19.2 mg, 16%), ArOCF2Cl (0.6 mg, 0.5%), and ArOCFCl2 (0.6 mg, 0.4%). Brown oil; 3q: 1H NMR (CDCl3, 600 MHz) δ 8.49 (d, J = 2.6 Hz 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.65 (dd, J = 8.8, 2.6 Hz, 1H), 3.14 (s, 3H), 3.11 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 167.7, 152.9, 146.3, 141.0, 129.1, 125.2, 120.4 (q, J = 260 Hz, CF3), 39.2, 36.1; 19F NMR (376 MHz, CDCl3) δ −58.0 (s); HRMS (APCI) calcd for C9H10F3N2O2+ (M + H)+ 235.0689, found 235.0691.
3-Trifluoromethoxyquinoline (3t).
General Procedure C was followed for the reaction of xanthate 2t (118 mg, 0.50 mmol). The reaction was conducted with dichloroethane (5 mL) for 24 h. The crude reaction mixture was purified by silica gel column chromatography to give a mixture of 3q (11.7 mg, 11%) and ArOCF2Cl byproduct (0.3 mg, 0.3%). 3q: 1H NMR (CDCl3, 600 MHz) δ 8.84 (d, J = 2.6 Hz, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.88–7.86 (m, 1H), 7.86 (d, J = 8.1 Hz, 1H), 7.77 (ddd, J = 8.4, 6.6, 1.1 Hz, 1H), 7.64 (ddd, J = 8.1, 6.6, 1.1 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 146.0, 144.6, 142.9, 130.3, 129.3, 128.3, 128.0, 127.9, 126.2, 120.7 (q, J = 260, CF3); 19F NMR (376 MHz, CDCl3) δ −58.1 (s); HRMS (EI) calcd for C10H6F3NO+ (M)+ 213.0401, found 213.0404.
7-Trifluoromethoxyquinoline (3u).
General Procedure C was followed for the reaction of xanthate 2u (118 mg, 0.50 mmol). The reaction was conducted with dichloroethane (5 mL) for 6 h. The crude reaction mixture was purified by silica gel column chromatography to give the product as a brown oil (41.6 mg, 39%). 3u: 1H NMR (CDCl3, 600 MHz) δ 8.95 (dd, J = 4.0, 1.8 Hz, 1H), 8.18–8.15 (m, 2H), 7.65 (d, J = 2.6 Hz, 1H), 7.58 (dd, J = 9.2, 2.6 Hz, 1H), 7.47 (dd, J = 8.4, 4.0 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 150.9, 147.1, 146.5, 136.2, 131.8, 128.6, 123.8, 122.2, 120.7 (q, J = 258, CF3), 117.9 (CH); 19F NMR (376 MHz, CDCl3) δ −57.7 (s); HRMS (APCI) calcd for C10H7F3NO+ (M + H)+ 214.0474, found 214.0467.
1-(4-Chlorophenyl)-4-(trifluoromethoxy)-1H-pyrazole (3w).
General Procedure C was followed for the reaction of xanthate 2w (142 mg, 0.50 mmol). The reaction was conducted with dichloroethane (5 mL) for 3 h. The crude reaction mixture was purified by silica gel column chromatography to give a mixture of 3w (39.4 mg, 30%) and ArOCF2Cl byproduct (1.8 mg, 1.3%). Brown oil; 3w: 1H NMR (CDCl3, 600 MHz) δ 7.89 (s, 1H), 7.66 (s, 1H), 7.61–7.58 (m, 2H), 7.45–7.42 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 138.5, 135.6, 133.3, 132.9, 129.8, 129.3, 120.6 (q, J = 258 Hz, CF3), 118.6; 19F NMR (376 MHz, CDCl3) δ −60.7 (s); HRMS (EI) calcd for C10H6ClF3N2O+ (M)+262.0121, found 262.0125.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful for support from Pfizer and the NIH (1R35GM130387). We also thank Dr. Vincent Mascitti for his helpful insight into this research.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b02717.
Additional experimental procedures and original spectral data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Betageri R; Zhang Y; Zindell RM; Kuzmich D; Kirrane TM; Bentzien J; Cardozo M; Capolino AJ; Fadra TN; Nelson RM; Paw Z; Shih D-T; Shih C-K; Zuvela-Jelaska L; Nabozny G; Thomson DS Trifluoromethyl group as a pharmacophore: Effect of replacing a CF3 group on binding and agonist activity of a glucocorticoid receptor ligand. Bioorg. Med. Chem. Lett 2005, 15, 4761–4769. [DOI] [PubMed] [Google Scholar]; (b) Wang J; Sánchez-Roselló M; Aceña J; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]; (c) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]; (d) Swallow S Fluorine in Medicinal Chemistry In Progress in Medicinal Chemistry; Elsevier: Amsterdam, Netherlands, 2015; Vol. 54, pp 65–133. [DOI] [PubMed] [Google Scholar]
- (2).(a) For reviews of fluorinations, see:Liang T; Neumann CN; Ritter T Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem., Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]; (b) Zhu Y; Han J; Wang J; Shibata N; Sodeoka M; Soloshonok VA; Coelho JAS; Toste FD Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev 2018, 118, 3887–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Remete AM; Nonn M; Fustero S; Fülöp F; Kiss L Synthesis of fluorinated amino acid derivatives through late-stagedeoxyfluorinations. Tetrahedron 2018, 74, 6367–6418 (and references cited therein). [Google Scholar]
- (3).(a) Serfaty I; Hodgins T; McBee E Halogen-containing substituents. II. Methoxy system. Reactivity parameters. Charge distribution and conformation of the anisoles. J. Org. Chem 1972, 37, 2651–2655. [Google Scholar]; (b) Herkes FE Halogenation of trifluoromethoxy and bis(trifluoromethoxy)benzene. J. Fluorine Chem 1977, 9, 113–126. [Google Scholar]; (c) Rose-Munch F; Khourzom R; Djukic J-P; Rose E; Langlois B; Vaisserman J Etude conformationnelle du complexe η6-(4-(trifluorométhoxy)aniline)tricarbonylchrome. J. Organomet. Chem 1994, 470, 131–135. [Google Scholar]; (d) Federsel D; Herrmann A; Christen D; Sander S; Willner H; Oberhammer H Structure and conformation of α,α,α-trifluoroanisol, C6H5OCF3. J. Mol. Struct 2001, 567, 127–136. [Google Scholar]; (e) Leroux FR; Manteau B; Vors J-P; Pazenok S Trifluoromethyl ethers – synthesis and properties of an unusual substituent. Beilstein J. Org. Chem 2008, 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Iijima A; Amii H Selective aromatic carbon–oxygen bond cleavage of trifluoromethoxyarenes: a trifluoromethoxy group as a convertible directing group. Tetrahedron Lett. 2008, 49, 6013–6015. [Google Scholar]
- (4).For example, OCF3Πx = 1.04, CF3 = 0.88, CH3 = 0.56, and OCH3 = −0.02. See: Hansch C; Leo A Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley: New York, 1979; part VII 339. [Google Scholar]
- (5).Liu J; Chen C; Chu L; Chen Z; Xu X; Qing F Silver-Mediated Oxidative Trifluoromethylation of Phenols: Direct Synthesis of Aryl Trifluoromethyl Ethers. Angew. Chem., Int. Ed 2015, 54, 11839–11842. [DOI] [PubMed] [Google Scholar]
- (6).(a) Huang C; Liang T; Harada S; Lee E; Ritter T Silver-Mediated Trifluoromethoxylation of Aryl Stannanes and Arylboronic Acids. J. Am. Chem. Soc 2011, 133, 13308–13310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang Y-M; Yao J-F; Yan W; Luo Z; Tang Z-Y Silver-Mediated Trifluoromethoxylation of (Hetero)aryldiazonium Tetrafluoroborates. Org. Lett 2019, 21, 8003–8007. [DOI] [PubMed] [Google Scholar]
- (7).(a) Hojczyk KN; Feng P; Zhan C; Ngai M Trifluoromethoxylation of Arenes: Synthesis of ortho-Trifluoromethoxylated Aniline Derivatives by OCF3 Migration. Angew. Chem., Int. Ed 2014, 53, 14559–14563. [DOI] [PubMed] [Google Scholar]; (b) Lee K; Lee J; Ngai M-Y Synlett 2016, 27, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zheng W; Morales-Rivera CA; Lee JW; Liu P; Ngai M Catalytic C-H Trifluoromethoxylation of Arenes and Heteroarenes. Angew. Chem., Int. Ed 2018, 57, 9645–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jelier BJ; Tripet PF; Pietrasiak E; Franzoni I; Jeschke G; Togni A Angew. Chem., Int. Ed 2018, 57, 13784–13789. [DOI] [PubMed] [Google Scholar]; (e) Yang S; Chen M; Tang P Visible-Light Photoredox-Catalyzed and Copper-Promoted Trifluoromethoxylation of Arenediazonium Tetrafluoroborates. Angew. Chem., Int. Ed 2019, 58, 7840–7844. [DOI] [PubMed] [Google Scholar]
- (8).(a) Zhang Q; Brusoe AT; Mascitti V; Hesp KD; Blakemore DC; Kohrt JT; Hartwig JF Fluorodecarboxylation for the Synthesis of Trifluoromethyl Aryl Ethers. Angew. Chem., Int. Ed 2016, 55, 9758–9762; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 9910–9914. [Google Scholar]; (b) Zhou M; Ni C; He Z; Hu J Synthesis of Trifluoromethoxylated (Hetero) Arenes via OCF3 Migration. Org. Lett 2016, 18, 3754–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chatalova-Sazepin C; Binayeva M; Epifanov M; Zhang W; Foth P; Amador C; Jagdeo M; Boswell BR; Sammis GM Xenon Difluoride Mediated Fluorodecarboxylations for the Syntheses of Di- and Trifluoromethoxyarenes. Org. Lett 2016, 18, 4570–4573. [DOI] [PubMed] [Google Scholar]
- (9).(a) For classical examples of synthetic methods of ArOCF3 using fluoride substitution, see:Sheppard WA α-Fluorinated Ethers. I. Aryl Fluoroalkyl Ethers. J. Org. Chem 1964, 29, 1–11. [Google Scholar]; (b) Feiring AE Chemistry in hydrogen fluoride. 7. A novel synthesis of aryl trifluoromethyl ethers. J. Org. Chem 1979, 44, 2907–2910. [Google Scholar]; (c) Kuroboshi M; Suzuki K; Hiyama T Oxidative desulfurization-fluorination of xanthates. A convenient synthesis of trifluoromethyl ethers and difluoro(methylthio)methyl ethers. Tetrahedron Lett. 1992, 33, 4173–4176. [Google Scholar]; (d) Kanie K; Tanaka Y; Suzuki K; Kuroboshi M; Hiyama T A Convenient Synthesis of Trifluoromethyl Ethers by Oxidative Desulfurization-Fluorination of Dithiocarbonates. Bull. Chem. Soc. Jpn 2000, 73, 471–484. [Google Scholar]; (e) Kuroboshi M; Kanie K; Hiyama T Oxidative Desulfurization-Fluorination: A Facile Entry to a Wide Variety of Organofluorine Compounds Leading to Novel Liquid-Crystalline Materials. Adv. Synth. Catal 2001, 343, 235–250. [Google Scholar]; (f) Manteau B; Genix P; Brelot L; Vors J-P; Pazenok S; Giornal F; Leuenberger C; Leroux FR A General Approach to (Trifluoromethoxy)pyridines: First X-ray Structure Determinations and Quantum Chemistry Studies. Eur. J. Org. Chem 2010, 2010, 6043–6066. [Google Scholar]; (g) Umemoto T; Singh RP; Xu Y; Saito N Discovery of 4-tert-Butyl-2,6-dimethylphenylsulfur Trifluoride as a Deoxofluorinating Agent with High Thermal Stability as Well as Unusual Resistance to Aqueous Hydrolysis, and Its Diverse Fluorination Capabilities Including Deoxofluoro-Arylsulfinylation with High Stereoselectivity. J. Am. Chem. Soc 2010, 132, 18199–18205. [DOI] [PubMed] [Google Scholar]
- (10).(a) For selected reviews of fluoroalkylation of alcohols and phenols, see:Tlili A; Toulgoat F; Billard T Synthetic Approaches to Trifluoromethoxy-Substituted Compounds. Angew. Chem., Int. Ed 2016, 55, 11726–11735; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 11900–11909. [Google Scholar]; (b) Lee KN; Lee JW; Ngai M-Y Catalytic trifluoromethoxylation reactions. Tetrahedron 2018, 74, 7127–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang XT; Tang P Recent advances in new trifluoromethoxylation reagents. Sci. China: Chem 2019, 62, 525–532. [Google Scholar]; (d) Lee JW; Lee KN; Ngai M-Y Synthesis of Tri- and Difluoromethoxylated Compounds by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2019, 58, 11171–11181 (and references cited therein). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) For synthetic examples of synthesizing hetero ArOCF3, see:Liang A; Han S; Liu Z; Wang L; Li J; Zou D; Wu Y; Wu Y Regioselective Synthesis of N-Heteroaromatic Trifluoromethoxy Compounds by Direct O-CF3 Bond Formation. Chem. - Eur. J 2016, 22, 5102–5106. [DOI] [PubMed] [Google Scholar]; (b) Zhang Q-W; Hartwig JF Synthesis of heteroaromatic trifluoromethyl ethers with trifluoromethyl triflate as the source of the trifluoromethoxy group. Chem. Commun 2018, 54, 10124–10127. [DOI] [PubMed] [Google Scholar]; Refs 5 and 7 also showed several examples of heteroaryl trifluoromethyl ethers. Although trifluoromethylation of 2-hydroxypyridine derivatives has been demonstrated in ref 11a, the trifluoromethylation of phenols by the Togni reagent usually proceeds at the carbon atom instead of the oxygen atom; see:; (c) Stanek K; Koller R; Togni A Reactivity of a 10-I-3 Hypervalent Iodine Trifluoromethylation Reagent With Phenols. J. Org. Chem 2008, 73, 7678–7685. [DOI] [PubMed] [Google Scholar]
- (12).Sun W; Hu J; Shi Y 1-(Methyldithiocarbonyl)imidazole: A Reagent of S-Methyldithiocarbonylation. Synlett 1997, 1997, 1279–1280. [Google Scholar]
- (13).Mohanta PK; Dhar S; Samal SK; Ila H; Junjappa H 1-(Methyldithiocarbonyl)imidazole: a Useful Thiocarbonyl Transfer Reagent for Synthesis of Substituted Thioureas. Tetrahedron 2000, 56, 629–637. [Google Scholar]
- (14).The pKa values of the conjugate acid of the leaving groups of 5, 6, and 7 are 8.4, 6.0, and 4.6, respectively. See: Buncel E; Joly HA; Jones JR Proton transfer from imidazole, benzimidazole, and their 1-alkyl derivatives. FMO analysis of the effect of methyl and benzo substitution. Can. J. Chem 1986, 64, 1240–1245. [Google Scholar]
- (15).(a) Beaulieu F; Beauregard L-P; Courchesne G; Couturier M; LaFlamme F; L’Heureux A Aminodifluorosulfinium Tetrafluoroborate Salts as Stable and Crystalline Deoxofluorinating Reagents. Org. Lett 2009, 11, 5050–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) L’Heureux A; Beaulieu F; Bennett C; Bill DR; Clayton S; LaFlamme F; Mirmehrabi M; Tadayon S; Tovell D; Couturier M Aminodifluorosulfinium Salts: Selective Fluorination Reagents with Enhanced Thermal Stability and Ease of Handling. J. Org. Chem 2010, 75, 3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kepp KP A Quantitative Scale of Oxophilicity and Thiophilicity. Inorg. Chem 2016, 55, 9461–9470. [DOI] [PubMed] [Google Scholar]
- (17).The addition of 2 or more equivalents of water totally prevented the desired trifluoromethylation and caused hydrolysis of xanthate to form the corresponding phenols. Since all the reactions were conducted under air, catalytic water is present. In the case of condition A, 1 equiv of water resulted in better reproducibility than the catalytic amount.
- (18).ArOCF2Cl and ArOCFCl2 byproducts have been obtained from several substrates. According to ref 11a, the chlorinated products might be converted to the corresponding trifluoromethyl ether by treatment of SbF3 and the catalytic amount of SbCl5.
- (19).(a) Moseley JD; Sankey RF; Tang ON; Gilday JP The Newman–Kwart rearrangement re-evaluated by microwave synthesis. Tetrahedron 2006, 62, 4685–4689. [Google Scholar]; (b) Lloyd-Jones G; Moseley J; Renny J Mechanism and Application of the Newman-KwartO→S Rearrangement of O-Aryl Thiocarbamates. Synthesis 2008, 2008, 661–689. [Google Scholar]; (c) Burns M; Lloyd-Jones GC; Moseley JD; Renny JS The Molecularity of the Newman-Kwart Rearrangement. J. Org. Chem 2010, 75, 6347–6353. [DOI] [PubMed] [Google Scholar]
- (20).Wang Y-Q; Ge Z-M; Hou X-L; Cheng T-M; Li R-T A Mild and Efficient Procedure for the Synthesis of 1-(Alkyldithiocarbonyl)-imidazoles. Synthesis 2004, 2004, 675–678. [Google Scholar]
- (21).Sun W; Hu J; Shi Y 1-(Methyldithiocarbonyl)imidazole: A Reagent of S-Methyldithiocarbonylation. Synlett 1997, 1997, 1279–1280. [Google Scholar]
- (22).Pedras MSC; Sarma-Mamillapalle VK Metabolism and Metabolites of Dithiocarbamates in the Plant Pathogenic Fungus Leptosphaeria maculans. J. Agric. Food Chem 2012, 60, 7792–7798. [DOI] [PubMed] [Google Scholar]
- (23).The ratio of ArOCF2Cl and ArOCFCl2 byproducts in the isolated mixture was increased from that of the crude mixture. This indicates that the boiling point of the desired product 3n/3o would be significantly lower than that of the byproducts.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.