

Abstract
Phenanthroline ligands and [Ir(cod)(OMe)]2 form complexes that catalyze the silylation of aromatic and aliphatic C–H bonds. However, no experimental data on the identity of complexes related to the mechanism of this process or the mechanisms by which they react to functionalize C–H bonds have been reported. Herein, we describe our studies on the mechanism of the iridium-catalyzed silylation of aryl C–H bonds. The resting state of the catalyst is an iridium disilyl hydride complex (phenanthroline)Ir(SiMe(OTMS)2)2(H)(L), in which L varies with the arene and additives. An iridium disilyl hydride complex was isolated, characterized, and allowed to react with arenes to form aryl silanes. The kinetics of the reactions of electron-rich and electron-poor arenes showed that the rate-limiting step varies with the electronic properties of the arene. Computational studies on related iridium silyl complexes revealed that the high activity of iridium complexes containing sterically encumbered phenanthroline ligands is due to a change in the number of silyl groups bound to iridium between the resting state of the catalyst containing the hindered phenanthroline and that containing less-hindered phenanthroline.
Graphical Abstract
INTRODUCTION
The iridium-catalyzed silylation of aromatic C–H bonds forms aryl silanes with high sterically derived regioselectivity and is conducted with silane reagents that are produced and consumed on large scales.1 In many cases, the heteroaryl silane and aryl silane products are more stable than the corresponding aryl boronic esters and undergo related subsequent reactions, such as hydroxylation,2 amination,3 halogenation,4 and arylation.5 In addition, the silylation of aryl and heteroaryl C–H bonds often occurs with higher sterically derived selectivity than the iridium-catalyzed borylation of the same arenes.4a,6
To facilitate access to silicon-containing organic compounds, our group and others have reported silylations of aryl4b,c,7 and alkyl C–H bonds,8 with a range of silane reagents and a series of iridium catalysts.4a,b,7a,c,e–g,9 Such efforts to develop new silylation reactions would be aided by a detailed understanding of the mechanism by which these reactions occur. Yet, little is known about the mechanism of these reactions or even the identity of the Ir complexes present during the reaction. This dearth of experimental mechanistic data has motivated several groups to conduct computational studies. Sunoj et al. reported DFT calculations on the iridium-catalyzed intramolecular silylation of sp3 C–H bonds that suggested that an Ir(I) silyl species containing a bidentate nitrogen ligand and an additional norbornene ligand can cleave C–H bonds and mediate C–Si bond formation with energetic barriers that are consistent with the observed rate of the reaction.10 However, in later computational studies, the groups of Li11 and Huang12 concluded that an Ir(III) silyl complex is the resting state of the catalyst and that the difference in free energy between this complex and the transition state for oxidative addition of the C-H bond to the Ir(III) complex was smaller than the difference in free energy between the Ir(III) complex and the transition state for for oxidative addition of a C–H bond to a related Ir(I) complex. These contrasting computational results indicate the need for experimental mechanistic studies to determine the identity of the iridium complexes that lie on the catalytic cycle for the silylation of C–H bonds.
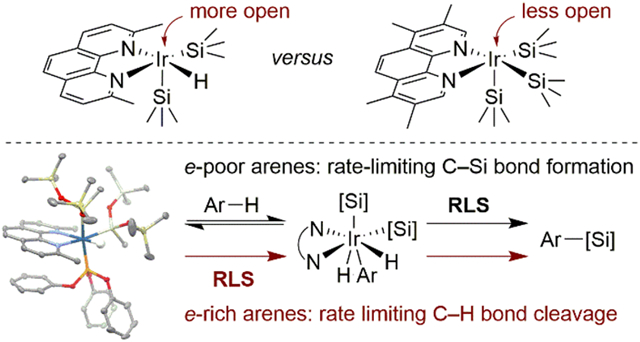
We recently reported a highly active catalyst for the silylation of aromatic C–H bonds that is formed from the combination of [Ir(cod)(OMe)]2 and 2,9-Me2-phenanthroline (2,9-Me2-phen, L1) (Figure 1).4b Two unusual features of this reaction motivated further study of the mechanism. First, the catalyst containing a phenanthroline ligand bearing methyl groups in the 2- and 9-positions was much more active than the catalysts containing phenanthrolines lacking substituents in one or both of those positions. Second, the rate of the reaction is ten times faster under an atmosphere of nitrogen than under an atmosphere of hydrogen.4b Consequently, the reactions of these highly active catalysts were conducted in an open system to remove the hydrogen byproduct with a stream of nitrogen. Thus, we sought to understand the origin of the high sensitivity of the rate of the reaction to the hydrogen byproduct and to the substitution pattern on the phenanthroline ligand. We also sought to provide a foundation for the aforementioned computational data by determining whether the iridium complex that mediates this reaction is an Ir(I) or Ir(III) complex and by identifying the number and type of silyl or hydride ligands bound to the iridium center.
Figure 1.
Catalytic silylation of aromatic C–H bonds.
To this end, we conducted a study that included both experimental and computational investigations. We generated and determined the identity of the resting state of the iridium catalyst in solution, isolated derivatives of this complex, and measured kinetic isotope effects for the silylation of arenes with varying electronic properties. Computational data are consistent with the intermediates and rate-limiting step in the catalytic cycle that we observed experimentally and reveal principles that dictate the activity and selectivity of these iridium complexes that catalyze the silylation of aryl and heteroaryl C–H bonds.
RESULTS AND DISCUSSION
1. Determination of the Resting State of the Catalyst.
To determine the identity of the iridium complexes in the silylation of aromatic C–H bonds, we first subjected a mixture of [Ir(cod)(OMe)]2, 2,9-Me2phen, and excess heptamethyltrisiloxane to analysis by high resolution ESI-mass spectrometry. The resulting spectrum contained a peak corresponding to the exact mass of a protonated iridium complex bearing 2,9-Me2phen, two silyl ligands, and one hydride (complex 1) (Scheme 1).
Scheme 1.
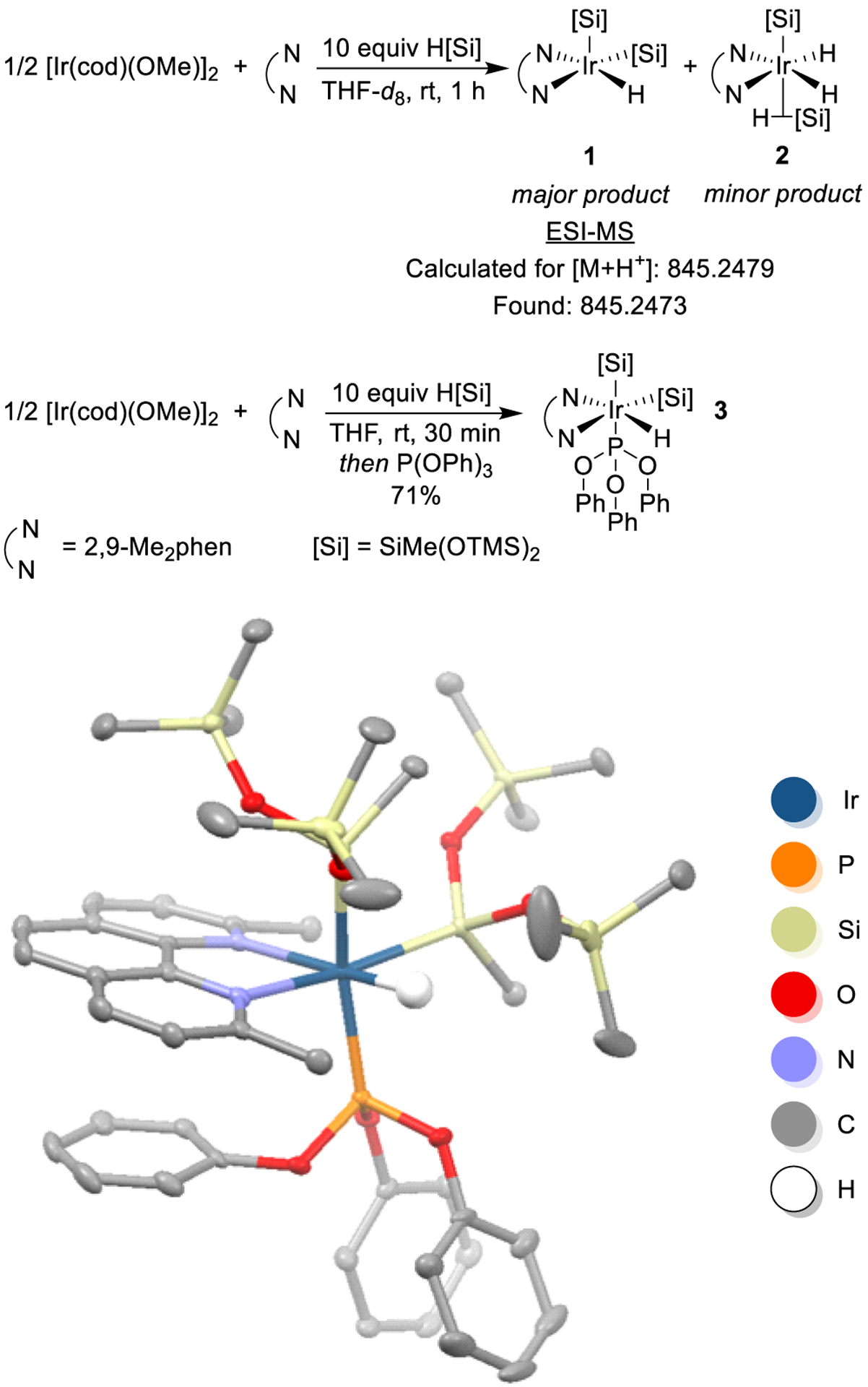
Synthesis of Catalyst Resting State and Solid-State Structure of Complex 3
To assess the validity of the MS data, we acquired 1H NMR spectra of the mixture of [Ir(cod)(OMe)]2, 2,9-Me2phen, and excess heptamethyltrisiloxane at room temperature and low temperatures. The 1H NMR spectrum acquired at room temperature contains a set of four signals corresponding to the phenanthroline ligand and a broad peak at −13 ppm that suggests the presence of a metal hydride. A large broad peak was also observed at 3.5 ppm that corresponds to the hydrogen attached to the silicon of free silane. This signal is located 1.2 ppm upfield of that for free silane alone (4.7 ppm), suggesting that the proton bound to silicon in the silane is exchanging with that in the hydride position of iridium.
The 1H NMR spectrum acquired at −86 °C contains peaks that are different from those in the spectrum acquired at room temperature. The peak for the hydrogen of free silane was observed as a quartet at 4.6 ppm, corresponding to the chemical shift observed in the absence of any metal complex. The peak corresponding to the metal hydride was observed as a sharp singlet at −16 ppm. The ratio of the integrals of the peaks for the bound 2,9-Me2phen and the hydride suggests that this complex contains only a single hydride ligand. Furthermore, a singlet at −0.5 ppm integrates to 36 hydrogens, indicating the presence of four TMS groups in the complex. These data imply that the resting state of the catalyst 1 contains a phenanthroline ligand, two silyl groups, and a hydride, and this conclusion matches that from mass spectrometry (Scheme 1). The 1H NMR spectrum also contains a set of peaks corresponding to a smaller amount of a second complex (2), which is the major species observed when this solution is placed under an atmosphere of hydrogen gas. The identity of this complex will be discussed in detail below.
To convert the resting state of the catalyst to a 6-coordinate species that would be more stable to isolation than the putative five-coordinate resting state 1, triphenylphosphite was added to the mixture of [Ir(cod)(OMe)]2, 2,9-Me2phen, and excess heptamethyltrisiloxane. The resulting phosphite complex 3 was isolated by column chromatography and characterized by NMR spectroscopy. The 1H NMR spectrum of 3 contains eight distinct signals that correspond to the protons of 2,9-Me2phen. The spectrum also contains six peaks in a 3:3:3:3:1:1 ratio between −1 and 1 ppm that correspond to the two inequivalent methyl groups on the two Ir-bound silyl groups and to two pairs of OTMS groups on the silyl groups, each of which are diastereotopic, due to the stereogenic iridium center in the complex. A doublet (J = 14 Hz) at −20 ppm indicates that 3 contains a metal hydride and is consistent with coupling to the phosphorus of a phosphite ligand. The signal at −20 ppm is observed as a singlet in the in the 1H{31P} NMR spectrum, and the proton-coupled 31P NMR spectrum contains a doublet at 103 ppm with the same coupling constant, confirming that the complex contains a hydride and a bound phosphine. The resonance in the 31P NMR spectrum contains 29Si satellites with a coupling constant (JP–Si = 303 Hz) that is larger than the typical range of one- or two-bond Si–P coupling constants (1JP–S = 16–256 Hz, 2JP–S = 0–44 Hz).13 We hypothesize that the coupling constant is large because the Si atom and the P atom of the phosphite are mutually trans across the Ir center.14 Overall, these data demonstrate that the combination of [Ir(cod)(OMe)]2, 2,9-Me2phen, and excess heptamethyltrisiloxane forms an iridium complex that contains one phenanthroline ligand, two heptamethyltrisiloxyl ligands, and one hydride (Scheme 1).
The identity and connectivity of phosphite adduct 3 was further assessed by single-crystal X-ray diffraction. The solid-state structure of 3 consists of an octahedral Ir(III) center containing mutually trans silyl and phosphite ligands. An additional silyl group and the hydride ligand are located in the same plane as the phenanthroline ligand. The silyl groups are geared to minimize steric interactions. The two siloxy groups of the axial silyl group lie over the phenanthroline so that the methyl group points toward the other large silyl group. One of the trimethylsiloxy substituents on the equatorial silyl ligand lies in the equatorial plane of the complex and is close to the hydride ligand, while the other two substituents lie above and below the methyl group of the 2,9-Me2phen. It is difficult to envision an orientation of the silyl groups that would allow three silyl groups, rather than two silyl groups and a hydride, to be present on iridium.
Lewis bases other than phosphites form similar iridium complexes that are less stable to isolation. Reaction of complex 1 with 4-dimethylaminopyridine (DMAP) yielded a six-coordinate complex 4 containing DMAP bound to iridium (Scheme 2). The 1H NMR spectrum of this complex contains a pattern of signals that are similar to those of phosphite adduct 3, including a signal corresponding to a metal hydride at −20 ppm that integrates to 1 proton. However, only one set of broad peaks was observed for the protons of DMAP when excess DMAP was present in solution, and the integration of these broad peaks corresponded to more than one equivalent of DMAP. This result suggests that free and bound DMAP undergo exchange on the NMR time scale.15
Scheme 2.

Synthesis of Pyridine Adduct of Catalyst
The rate of the silylation of arenes is ten times lower under an atmosphere of hydrogen than under nitrogen. To determine the mechanism by which hydrogen gas inhibits the silylation reaction, Complex 1 generated in situ was treated with 1 atm of H2.
The 1H NMR spectrum of this solution at −86 °C contains peaks that are different from those in the spectrum acquired under nitrogen. Under nitrogen, a single hydride peak that integrates to one proton is observed at −16 ppm. Under hydrogen and at −86 °C, two sharp peaks corresponding to metal hydrides, which integrate to one and two protons, were observed at −2.7 ppm and at −18.4 ppm. The peak at −2.7 ppm contains satellites from coupling to 29Si (JH–Si = 61 Hz). This coupling constant and the downfield chemical shift of this peak, relative to that of the other hydride, indicate that this resonance corresponds to the proton of a silane σ-complex.16 Thus, we assign complex 2 as an iridium complex bearing a phenanthroline unit, two hydride ligands, a silyl ligand, and an Si–H-bound silane (Scheme 3). This complex is the second, minor species formed from the combination of [Ir(cod)(OMe)]2, 2,9-Me2phen, and excess heptamethyltrisiloxane discussed earlier in this section.
Scheme 3.

Synthesis of Iridium Dihydride Complexes
The formation of σ-silane complex 2 in the presence of hydrogen implies that the inhibition of the catalytic silylation reaction by hydrogen gas results from a difference in the resting state of the catalyst in the presence and absence of hydrogen. Complex 1 contains an open coordination site that binds the C–H bond of an arene, whereas complex 2 is coordinatively saturated and must dissociate silane or hydrogen to react with an arene.
To further assess the effect of hydrogen gas on the iridium complexes formed from silane, phosphite adduct 3 was allowed to react with hydrogen gas (Scheme 3). The reaction of 3 with 1 atm of H2 formed complex 5, which contains two hydrides and one silyl group, in addition to the phosphite. This complex was characterized by 1H, 13C, and 31P NMR spectroscopy. The identity of 5 was clear from the doublet at −19 ppm in the 1H NMR spectrum integrating to two hydrogens and the triplet at 108 ppm in the 31P NMR spectrum.
2. Reactivity of Isolated Iridium Complex.
To assess the relevance of the iridium complexes bearing phenanthroline, hydride, and silyl ligands to the silylation of C–H bonds, phosphite-adduct 3 was allowed to react with a series of arenes. Reaction of 3 with 2-Cl-thiophene conducted in the presence of varied amounts of triphenyl phosphite showed that this reaction is inverse first order in the concentration of phosphite (see page 15 of the Supporting Information for details). These data indicate that 3 reacts with arenes after initial, reversible dissociation of the phosphite ligand.
The reaction of phosphite-ligated disilyl hydride 3 with 3-Cl-thiophene produced a 2.8 to 1 ratio of the two isomeric thienyl silanes 6 and 7 from the silylation of the C–H bonds alpha to sulfur (Scheme 4). The catalytic reaction of heptamethyltrisiloxane with 3-Cl-thiophene in the presence of the combination of [Ir(cod)(OMe)]2 and 2,9-Me2phen also produced 6 and 7 in the ratio of 2.8 to 1. These identical ratios imply that the catalytic silylation occurs through the five-coordinate disilyl monohydride complex 1 formed by dissociation of phosphite and imply that the observed resting state 1 lies directly on the catalytic cycle.
Scheme 4.
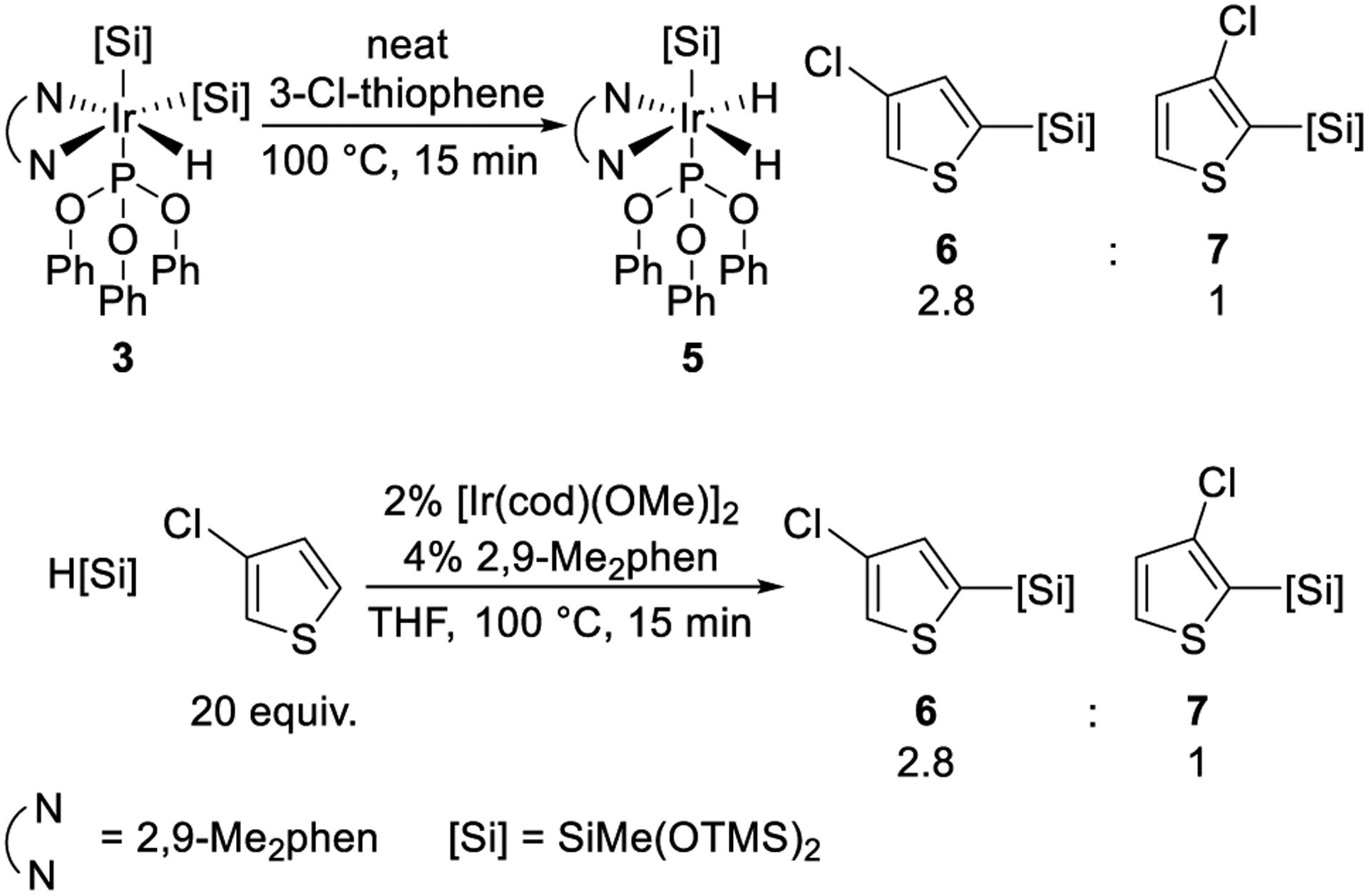
Reactions of 3 or Silane with Heteroarene
Iridium complex 3 reacted with both electron-rich and electron-poor deuterated arenes to produce deutero-aryl silanes (Scheme 5). The reaction of o-xylene-d4 with 3 produced deutero-aryl silane 8 and iridium complex 5 that contains a mixture of hydride and deuteride ligands. At partial conversion of complex 3, 1H NMR spectroscopy indicated the lack of exchange of deuterium into the hydride position of 3. This result implies that cleavage of the C–D bond of the electron-rich arene o-xylene-d4 is effectively irreversible, due to faster formation of the Si–C bond from the aryliridium complex than reformation of the C–H bond.
Scheme 5.

Reactions of 3 with Deuterated Arenes
In contrast, the reaction of 3 with the electron-poor deuteroarene o-Cl2-benzene-d4 led to exchange of deuterium into 3. The reaction of 3 with o-Cl2-benzene-d4 produced deutero-aryl silane and complex 5 containing a mixture of hydride and deuteride ligands. However, at partial conversion of complex 3 to product, 1H NMR spectroscopy indicated significant substitution of deuterium from the arene for the hydride of 3. These results indicate that the cleavage of the C–D bond of electron-poor arene Cl2-benzene-d4 is reversible.
3. Kinetic Analysis of the Catalytic Process.
The dependence of rate of the catalytic silylation reaction on the concentration of each reagent was measured to reveal the rate limiting step of the catalytic process. The reaction orders in silane, catalyst, and arene were determined by measuring initial rates of the silylation reaction at 60–100 °C with varied concentrations of each reagent. Reactions with concentrations of catalyst ranging from 1.25 to 10 mM showed that the reaction is first order in catalyst. Reactions with concentrations of silane varying from 0.5 to 4 M showed that the reaction is zero order in silane (see pages 16–17 of the Supporting Information for details).
The order in arene depended on the electronic character of the C–H bonds of the arene. The rates of reactions of silane with an electron-poor arene, methyl 3-CF3-benzoate, and an electron-rich arene 1,3-(OMe)2-benzene were determined (see pages 16–19 of the Supporting Information for details). Reactions conducted with concentrations of methyl 3-CF3-benzoate varying from 0.25 to 2 M were first order in the concentration of arene.
However, the order of the reaction in the electron-rich arene, 1,3-(OMe)2-benzene, was not straightforward. The increase in rates of reactions conducted with concentrations of 1,3-(OMe)2-benzene varying from 0.25 to 2 M in an open system diminished as the concentration of 1,3-(OMe)2-benzene increased (Figure 2, top). We hypothesized that this saturation of the rate resulted from a change in resting state from five-coordinate 1 to a complex with arene bound at high concentrations of the arene. To test this hypothesis, we measured the rate of the same reaction in the presence of 0.2 equiv of 3,5-lutidine. We hypothesized that lutidine would bind iridium more tightly than an arene, prevent binding of the arene, and simplify the kinetic behavior. Indeed, reactions conducted with concentrations of 1,3-(OMe)2-benzene varying from 0.25 to 2 M in the presence of 3,5-lutidine were first order in 1,3-(OMe)2-benzene (Figure 2, bottom). These results suggest that electron-rich arenes bind to the catalyst in the absence of strong Lewis bases.
Figure 2.

Dependence of rate on arene concentration.
The kinetic isotope effect (KIE) of the catalytic process was measured for three electronically varied arenes to gain further information on the reversibility of the C–H bond cleavage (Figure 3). The rates of the reaction of the protio and deutero substrates were measured independently. A kinetic isotope effect of 3.2 was obtained from the rate of formation of silylarene from o-xylene and o-xylene-d4 side by side (7.3 × 10−5 and 2.3 × 10−5 M/s). This KIE is similar in magnitude to those measured for the borylation of protio and deutero o-Cl2-benzene catalyzed by an Ir(III) boryl complex (KIE = 3.3).17 On the basis of these data, C–H bond cleavage of the arene is irreversible and rate limiting for the silylation of electron-rich arenes. These data are consistent with the observed irreversibility of C–H bond cleavage of o-xylene-d4 by complex 3 (vide supra).
Figure 3.

Determination of kinetic isotope effects. See reference 13.
The KIEs measured for reactions of electron-poor and electron-neutral arenes were different from that observed for o-xylene. The KIE for the analogous silylation of benzene and benzene-d6 was reported previously to be 1.3.4b The KIE from the rate of formation of silylarene from o-Cl2-benzene and o-Cl2-benzene-d4 side by side was 1.6 (kH = 6.3 × 10–5, kD = 3.9 × 10–5 M/s). These KIE values are smaller than that of the reaction of electron-rich o-xylene and do not suggest that the KIE is primary and resulting from rate-limiting C–H bond cleavage. Instead, these data are more consistent with reversible C–H bond cleavage during reactions of electron-poor or electron-neutral substrates. Such equilibrium isotope effects suggest that reductive elimination of the C–Si bond is rate-limiting for reactions of electron-poor and electron-neutral arenes.
The low KIE values measured for the silylation of protiated and deuterated electron-neutral and electron-poor arenes indicate that the transition state for a step following cleavage of the C–H bond is the highest-energy transition state during the silylation of these arenes. This transition state could be the one for reductive elimination of the C–Si bond or for an isomerization of the Ir complex that results from addition of the C–H bond. Such an isomerization has been computed to occur during the borylation of benzylic methyl groups catalyzed by iridium complexes of phenanthroline ligands. On the basis of a modest, but positive ρ value for the borylation of benzylic C–H bonds with different electronic character (ρ = 2.1) isomerization was concluded to be the rate-limiting step of the borylation of benzylic methyl groups.18
We considered that rate-limiting isomerization and rate-limiting reductive elimination could be distinguished by the effects of the electronic properties of the arene on the outcome of the silylation reaction. If an isomerization of the Ir complex that results from addition of the C–H bond is the rate-limiting step, then the effect of the electronic properties of the arene on the silylation should be large because the starting material is a free arene and the transition-state for the rate-limiting step contains a metal-carbon bond. However, if reductive elimination is rate-limiting, then the effect of the electronic properties of the arene on the silylation should be smaller. The effect should be smaller because the amount of negative charge on the ipso carbon in the transition state for reductive elimination is less than that of the complex that immediately precedes reductive elimination.
A Hammett plot of the effect of the electronics of the arene on the rate of the silylation is shown in Figure 4. The ρ value for this plot is 1.3. This positive ρ value indicates the accumulation of negative charge on the ipso carbon in the transition state of the rate-limiting step of the reaction. The magnitude of this ρ value is inconsistent with the values obtained from related borylation reactions that occur with rate-limiting cleavage of the C–H bond (ρ = 3.3) or isomerization of the complex that results from oxidative addition (ρ = 2.1).18 Instead, the measured ρ value is most consistent with the silylation of electron-poor arenes occurring by reversible cleavage of the C–H bond followed by rate-limiting reductive elimination of the C–Si bond.
Figure 4.

Hammett plot for silylation of arenes.
4. Proposed Catalytic Cycles.
On the basis of the orders in arene, silane, and catalyst, kinetic isotope and electronic effects, isolated adducts of the resting state, and iridium complexes observed in solution, we conclude that the iridium-catalyzed silylation of aromatic C–H bonds occurs by the catalytic cycle in Scheme 6. The silylation of electron-rich, electron-neutral, and electron-poor C–H bonds occurs by the same intermediate iridium complexes, but reactions of these arenes differ in the rate-limiting step and the resting state of the catalyst.
Scheme 6.
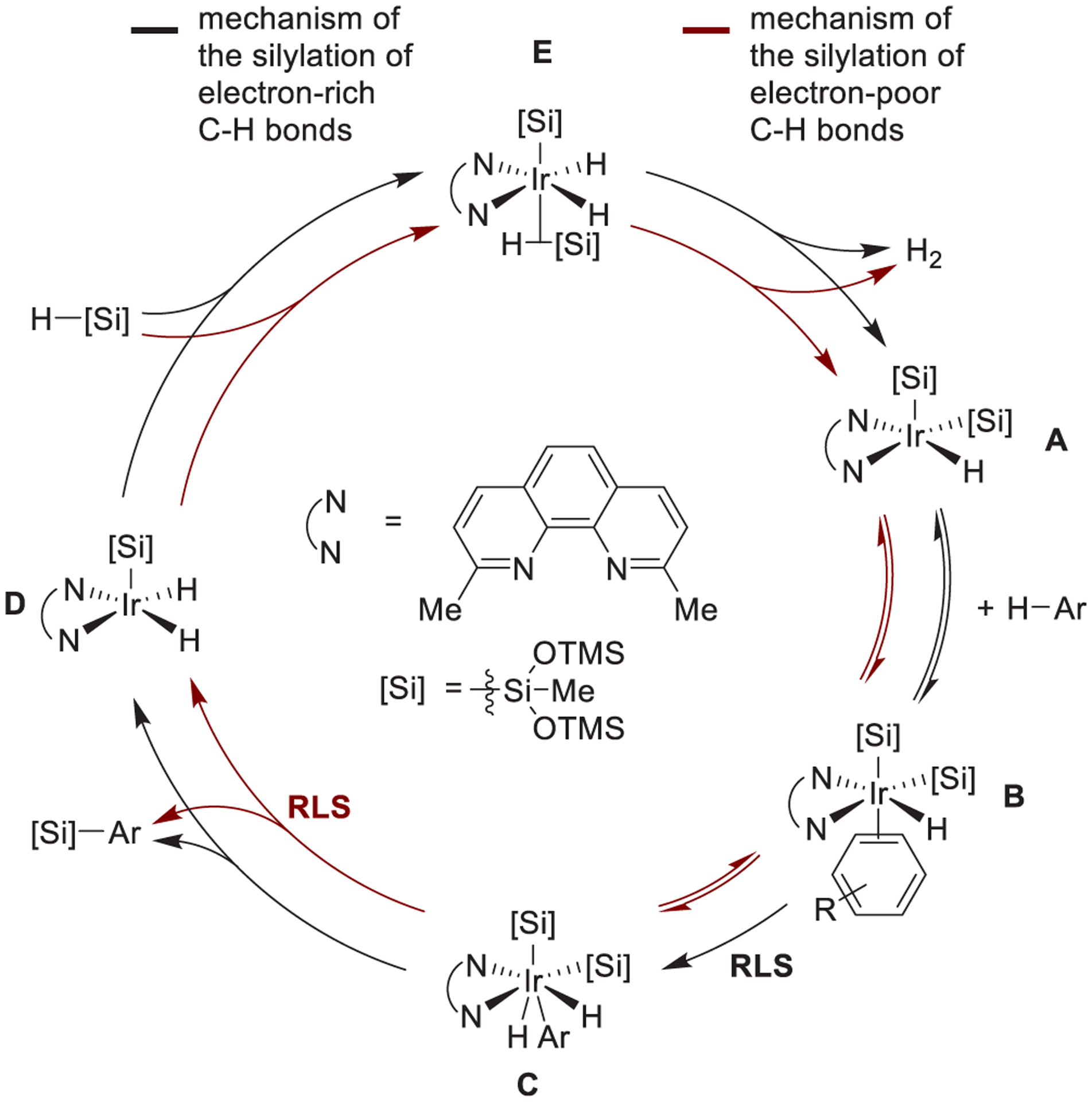
Proposed Catalytic Cycles for the Silylation of Electron-Rich or Electron-Poor Aromatic C–H Bonds
For reactions of electron-poor or electron-neutral arenes (Scheme 6, red), the resting state of the catalyst is the 5-coordinate iridium disilyl hydride complex (A). Complex A reversibly binds the arene to form complex B. Complex B undergoes reversible C–H bond cleavage to form an Ir(V) complex (C). Reductive elimination of the C–Si bond from C forms the aryl silane product and complex D. Complex D undergoes oxidative addition of the silane silane to produce complex E. For reactions conducted in an open system, complex E undergoes irreversible loss of H2 to regenerate resting state A.
For reactions of electron-rich arenes (Scheme 6, black), the resting state of the catalyst is the coordinatively saturated arene-bound iridium disilyl hydride complex (B). Complex B undergoes irreversible and rate-limiting C–H bond cleavage to form complex C. Reductive elimination of a C–Si bond from C forms aryl silane product and complex D. Complex D undergoes oxidative addition of the silane and reductive elimination of H2 to generate the coordinatively unsaturated complex A. A binds arene to regenerate the resting state of the catalyst, B.
5. Origin of Selectivity.
The silylation of o-Cl2-benzene-d4 provided information on the origin of the high selectivity for functionalization of the least sterically hindered C–H bond. During the silylation of o-Cl2-benzene-d4, the substitution of deuterium in the arene for hydrogen from the silane occurred 60 times faster than the silylation of the arene (Scheme 7). However, no incorporation of hydrogen is observed at the positions proximal to chlorine. These data demonstrate that the origin of high selectivity for the silylation of the least sterically hindered C–H bond in six-membered arenes is the high barrier to cleavage of the C–H bonds proximal to existing substituents.
Scheme 7.
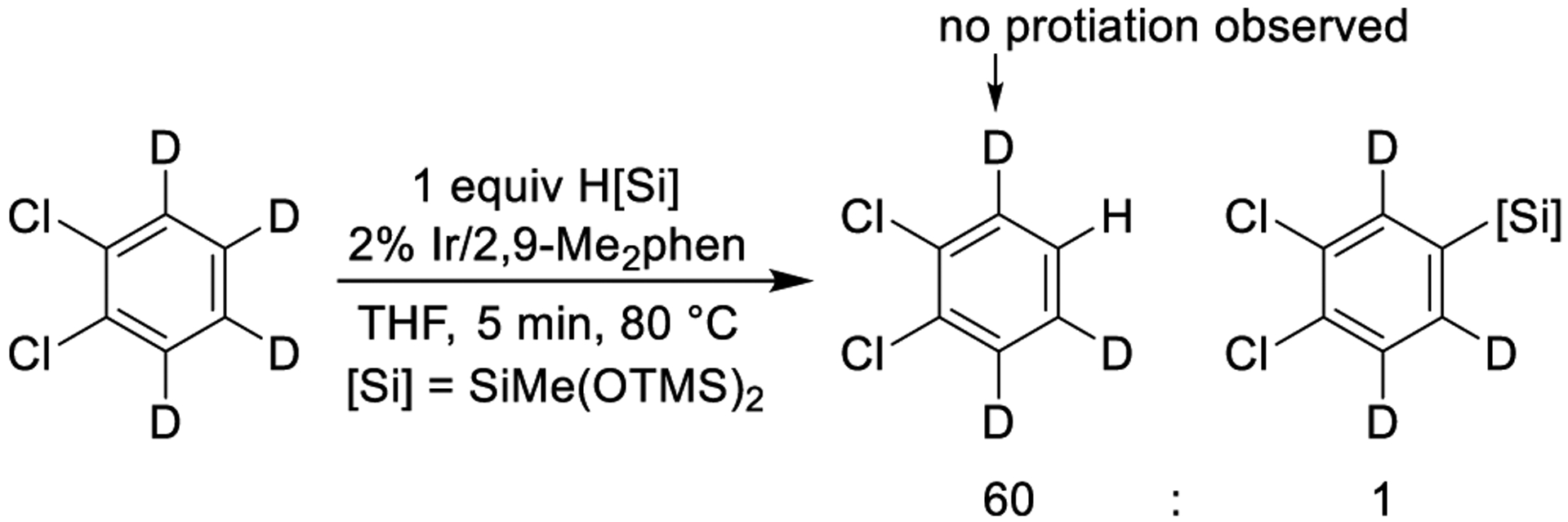
Observation of H/D Exchange Reaction
This origin of selectivity contrasts with that we reported recently for the silylation of five-membered ring heteroarenes.6 In our study of H/D exchange between the silane and these heteroarenes, the selectivity for H/D exchange at the least hindered C–H bond was lower than the selectivity for silylation at the least sterically hindered C–H bond. These data imply that the high selectivity for reactions of these less hindered substrates results from a slower rate for reductive elimination from a more hindered arylsilane than from a less hindered arylsilane.
6. Computational Model of Reaction Mechanism.
Experimental mechanistic studies enabled us to identify the iridium complexes that catalyze the silylation of aromatic C–H bonds and probe some of the elementary steps of the reaction. However, our experiments did not reveal why complexes containing two silyl ligands and one hydride ligand formed in the reaction, rather than complexes containing three silyl ligands. Our structural data from single-crystal X-ray diffraction of the disilyl hydride complex 3 began to show the severe steric hindrance that would be created by replacing the hydride with a third silyl group. The related iridium-catalyzed borylation reaction occurs by generation of triboryl compounds containing a phenanthroline or bipyridine ligand and reaction of this complex with arenes.17 The similarities in conditions and catalysts for the silylation and borylation reactions alone make it unclear why iridium complexes that contain three silyl ligands do not form during the silylation reaction and react with arenes.
However, our experimental studies do not reveal why the silylation reaction is faster with catalysts containing more sterically encumbered ligands. Like the difference in number of main-group ligands on the metal, this trend is not observed for the borylation of aromatic C–H bonds. One might imagine that the sterically encumbered 2,9-Me2phen could make low coordinate, Ir(I) silyl complexes thermodynamically accessible. We sought to use computational modeling to assess the effect of the steric properties of the phenanthroline ligand on the identity of the iridium species that form and the rates by which they react with the C–H bonds of arenes.
To understand the importance of the identity of the ancillary ligands on the rate and outcome of the reaction, we modeled iridium silyl complexes ligated by 3,4,7,8-tetramethylphenan-throline (Me4phen, L2) and by the sterically encumbered 2,9-Me2phen. We chose to simplify our model of the silane from SiMe(OTMS)2 to SiMe(OMe)2 (henceforth abbreviated as “[Si]”) to reduce the required computational time. Geometry optimizations of each structure were conducted with the Gaussian 09 software package, B3LYP functional (with gd3 dispersion correction), and LANL2DZ basis set for Ir and 6–31g(d,p) basis set for all other atoms. The energy of each geometry-optimized structure was further refined with single-point energy calculations that were conducted with the PBE functional (with g2 dispersion correction) and LANL2TZ basis set for Ir and the 6–31++g** basis set for all other atoms, along with the SMD THF solvent correction. This combination of basis set and functional was identified as particularly accurate by Hopmann for reactions of complexes of iridium.19
To assess whether an Ir(I) silyl complex is a thermodynamically accessible intermediate in the catalytic cycle of this reaction, we modeled the thermodynamics of the addition of two molecules of silane to (L)Ir[Si] complexes (L = Me4phen or 2,9-Me2phen) to produce (L)Ir[Si]3 complexes and one molecule of hydrogen (Figure 5). (L2)Ir[Si] and two molecules of silane were found to be 62 kcal/mol higher in energy than (L2)Ir[Si]3 and hydrogen. Because the Me4phen-Ir(I) species lies uphill of Ir(III)-silyl complexes in the presence of silane by such a large amount of energy, Ir(I) intermediates are unlikely to be part of the catalytic cycle.
Figure 5.

Computed energies for formation of Ir(III) complexes from Ir(I) complexes.
However, it is possible that the two methyl groups ortho to the nitrogen atoms in 2,9-Me2phen would destabilize Ir(III) intermediates. Thus, we calculated the thermodynamics for the same equation with 2,9-Me2phen as ligand. While less uphill, the reaction of (L1)Ir[Si] with two molecules of silane to form (L1)Ir[Si]3 and H2 remains exergonic by 53 kcal/mol. This result shows that the Ir(I) complex ligated by 2,9-Me2phen remains too far uphill thermodynamically to be a plausible intermediate in the catalytic reaction.
We modeled six Ir(III) complexes that are potential intermediates in the silylation of aromatic C–H bonds: (ligand)Ir[Si]3, (ligand)Ir[Si]2H, and (ligand)Ir[Si]H2 (Figures 6 and 7). Each complex was modeled with 2,9-Me2phen (L1) or Me4phen (L2) as ligand and was posed in a square pyramidal geometry with a silyl group in the axial position. This disposition of silyl and hydride ligands is based on the observed solid-state structure of complex 3, most sterically accessible geometry, and strong trans influence of a silyl ligand.20 For each combination of silyl and hydride ligands, we also modeled the transition state for cleavage of the C–H bond of benzene to the iridium and the ground-state energy of the seven-coordinate Ir(V) complex that is the product of this reaction (Figures 9 and 10).
Figure 6.

Computed energies of Ir(III) complexes of Me4phen and silyl and hydride Ligands.
Figure 7.

Computed energies of Ir(III) complexes of 2,9-Me2phen and silyl and hydride ligands.
Figure 9.
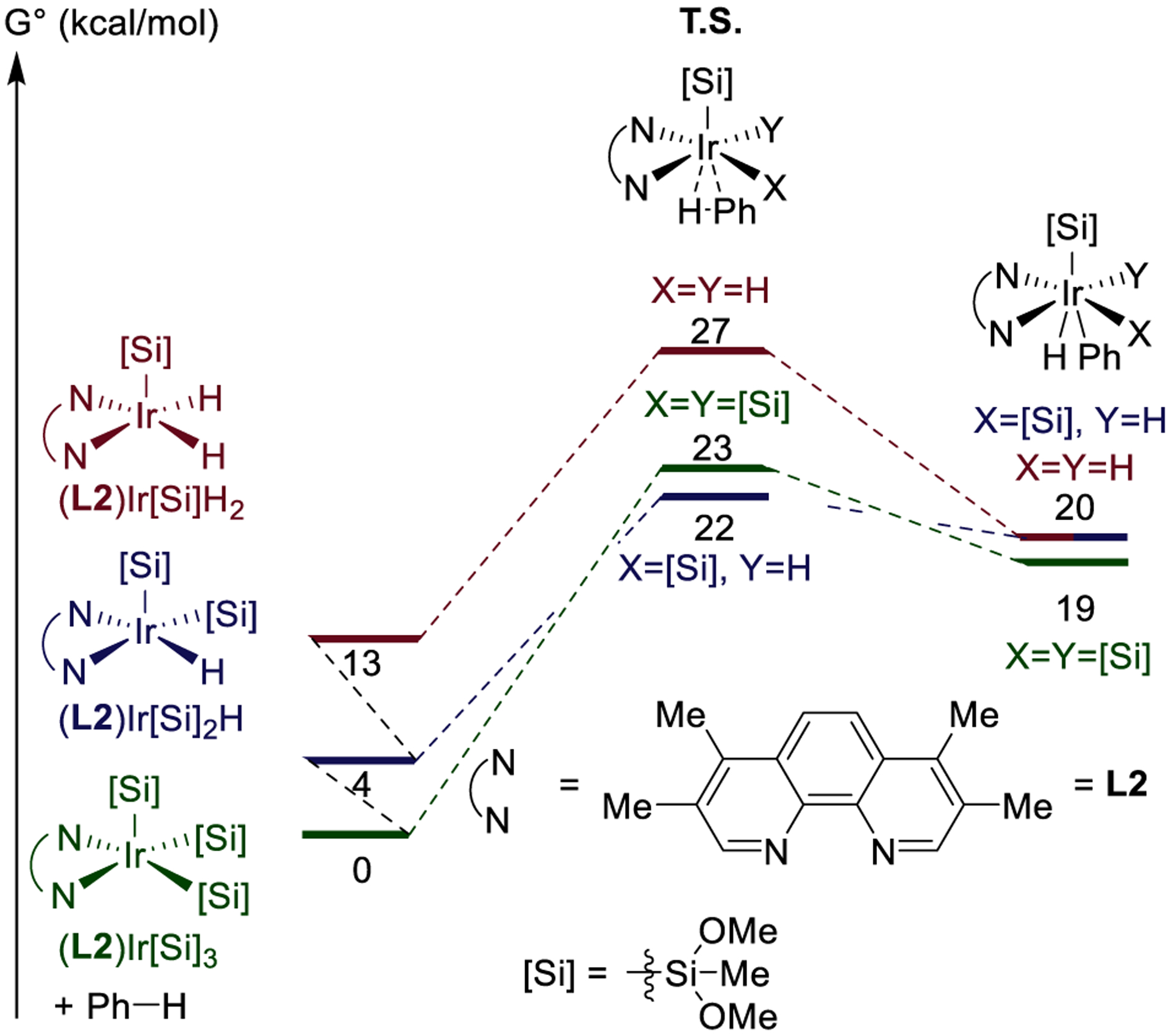
Computed energies of barriers to oxidative addition of benzene to Ir(III) complexes of Me4phen.
Figure 10.
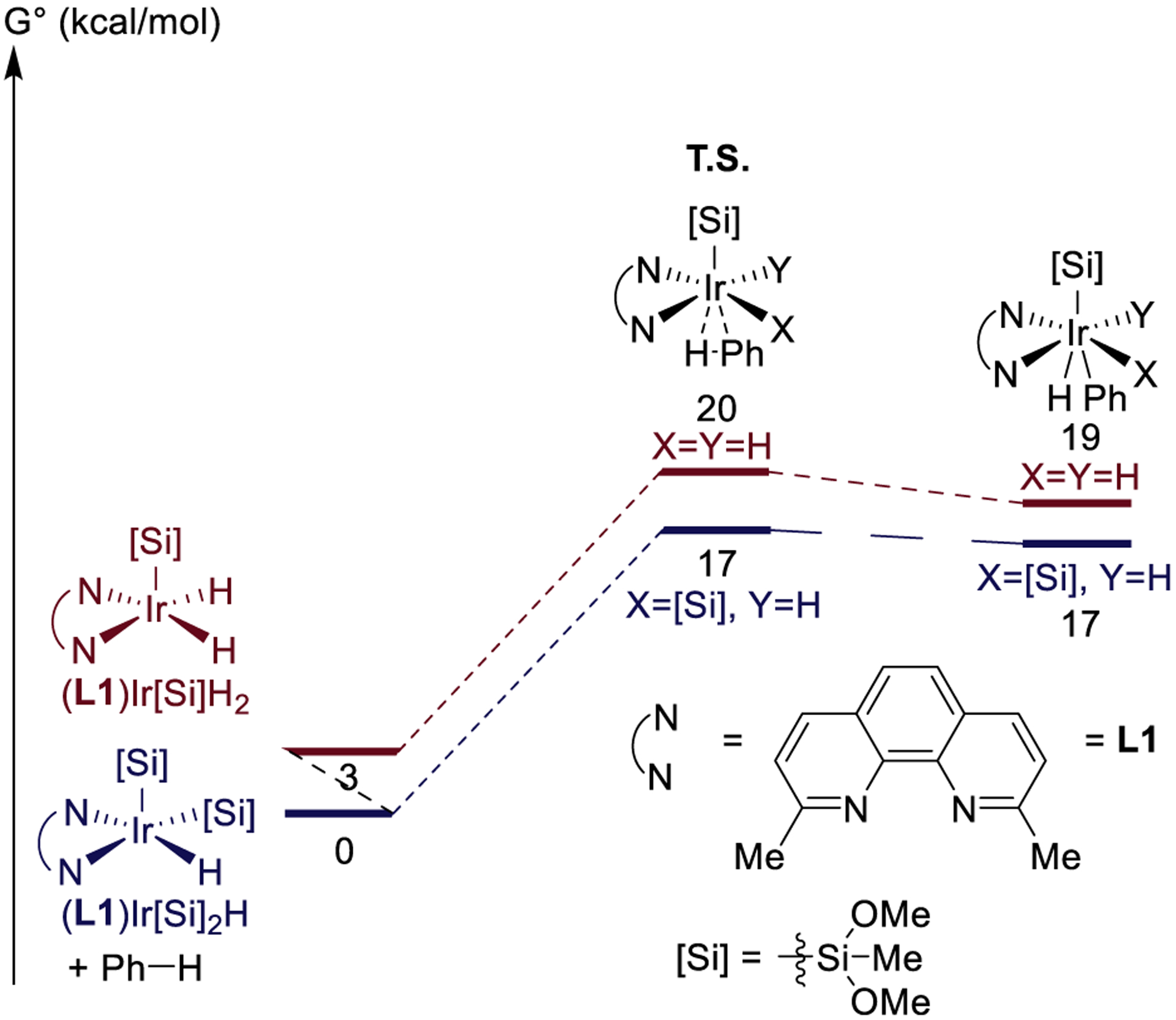
Computed energies of barriers to oxidative addition of benzene to Ir(III) complexes of 2,9-Me2phen.
For iridium complexes ligated by Me4phen, we found that the addition of silane to (L2)Ir[Si](H)2 to form hydrogen and (L2)Ir[Si]2(H) is exergonic by 9 kcal/mol (Figure 6). The addition of silane to (L2)Ir[Si]2(H) to form hydrogen and (L2)Ir[Si]3 is exergonic by a further 4 kcal/mol. These results predict that (L2)Ir[Si]3 would be the resting state of a C–H silylation reaction catalyzed by the combination of iridium precatalyst and Me4phen. This predicted resting state contains a different number of silyl and hydride ligands than the observed resting state of reactions catalyzed by iridium complexes containing the more sterically encumbered 2,9-Me2phen ligand (complex 1).21
The thermodynamics of the ground-state iridium complexes ligated by 2,9-Me2phen were much different from those of the complexes ligated by tmphen. For iridium complexes ligated by 2,9-Me2phen, the addition of silane to (L1)Ir[Si](H)2 to form hydrogen and (L1)Ir[Si]2(H) was computed to be exergonic by only 3 kcal/mol (Figure 7). This value is 6 kcal/mol less favorable than that for the reaction of silane with (L2)Ir[Si](H)2, in which the complex contains an unhindered ligand, Me4phen. Moreover, the addition of silane to (L1)Ir[Si]2(H) to form hydrogen and (L1)Ir[Si]3 was computed to be endergonic by 3 kcal/mol. This value is 7 kcal/mol less favorable than that for the reaction of silane with (L2)Ir[Si](H)2, which contains the unhindered ligand Me4phen, and suggests that (L2)Ir[Si]2(H), not the trisilyl analog, will be the resting state of a C–H silylation reaction catalyzed by the combination of iridium precatalyst and 2,9-Me2phen. This computational prediction is consistent with the experimentally observation of complexes 1 and 3 in the catalytic and stoichiometric reactions.
The geometry of (L1)Ir[Si]3 is different from that of the other Ir(III) complexes we computed. For all complexes except (L1)Ir[Si]3, the phenanthroline ligand lies in the same plane as the basal plane of the iridium complex. In contrast, for (L1)Ir[Si]3, the phenanthroline ligand lies 64° out of the basal plane of the iridium complex (Si(1)–N(1)–C(3) = 116°), causing the methyl groups to lie further from the silyl substituents (Figure 8). For comparison, this distortion is 16° for (L1)Ir[Si]2(H) and 7° for (L2)Ir[Si]3. A comparison of the structures of (L1)Ir[Si]3 to that of (L1)Ir[Si]2(H) and (L2)Ir[Si]3 shows that the distortions of the ligand in (L1)Ir[Si]3 reduce clashes between the methyl groups of the ligand and the silyl groups. This distortion hinders approach of the arene to the open coordination site of the iridium complex.
Figure 8.
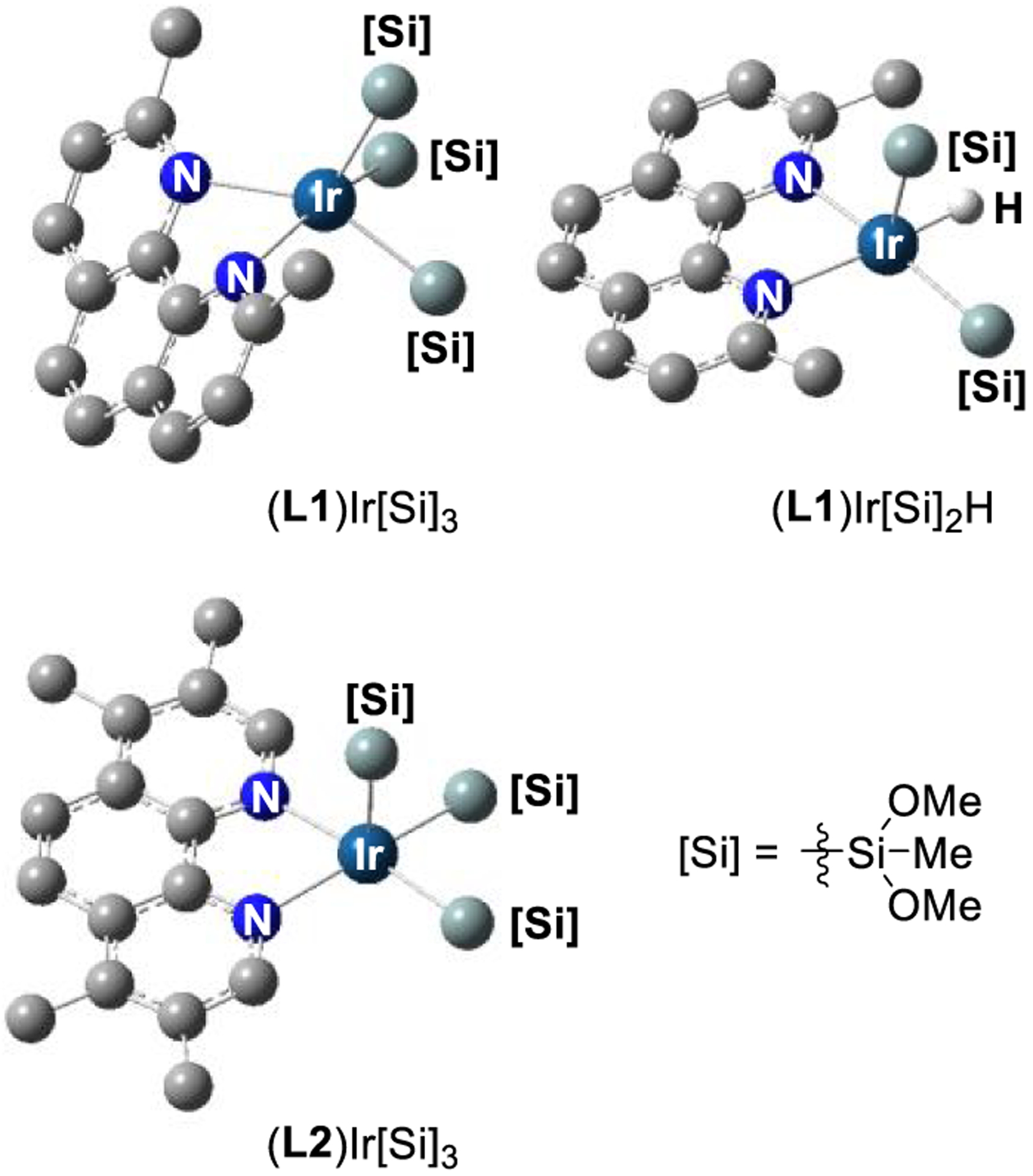
Computed structures of Ir(III) complexes. Silyl groups are truncated for visual clarity.
We also computed the transition state for cleavage of the C–H bond. This process is best described as an oxidative addition to form an iridium(V) complex containing a hydride and aryl unit. However, the addition to related boryl complexes occurs with association of the hydrogen of the C–H bond to the boryl group and addition of the silane occurs with partial association of the silane with the same hydrogen. This process resembles a σ-CAM mechanism, but the resulting Ir complex is better described as an Ir(V) complex than an Ir(III) silyl complex. More details on these relationships will be published elsewhere, but the Si–H bond orders determined by NBO analysis in the transition state and product are 0.1 and 0.2. Previously, iridium complexes with bond orders of this magnitude between a hydride and silyl group have been assigned as iridium silyl complexes, rather than iridium silane complexes.22 The computed bond order of classic Mn silane complexes were greater than 0.3.23
The barrier for oxidative addition of benzene to the Ir(III) silyl complexes depended on the number of silyl and hydride ligands. The barrier for oxidative addition to (L2)Ir[Si](H)2 (L2= Me4phen) was computed to be just 14 kcal/mol, that to (L2)Ir[Si]2(H) was computed to be 18 kcal/mol, and that to (L2)Ir[Si]3 was computed to be 23 kcal/mol (Figure 9). Thus, each substitution of a small hydride ligand for a bulky silyl ligand led to a 4–5 kcal/mol increase in the barrier to C–H activation. These results show that a catalyst containing fewer silyl ligands and more hydride ligands cleaves C–H bonds with higher rates than a catalyst containing more silyl ligands and fewer hydride ligands.
The computed relative energies of the barriers to oxidative addition of the C–H bond of benzene to 2,9-Me2phen-ligated iridium complexes were different than those to oxidative addition to Me4phen-ligated complexes. The barrier to oxidative addition of the C–H bond of benzene was computed to be 17 kcal/mol for both (L1)Ir[Si]2(H) and (L1)Ir[Si](H)2 (Figure 10). In contrast, we could not even locate a transition state geometry for the oxidative addition of the C–H bond of benzene to (L1)Ir[Si]3; attempts to model this transition state showed that the open coordination site was too hindered to accommodate approach of benzene to the Ir center. These calculations corroborate our conclusion that the resting state of the C–H silylation reactions catalyzed by the combination of iridium and 2,9-Me2phen contains two silyl ligands and one hydride and that C–H bond cleavage occurs by oxidative addition to this species. These results also are consistent with the experimentally observed reactivity of isolated complex 3 with arenes and heteroarenes.
The calculated free energy of activation for oxidative addition of the C–H bond of benzene to (L1)Ir[Si]2(H) (17 kcal/mol) is lower than the experimental value of 27 kcal/mol for the catalytic process. However, this difference likely results from the difference in the silane used in the calculations. The silane used, HSiMe(OMe)2, contains alkoxy substituents that are much smaller than the siloxy substituents in the heptamethyltrisiloxane used in the catalytic reaction. It is likely that the larger silyl groups present in the catalytic reaction would hinder the approach of the arene to the Ir center and increase the barrier to oxidative addition. Indeed, the calculated free energy of activation for oxidative addition of the C–H bond of benzene to complex 1 is 21 kcal/mol. While lower than the experimental value, this calculated barrier is more consistent with the observed rates of the catalytic reaction.
To understand the full catalytic cycle, we modeled other iridium complexes that are intermediates in the catalytic process, including the transition-state geometry for the carbon-silicon bond-forming reductive elimination and the Ir(V) complex that contains two silyl ligands and three hydride ligands. The barrier to reductive elimination of the phenylsilane derivative from (L1)Ir[Si]2(H)2(Ph) lies 17 kcal/mol higher in energy than the combination of (L1)Ir[Si]2(H) and benzene (Figure 11, left). This barrier to reductive elimination to form the C–Si bond is almost identical to the barrier to oxidative addition of the C–H bond. The similarities in the barriers for oxidative addition of the C–H bond of benzene and reductive elimination of the C–Si bond of the phenylsilane derivative are consistent with the experimental observation that differences in the electronic properties of the arene can cause the oxidative addition of the C–H bond or the reductive elimination to form the C–Si bond to be the step with the highest-energy transition state or “rate-limiting step.”
Figure 11.
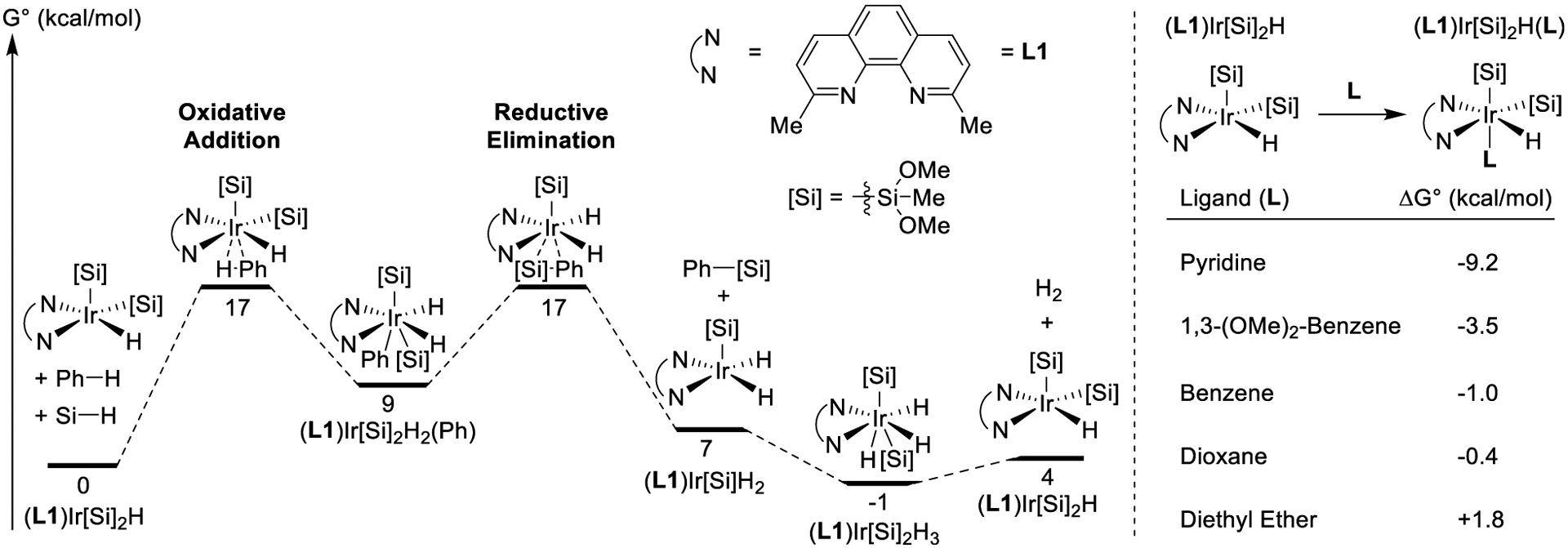
Computed energies for silylation of benzene and energies of binding of ligands to (L1)Ir[Si]2H.
The energies of the products of reductive elimination, (L1)Ir[Si](H)2, and the phenylsilane derivative are 7 kcal/mol higher than those of the starting molecules, (L1)Ir[Si]2(H) and benzene. The energy of (L1)Ir[Si]2(H)3 and the phenylsilane derivative is 1 kcal/mol lower than the energy of (L1)Ir[Si]2(H), silane, and benzene. (L1)Ir[Si]2(H)3, which is similar to the observed iridium complex with one silyl, two hydride, and one silane ligands (complex 2) is calculated to be 5 kcal/mol more stable than (L1)Ir[Si]2(H) and H2. The overall reaction of benzene with silane to form the phenylsilane derivative and H2 is computed to be 4 kcal/mol uphill in free energy at standard state. These results suggest that the experimental observation of the continuous removal of hydrogen from the reaction, necessary to obtain high rates and yields of aryl silane formation, is due to the unfavorable thermodynamics of the overall process and is manifested in the unfavorable regeneration of resting state complex 1 from hydrogen adduct 2 in a closed system.
The potential catalyst resting states that were modeled are all five-coordinate iridium complexes with open coordination sites. Having identified the most stable combination of silyl and hydride ligands, we modeled complexes of (L1)Ir[Si]2(H) with 1,4-dioxane, Et2O, benzene, 1,3-dimethoxybenzene, or pyridine as a sixth ligand. We found that the difference in the free energy of (L1)Ir[Si]2(H)(L) and of the combination of (L1)Ir[Si]2(H) and L was small for Et2O, dioxane, and benzene (Figure 11, right). However, the free energy of (L1)Ir[Si]2(H)(L) was significantly lower than that of (L1)Ir[Si]2(H) and L for L= 1,3-dimethoxybenzene and pyridine (−3.5 and −9.2 kcal/mol, respectively). These results predict that electron-rich arenes and pyridines should be bound to iridium in the resting state of the catalyst, and this prediction is consistent with the observation of signals in the 1H NMR spectrum of the solution of Ir, silane, 2,9-Me2phen, and pyridine indicating that the pyridine binds to iridium and the saturation of the rates of the reactions of the electron-rich arene, 1,3-dimethoxybenzene as a function of the concentration of arene.
The computational model described here suggests that the rate of oxidative addition of aromatic C–H bonds is slower to iridium complexes that contain more bulky silyl ligands and fewer small hydride ligands than to complexes that contain fewer silyl ligands and more hydride ligands. The substituents ortho to nitrogen on the phenanthroline ligand prevent the formation of Ir complexes containing three silyl ligands that are the least reactive. This destabilization of the Ir complex containing three silyl ligands changes the resting state of the reaction from a trisilyl complex with the less hindered tmphen ligand to a disilyl hydride complex with the more hindered 2,9-Me2phen ligand. The disilyl hydride complex undergoes more rapid oxidative addition of C–H bonds than the corresponding iridium complex that contains three silyl groups.
CONCLUSION
At the outset of this study, no experimental data had been published on iridium complexes that are likely intermediates in the silylation of aromatic C–H bonds. Only computational studies were reported on the mechanism of the silylation of any type of C–H bond. Through the combination of high-resolution mass spectrometry, low-temperature NMR spectroscopy, and trapping experiments with several dative ligands, we showed that the resting state of the iridium catalyst is (2,9-Me2phen)Ir(SiMe(OTMS)2)2(H). By studying the reactions of deuterated arenes with an isolated iridium complex and silane in the catalytic reaction, we determined that C–H bond cleavage occurs to the five-coordinate, Ir(III) complex (2,9-Me2phen)Ir(SiMe(OTMS)2)2(H). This C–H bond cleavage is the rate limiting step of the silylation of electron-rich arenes, and C–Si bond formation is the rate limiting step of the silylation of electron-neutral and electron-poor arenes. In addition, the combination of kinetic data and computational modeling revealed the propensity of electron-rich arenes to bind the catalyst. Finally, our computational model rationalizes the high rates observed for the silylation of arenes catalyzed by iridium complexes of sterically encumbered phenanthrolines. Steric clashes between the methyl groups on the phenanthroline and the silyl groups on Ir prevent the formation of the less reactive (L1)Ir[Si]3 complexes and favor the formation of the more reactive (L1)Ir[Si]2(H) complexes.
The mechanistic data described here provide a new model for understanding the silylation of C–H bonds. The knowledge that the catalyst contains an unsymmetrical disposition of silicon and hydrogen ligands informs the design of ligands for new silylation reactions and lays the groundwork for a new generation of catalysts for the functionalization of C–H bonds that are currently being developed in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the NIH (R35 GM130387) (J.F.H.). We thank the College of Chemistry’s NMR facility for resources provided and the staff for their assistance. Instruments in CoC-NMR are supported in part by the NIH (S10OD024998). We thank the College of Chemistry’s Molecular Graphics and Computing facility for resources provided and staff for their assistance. The COC-MGCF is supported in part by the NIH (NIH S10OD023532). We thank Dr. Nicholas Settineri of Berkeley X-ray Crystallography Facility for solving the crystal structure of complex 3. C.K. thanks Zhewei Chen, Dr. Noam Saper, and Dr. Ala Bunescu for helpful discussions. C.K. thanks Eric Kalkman for assistance in the preparation of the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c03301.
Experimental procedures and spectroscopic data on the reaction products (PDF)
(CIF)
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Hartwig JF Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res 2012, 45, 864. [DOI] [PubMed] [Google Scholar]; (b) Cheng C; Hartwig JF Catalytic Silylation of Unactivated C–H Bonds. Chem. Rev 2015, 115, 8946. [DOI] [PubMed] [Google Scholar]; (c) Obligacion JV; Chirik PJ Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem 2018, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Bracegirdle S; Anderson EA Arylsilane oxidation—new routes to hydroxylated aromatics. Chem. Commun 2010, 46, 3454. [DOI] [PubMed] [Google Scholar]; (b) Rayment EJ; Summerhill N; Anderson EA Synthesis of Phenols via Fluoride-free Oxidation of Arylsilanes and Arylmethoxysilanes. J. Org. Chem 2012, 77, 7052. [DOI] [PubMed] [Google Scholar]; (c) Rayment EJ; Mekareeya A; Summerhill N; Anderson EA Mechanistic Study of Arylsilane Oxidation through 19F NMR Spectroscopy. J. Am. Chem. Soc 2017, 139, 6138. [DOI] [PubMed] [Google Scholar]
- 3.Morstein J; Kalkman ED; Bold C; Cheng C; Hartwig JF Copper-Mediated C–N Coupling of Arylsilanes with Nitrogen Nucleophiles. Org. Lett 2016, 18, 5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Cheng C; Hartwig JF Iridium-Catalyzed Silylation of Aryl C–H Bonds. J. Am. Chem. Soc 2015, 137, 592. [DOI] [PubMed] [Google Scholar]; (b) Karmel C; Chen Z; Hartwig JF Iridium-Catalyzed Silylation of C–H Bonds in Unactivated Arenes: A Sterically Encumbered Phenanthroline Ligand Accelerates Catalysis. J. Am. Chem. Soc 2019, 141, 7063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karmel C; Rubel CZ; Kharitonova EV; Hartwig JF Iridium-Catalyzed Silylation of Five-Membered Heteroarenes: High Sterically Derived Selectivity from a Pyridyl-Imidazoline Ligand. Angew. Chem., Int. Ed 2020, 59, 6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Nakao Y; Hiyama T Silicon-based cross-coupling reaction: an environmentally benign version. Chem. Soc. Rev 2011, 40, 4893. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE; Ambrosi A Why You Really Should Consider Using Palladium-Catalyzed Cross-Coupling of Silanols and Silanolates. Org. Process Res. Dev 2015, 19, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karmel C; Rubel CZ; Kharitonova EV; Hartwig JF Iridium-Catalyzed Silylation of Five-Membered Heteroarenes: High Sterically Derived Selectivity from a Pyridyl-Imidazoline Ligand. Angew. Chem., Int. Ed 2020, 59, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ishiyama T; Sato K; Nishio Y; Miyaura N Direct Synthesis of Aryl Halosilanes through Iridium(I)-Catalyzed Aromatic C–H Silylation by Disilanes. Angew. Chem., Int. Ed 2003, 42, 5346. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T; Sato K; Nishio Y; Saiki T; Miyaura N Regioselective aromatic C-H silylation of five-membered heteroarenes with fluorodisilanes catalyzed by iridium(I) complexes. Chem. Commun 2005, 5065. [DOI] [PubMed] [Google Scholar]; (c) Saiki T; Nishio Y; Ishiyama T; Miyaura N Improvements of Efficiency and Regioselectivity in the Iridium(I)-Catalyzed Aromatic C-H Silylation of Arenes with Fluorodisilanes. Organometallics 2006, 25, 6068. [Google Scholar]; (d) Simmons EM; Hartwig JF Iridium-Catalyzed Arene Ortho-Silylation by Formal Hydroxyl-Directed C–H Activation. J. Am. Chem. Soc 2010, 132, 17092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ishiyama T; Saiki T; Kishida E; Sasaki I; Ito H; Miyaura N Aromatic C–H silylation of arenes with 1-hydrosilatrane catalyzed by an iridium(i)/2,9-dimethylphenanthroline (dmphen) complex. Org. Biomol. Chem 2013, 11, 8162. [DOI] [PubMed] [Google Scholar]; (f) Choi G; Tsurugi H; Mashima K Hemilabile N-Xylyl-N′-methylperimidine Carbene Iridium Complexes as Catalysts for C–H Activation and Dehydrogenative Silylation: Dual Role of N-Xylyl Moiety for ortho-C–H Bond Activation and Reductive Bond Cleavage. J. Am. Chem. Soc 2013, 135, 13149. [DOI] [PubMed] [Google Scholar]; (g) Su B; Zhou TG; Li XW; Shao XR; Xu PL; Wu WL; Hartwig JF; Shi ZJ A Chiral Nitrogen Ligand for Enantioselective, Iridium-Catalyzed Silylation of Aromatic C-H Bonds. Angew. Chem., Int. Ed 2017, 56, 1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Simmons EM; Hartwig JF Catalytic functionalization of unactivated primary C-H bonds directed by an alcohol. Nature 2012, 483, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Frihed TG; Heuckendorff M; Pedersen CM; Bols M Easy Access to L-Mannosides and L-Galactosides by Using C–H Activation of the Corresponding 6-Deoxysugars. Angew. Chem., Int. Ed 2012, 51, 12285. [DOI] [PubMed] [Google Scholar]; (c) Li BJ; Driess M; Hartwig JF Iridium-Catalyzed Regioselective Silylation of Secondary Alkyl C-H Bonds for the Synthesis of 1,3-Diols. J. Am. Chem. Soc 2014, 136, 6586. [DOI] [PubMed] [Google Scholar]; (d) Frihed TG; Pedersen CM; Bols M Synthesis of All Eight L-Glycopyranosyl Donors Using C–H Activation. Angew. Chem., Int. Ed 2014, 53, 13889. [DOI] [PubMed] [Google Scholar]; (e) Hua Y; Jung S; Roh J; Jeon J Modular Approach to Reductive Csp2–H and Csp3–H Silylation of Carboxylic Acid Derivatives through Single-Pot, Sequential Transition Metal Catalysis. J. Org. Chem 2015, 80, 4661. [DOI] [PubMed] [Google Scholar]; (f) Manna K; Zhang T; Greene FX; Lin W Bipyridine- and Phenanthroline-Based Metal-Organic Frameworks for Highly Efficient and Tandem Catalytic Organic Transformations via Directed C–H Activation. J. Am. Chem. Soc 2015, 137, 2665. [DOI] [PubMed] [Google Scholar]; (g) Su B; Hartwig JF Ir-Catalyzed Enantioselective, Intramolecular Silylation of Methyl C-H Bonds. J. Am. Chem. Soc 2017, 139, 12137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Su B; Lee T; Hartwig JF Iridium-Catalyzed, β-Selective C(sp3)–H Silylation of Aliphatic Amines To Form Silapyrrolidines and 1,2-Amino Alcohols. J. Am. Chem. Soc 2018, 140, 18032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ma X; Kucera R; Goethe OF; Murphy SK; Herzon SB Directed C–H Bond Oxidation of (+)-Pleuromutilin. J. Org. Chem 2018, 83, 6843. [DOI] [PubMed] [Google Scholar]; (j) Bian L; Cao S; Cheng L; Nakazaki A; Nishikawa T; Qi J Semi-synthesis and Structure-Activity Relationship of Neuritogenic Oleanene Derivatives. ChemMedChem 2018, 13, 1972. [DOI] [PubMed] [Google Scholar]; (k) Bunescu A; Butcher TW; Hartwig JF Traceless Silylation of β-C(sp3)–H Bonds of Alcohols via Perfluorinated Acetals. J. Am. Chem. Soc 2018, 140, 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Hung K; Condakes ML; Novaes LFT; Harwood SJ; Morikawa T; Yang Z; Maimone TJ Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes. J. Am. Chem. Soc 2019, 141, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons EM; Hartwig JF Iridium-Catalyzed Arene Ortho-Silylation by Formal Hydroxyl-Directed C-H Activation. J. Am. Chem. Soc 2010, 132, 17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parija A; Sunoj RB Mechanism of Catalytic Functionalization of Primary C–H Bonds Using a Silylation Strategy. Org. Lett 2013, 15, 4066. [DOI] [PubMed] [Google Scholar]
- 11.Li Z; Xia M; Boyd RJ Theoretical study on the mechanism of iridium-catalyzed γ-functionalization of primary alkyl C–H bonds. Can. J. Chem 2016, 94, 1028. [Google Scholar]
- 12.Zhang M; Liang J; Huang G Mechanism and Origins of Enantioselectivity of Iridium-Catalyzed Intramolecular Silylation of Unactivated C(sp3)–H Bonds. J. Org. Chem 2019, 84, 2372. [DOI] [PubMed] [Google Scholar]
- 13.(a) Marsmann H In 29Si-NMR Spectroscopic Results, Springer Berlin Heidelberg: Berlin, Heidelberg, 1981; pp 65. [Google Scholar]; (b) Smith PW; Tilley TD Base-Free Iron Hydrosilylene Complexes via an α-Hydride Migration that Induces Spin Pairing. J. Am. Chem. Soc 2018, 140, 3880. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bertrand RD; Ogilvie FB; Verkade JG Signs of phosphorus-phosphorus coupling constants in coordination compounds. J. Am. Chem. Soc 1970, 92, 1908. [Google Scholar]; (b) Brown JM; Dayrit FM; Lightowler D Interconversion of cis-and trans-dihydrides derived from chelate biphosphine iridium cations. J. Chem. Soc., Chem. Commun 1983, 414. [Google Scholar]
- 15.The reaction of excess 3,5-lutidine with complex 1 yielded a six-coordinate complex containing lutidine bound to iridium. However, the 1H NMR of the solution contains signals that are more similar in pattern to those of complex 1 than to those of the phosphite adduct 3. As in the 1H NMR spectra of 3 and DMAP-adduct 4, a signal corresponding to a metal hydride is present at −20 ppm, which integrates to 1 proton. These data indicate that exchange of the positions of the hydride and silyl ligands on this complex is much faster than that in DMAP adduct 4, due to the weaker binding constant of lutidine.
- 16.(a) Lipke MC; Liberman-Martin AL; Tilley TD Electrophilic Activation of Silicon-Hydrogen Bonds in Catalytic Hydrosilations. Angew. Chem., Int. Ed 2017, 56, 2260. [DOI] [PubMed] [Google Scholar]; (b) Lipke MC; Poradowski M-N; Raynaud C; Eisenstein O; Tilley TD Catalytic Olefin Hydrosilations Mediated by Ruthenium η3-H2Si σ Complexes of Primary and Secondary Silanes. ACS Catal. 2018, 8, 11513. [Google Scholar]; (c) Handford RC; Smith PW; Tilley TD Activations of all Bonds to Silicon (Si–H, Si–C) in a Silane with Extrusion of [CoSiCo] Silicide Cores. J. Am. Chem. Soc 2019, 141, 8769. [DOI] [PubMed] [Google Scholar]
- 17.Boller TM; Murphy JM; Hapke M; Ishiyama T; Miyaura N; Hartwig JF Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Tris Boryl Complexes. J. Am. Chem. Soc 2005, 127, 14263. [DOI] [PubMed] [Google Scholar]
- 18.Larsen MA; Wilson CV; Hartwig JF Iridium-Catalyzed Borylation of Primary Benzylic C-H Bonds without a Directing Group: Scope, Mechanism, and Origins of Selectivity. J. Am. Chem. Soc 2015, 137, 8633. [DOI] [PubMed] [Google Scholar]
- 19.Hopmann KH How Accurate is DFT for Iridium-Mediated Chemistry? Organometallics 2016, 35, 3795. [Google Scholar]
- 20.Zhu J; Lin Z; Marder TB Trans Influence of Boryl Ligands and Comparison with C, Si, and Sn Ligands. Inorg. Chem 2005, 44, 9384. [DOI] [PubMed] [Google Scholar]
- 21.When the crude mixture of [Ir(cod)(OMe)]2, L2, and excess heptamethyltrisiloxane was treated with triphenyl phosphite, the 1H NMR spectrum indicated that multiple phenanthroline containing and phosphite containing products formed. A doublet at −18.3 ppm, and the presence of 4 singlets between 0.5 and −1 ppm indicated the formation of a complex similar to 3. In addition, the 31P NMR spectrum contained a doublet at 102 ppm with the same coupling constant at the hydride in the 1H NMR spectrum. However, the signal intensities indicated that the doublet at 102 ppm corresponded to a minor species. Instead, a singlet at 128 ppm corresponded to the major product. Although these data are far from conclusive, we hypothesize that, in the presence of unhindered ligand L2, both trisilyl and disilyl hydride complexes are formed. The reaction catalyzed by complexes of L2 occur with both low rates and low catalyst lifetime; thus, it is unclear whether the formation of trisilyl complexes leads to less active catalysts for the silylation of aromatic C–H bonds or to catalyst deactivation.
- 22.Hamdaoui M; Ney M; Sarda V; Karmazin L; Bailly C; Sieffert N; Dohm S; Hansen A; Grimme S; Djukic J-P Evidence of a Donor-Acceptor (Ir–H)→SiR3 Interaction in a Trapped Ir(III) Silane Catalytic Intermediate. Organometallics 2016, 35, 2207. [Google Scholar]
- 23.McGrady GS; Sirsch P; Chatterton NP; Ostermann A; Gatti C; Altmannshofer S; Herz V; Eickerling G; Scherer W Nature of the Bonding in Metal-Silane σ-Complexes Inorg. Chem 2009, 48, 1588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.