

Abstract
Background
We examined feasibility of a unique approach towards gaining insight into heritable risk for early atherosclerosis: surveying gene expression by endothelial cells from living subjects.
Methods and Results
Subjects aged <50 years (mean age, 37; range, 22–49) without obstructive coronary artery disease underwent coronary reactivity testing that identified them as having normal or abnormal coronary endothelial function. Cultures of Blood Outgrowth Endothelial Cells (BOEC) from 6 normal and 13 abnormal subjects passed rigorous quality control and were used for microarray assessment of gene expression. Of 9 genes differentially expressed at false discovery rate <0.1%, we here focus upon abnormal subjects having elevated expression of HMGB1 (high mobility group box 1) which we unexpectedly found to be linked to low LAMC1 (laminin gamma 1) expression. This linkage was corroborated by 3 of our past studies and confirmed bio‐functionally. Compared with normal BOEC, abnormal BOEC released 13±3‐fold more HMGB1 in response to lipopolysaccharide; and they deposited one tenth as much LAMC1 into collagen subendothelial matrix during culture. Clinical follow‐up data are provided for 4 normal subjects (followed 13.4±0.1 year) and for 12 abnormal subjects (followed 9.1±4.5 years).
Conclusions
The known pathogenic effects of high‐HMGB1 and low‐LAMC1 predict that the combination would biologically converge upon the focal adhesion complex, to the detriment of endothelial shear responsiveness. This gene expression pattern may comprise a heritable risk state that promotes early coronary atherosclerosis. If so, the testing could be applied even in childhood, enabling early intervention. This approach offers a way to bridge the information gap between genetics and clinical phenotype.
Keywords: atherosclerosis, endothelial function, focal adhesion complex, focal adhesion kinase, HMGB1, laminin, risk factor, shear stress
Subject Categories: Cell Biology/Structural Biology, Pathophysiology, Genetics, Endothelium/Vascular Type/Nitric Oxide, Atherosclerosis
Nonstandard Abbreviations and Acronyms
- BOEC
blood outgrowth endothelial cells
- HMGB1
high mobility group box 1
- LAMC1
laminin gamma 1
Clinical Perspective
What Is New?
Using subjects aged <50 years shown by clinically‐indicated coronary reactivity testing to have either normal or abnormal coronary endothelial function, this feasibility study surveyed gene expression by blood outgrowth endothelial cells (a stable endothelial type that can identify heritable differences in gene expression) to bridge the gap between genomics and clinical phenotype—and has thereby provisionally identified a heritable risk factor for early development of atherosclerosis.
Compared with blood outgrowth endothelial cells from subjects having normal coronary endothelial function, blood outgrowth endothelial cells from subjects with abnormal coronary endothelial function exhibited abnormally elevated expression of HMGB1 (high mobility group box 1) that was linked to abnormally depressed expression of LAMC1 (laminin gamma 1).
What Are the Clinical Implications?
This combination may be a heritable risk factor for early atherosclerosis, since known effects and functions of HMGB1 and LAMC1 predict that their abnormal expression in the observed directions would biologically converge at the focal adhesion complex and endothelial cell membrane in a manner detrimental to the vascular endothelial cell's normal responsiveness to shear stress.
Clinical atherosclerosis emerges from complexity involving multiple promotive influences, cell types, and biologic systems. Although heritable factors are believed to account for ≥50% of disease risk, 1 only rarely does this involve a single gene exerting a large influence. Rather, the heritable component of risk most likely involves multiple genes that individually exert smaller effects. Identifying these has been a formidable challenge. We here demonstrate the feasibility of using a unique approach to bridging the information gap between genomics and clinical phenotype: assessing gene expression by endothelial cells obtained from living patients.
For this we use blood outgrowth endothelial cells (BOEC) from cultures of peripheral blood. Unlike cell types often labeled "EPC," BOEC are fully differentiated, bona fide endothelial cells that are the in vitro progeny of a circulating, marrow‐derived, transplantable endothelial progenitor. 2 , 3 , 4 , 5 Importantly, BOEC themselves have never been exposed to inflammatory or tissue‐specific influences of the in vivo environment. Even at high‐fold expansion they are far more stable than other endothelial types, and they are generic endothelial reporter cells uniquely suitable for a study such as this. 3 Since they can be obtained from a known donor, BOEC enable matching of donor characteristics to endothelial features of interest, in the context of measured heritable gene expression. Indeed, we previously applied this approach to identify underlying risk for a clinical stroke phenotype affecting some children with sickle cell anemia 4 and, separately, to suggest an influence of ancestral continent‐of‐origin on endothelial shear stress responsiveness. 5
The present study addresses the feasibility of using BOEC gene expression to identify risk for developing atherosclerosis at a young age. Operationally, the phenotype we focused on here is coronary endothelial dysfunction in subjects aged <50 years. Coronary endothelial dysfunction is the earliest clinically detectable form of atherosclerosis, providing gold‐standard evidence for presence of atheropromotive pathobiology. 6 , 7 , 8 , 9 , 10 Coronary endothelial dysfunction is associated with plaque progression and increased risk of major adverse cardiovascular events. 6 , 7 , 8 , 9
METHODS
This gene expression study was done in 2005 to 2007 and, hence, reflects technologies then extant. The delay in submission for publication was because the project leader paused from work for a decade because of a family medical catastrophe.
Methods described herein are sufficient to enable replication of the study. The new gene expression data underlying this report are deposited and available at Gene Expression Omnibus, series GSE132651; previously reported data extracted for use herein were previously deposited as series GSE22688 and GSE9877. Aliquots of the BOEC samples studied herein are present in our BOEC bio‐bank and may be accessible by contacting the corresponding author.
Subjects
This study was approved by the Institutional Review Boards at the University of Minnesota and the Mayo Clinic. Subjects were adults, aged <50 years (all but 1 was <46 years), undergoing clinically indicated invasive angiography (at the Mayo Clinic Catheterization Laboratory) for signs and/or symptoms suggestive of angina plus risk factors. Subjects gave written informed consent.
Coronary Reactivity Testing
Patients withheld all vasoactive prescription medications for at least 24 to 48 hours, and fasted for 12 hours, before coronary angiography and coronary reactivity testing. 8 , 10 Following diagnostic angiography and exclusion of obstructive coronary artery disease, we positioned a Doppler guidewire (Flowire, Volcano Therapeutics Inc, Rancho Cordova, CA) within a coronary‐infusion catheter into the mid‐left anterior descending (LAD) coronary artery. 11 , 12 We gave escalating intracoronary doses of acetylcholine (10−6, 10−5, and 10−4 mol/L for 3 minutes at each concentration), infused selectively into the mid‐LAD. We measured coronary artery diameter offline (by an independent investigator) in the segment 5 mm distal to the tip of the Doppler wire, using a quantitative coronary angiography program (Medis Corp, Leiden, the Netherlands) as previously described. 11 We calculated coronary blood flow in the LAD from the Doppler derived time velocity integral and vessel diameter, where coronary blood flow =π×(coronary artery diameter/2)2×(average peak velocity/2). Coronary endothelial dysfunction was defined as >20% decrease in mid‐LAD diameter and/or <50% increase in coronary blood flow in response to acetylcholine infusion. 13 , 14 , 15 Subjects were thus identified as having normal or abnormal coronary endothelial dysfunction. These data for individual subjects are summarized in Table S1.
BOEC Culture
At time of angiography, before heparin administration, we drew 50‐ to 100‐mL venous blood into citrate anti‐coagulant and sent it to the University of Minnesota. Within 4 hours of venipuncture, we established BOEC cultures using an updated version of the long‐term culture method we originally developed. 2 Using Histopaque‐1077 and blood diluted with Ca++/Mg++ free Hanks buffered salt solution, we obtained blood mononuclear cells. These were washed twice with BOEC culture medium (EBM2 basal medium plus EGM‐2 SingleQuot and 10% fetal bovine serum) and resuspended in 4 mL of the same. All 4 mL of cell suspension was added to a single well of a 6‐well culture plate previously coated with collagen I. Cultures were incubated at 37°C in a humidified environment having 5% CO2. After 16 hours, a gentle wash with culture medium removed debris and unattached cells. Thereafter, culture medium was changed daily for 7 days and thereafter on every other day. Subcultures were established on collagen I whenever cells reached 60% to 70% confluence. An important aspect of the method is fastidious application and meticulous performance of each of the extraordinary precautions we originally adopted (Data S1).
We harvested cells at a nominal 106‐fold expansion to collect ≈3×107 BOEC. This degree of expansion falls well within a broad safe expansion window wherein (deliberately induced) acquired effects have washed out, yet long before onset of gene expression instability. 2 For the resulting 28 unique‐patient BOEC cultures, we used fresh cells for quality control and RNA preparation, and we cryopreserved aliquots for later experimental use.
Nineteen BOEC cultures passed our multi‐parameter quality control testing requiring: cobblestone morphology; staining positive for VE‐cadherin, von Willebrand Factor, and P1H12 (CD146); staining negative for CD45 and CD14; a single population of cells at the sensitivity of light microscopy; and normal cytogenetics. Our previous studies documented that cultures meeting all these criteria additionally: are negative for CD133 and positive for multiple additional endothelial antigens; are 100% endothelial by FACS; display typical endothelial features such as VCAM‐1 upregulation in response to tumor necrosis factor/interleukin‐1, uptake of acetylated low‐density lipoprotein, and tube formation in Matrigel; exhibit presence of Weibel Palade bodies; and display endothelial lineage fidelity by gene expression. 2 , 3 Each of the 9 quality control failures was because of cytogenetics analysis returned as being abnormal for culture acquired abnormalities. This left us with BOEC from the 6 normal and 13 abnormal subjects reported herein (Table 1).
Table 1.
Subjects at Time of Enrollment
| Coronary Endothelial Function | ||
|---|---|---|
| Normal (n=6) | Abnormal (n=13) | |
| Age, y | 36.8±10.4 (24–49) | 37.4±6.1 (22–45) |
| BMI, kg/m2 | 23.5±4.1 (20.1–31.0) | 27.7±3.9 (22.8–35.2) |
| C‐reactive protein, nmol/L | 4±1 (3–6) | 18±23 (1–76) |
| C‐reactive protein, mg/L | 0.4±0.1 (0.3–0.6) | 1.8±2.3 (0.1–7.6) |
| Men | 2/6 | 7/13 |
| White | 6/6 | 13/13 |
| Hypertension | 0/6 | 2/13 |
| Diabetes mellitus | 0/6 | 0/13 |
| Hyperlipidemia | 0/6 | 10/13 |
| Smoker, active | 2/6 | 3/13 |
| Smoker, never | 3/6 | 7/13 |
| Family history positive | 3/6 | 11/13 |
| History chest pain | 6/6 | 13/13 |
BMI indicates body mass index.
Gene Expression
From each culture we isolated total RNA that was then reverse transcribed, quality verified, labeled, fragmented, and applied to Affymetrix U133A microarrays (assay for 14 500 well characterized genes and 18 400 transcripts/variants). To minimize possible batch effects, all samples for gene expression were profiled in a single batch at the University of Minnesota Microarray Core facility. As previously described, 4 , 5 we used the robust multi‐array average method to background‐adjust, quantile‐normalize, and summarize expression using median polish algorithm, as implemented in the software Genedata Expressionist Pro3.1PP (Basel, Switzerland). Our analysis applied the R function “t test” for the Welch t test (we report uncorrected P values) and the R package “samr” for Significance Analysis of Microarrays that reports false discovery rate (FDR) with 500 permutations and a delta value of 0.719 16 , 17 ; the code used is provided in Data S1. We thus used the permutation‐based Significance Analysis of Microarrays method and its associated FDR q‐values as the primary criteria for statistical inference, while using the parametric, yet robust, Welch test 18 as secondary evidence of statistical significance.
Hierarchical Clustering
We conducted 2 clustering analyses,1 using only the universe of 43 transcripts exhibiting differential expression at P<0.001, and 1 using the universe of 9 transcripts exhibiting FDR <0.1%. We used R function “hclust” for unsupervised hierarchical clustering, using normalized gene expression, complete linkage, and 1 minus Pearson correlation as the distance measure. Expression level per probe was centered and normalized to have variance 1 before clustering. To assess relative discriminatory importance amongst these 43 transcripts, we constructed a random forest using normal/abnormal expression ratio as the response and each of the 43 transcripts as predictors. We determined variable importance by the mean decrease in the Gini coefficient in R package “randomForest.”
Informatics
To identify biological inter‐relationships we searched using databases: Enrichr, Ensembl, DIANA‐TarBase v.8, miRbase, PathwayCommons, genomatix, Ingenuity Pathway Analysis, and the cardiovascular literature. BOEC gene expression data for the present 19 study subjects have been deposited in the NCBI Gene Expression Omnibus repository with accession number GSE132651 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132651). Other data sets used herein were deposited in the past: GSE22688 and GSE9877.
Bio‐Functional Validation Testing
From cryopreserved aliquots, we re‐established BOEC cultures from normal and abnormal subjects to seek bio‐functional confirmatory data.
To assay HMGB1 (high mobility group box 1) content of culture medium, we used 1.9×105 BOEC at 85% confluence, switched to serum‐free medium, and incubated ±100 ng/mL lipopolysaccharide. After 24 hours an ELISA measured HMGB1 released into medium.
To assay LAMC1 (laminin gamma 1) deposition into subendothelial matrix, we plated BOEC onto collagen I coated wells in sufficient number to reach confluence in 2 days. We then maintained them for 14 days, changing culture medium 3 times a week. Then, after rinsing with PBS, we treated culture wells with 0.5% Triton for 10 minutes to eliminate cell bodies but leave behind extracellular matrix on the well. 19 An in situ ELISA measured LAMC1 in matrix.
RESULTS
Characteristics of the 6 normal and 13 abnormal study subjects at time of coronary and gene studies reveal that, on average, the abnormal subjects had somewhat higher body mass index, C‐reactive protein, and risk factor burden (Table 1). All but one subject was aged <46 years.
The reporter cells used here, BOEC, have themselves never been exposed to in vivo signaling effects. 2 , 3 , 4 , 5 Rather, they are the in vitro outgrowth progeny of a circulating, marrow‐derived progenitor cell. Further, the necessary degree of their expansion in culture is sufficient for (deliberately) induced inflammatory responses to fully wash out yet is still many logs below the expansion degree at which phenotypic or gene expression drift is seen. Unlike other endothelial cell types, BOECs are very stable.
Single Gene Expression
At the significance threshold of FDR <10%, 29 transcripts exhibited differential expression for abnormal versus normal subjects (Table 2). Among the 9 transcripts exhibiting differential expression at the highly stringent threshold of FDR <0.1%, there was little overlap in the expression value ranges for normal versus abnormal groups (Figure 1).
Table 2.
Differentially Expressed Transcripts at Threshold of False Discovery Rate <10%, Listed in Order of False Discovery Rate and Then by P Value
| False Discovery Rate | FOLD | ||||
|---|---|---|---|---|---|
| Probe Set | Gene | (%) | P Value | (Abnormal/Normal) | NAME |
| 209041_s_at | UBE2G2 | ≤0.1 | 2.9×10−6 | 1.28 | Ubiquitin conjugating enzyme E2 G2 |
| 209181_s_at | RABGGTB | ≤0.1 | 8.5×10−6 | 1.27 | Rab geranylgeranyltransferase subunit beta |
| 203622_s_at | PNO1 | ≤0.1 | 1.3×10−5 | 1.42 | Partner of NOB1 homolog |
| 202855_s_at | SLC16A3 | ≤0.1 | 2.0×10−5 | 1.84 | Solute carrier family 16 member 3 (MCT4) |
| 208996_s_at | POLR2C | ≤0.1 | 4.3×10−5 | 1.34 | RNA polymerase II, subunit C |
| 214938_x_at | HMGB1 | ≤0.1 | 5.7×10−5 | 1.40 | High mobility group box 1 |
| 212714_at | LARP4 | ≤0.1 | 1.1×10−4 | 1.20 | La ribonucleoprotein 4 |
| 213825_at | OLIG2 | ≤0.1 | 1.3×10−4 | 1.11 | Oligodendrocyte transcription factor 2 |
| 219082_at | AMDHD2 | ≤0.1 | 1.3×10−4 | 0.82 | Amidohydrolase domain containing 2 |
| 218447_at | CMC2 | 6.75 | 3.2×10−5 | 1.35 | C‐X9‐C containing motif containing 2 |
| 220890_s_at | DDX47 | 6.75 | 4.1×10−5 | 1.22 | DEAD box helicase 47 |
| 216149_at | LRRC37BP1 | 6.75 | 5.7×10−5 | 1.13 | Leucine rich repeat containing 37B pseudogene 1 |
| 220016_at | AHNAK | 6.75 | 2.3×10−4 | 1.15 | AHNAK nucleoprotein |
| 211999_at | H3F3B | 6.75 | 3.7×10−4 | 1.24 | H3 histone family member 3B |
| 208672_s_at | SFRS3 | 6.75 | 5.7×10−4 | 1.30 | Serine and arginine rich splicing factor 3 |
| 212394_at | EMC1 | 6.75 | 6.1×10−4 | 1.13 | ER membrane protein complex subunit 1 |
| 202856_s_at | MCT4 | 6.75 | 7.9×10−4 | 1.72 | Solute carrier family 16 member 3 |
| 200700_s_at | KDELR2 | 6.75 | 8.1×10−4 | 1.17 | KDEL endoplasmic reticulum protein retention receptor 2 |
| 201574_at | ETF1 | 6.75 | 8.2×10−4 | 1.22 | Eukaryotic translation termination factor 1 |
| 201862_s_at | LRRFIP1 | 6.75 | 9.7×10−4 | 1.49 | LRR binding FLII interacting protein 1 |
| 207094_at | IL8RA | 6.75 | 1.3×10−3 | 1.11 | C‐X‐C motif chemokine receptor 2 |
| 214058_at | MYCL1 | 6.75 | 1.6×10−3 | 1.11 | MYCL proto‐oncogene, bHLH transcription factor |
| 201862_s_at | LRRFIP1 | 6.75 | 1.8 ×10−3 | 1.49 | LRR binding FLII interacting protein 1 |
| 218948_at | QRSL1 | 6.75 | 3.3×10−3 | 1.26 | Glutaminyl‐tRNA synthase (glutamine‐hydrolyzing)‐like 1 |
| 216302_at | HNRPC | 6.75 | 3.9×10−3 | 1.11 | Heterogeneous nuclear ribonucleoprotein C (C1/C2) |
| 202564_x_at | SNX15 | 8.64 | 1.1×10−4 | 0.86 | Sorting nexin 15 |
| 200770_s_at | LAMC1 | 8.64 | 1.5×10−4 | 0.71 | Laminin subunit gamma 1 |
| 214150_x_at | ATP6V0E | 8.64 | 4.2×10−4 | 0.80 | ATPase H+ transporting V0 subunit e1 |
| 221097_s_at | KCNMB2 | 8.64 | 5.8×10−3 | 1.08 | K+ Ca‐activated channel subfamily regulatory beta subunit |
Figure 1.
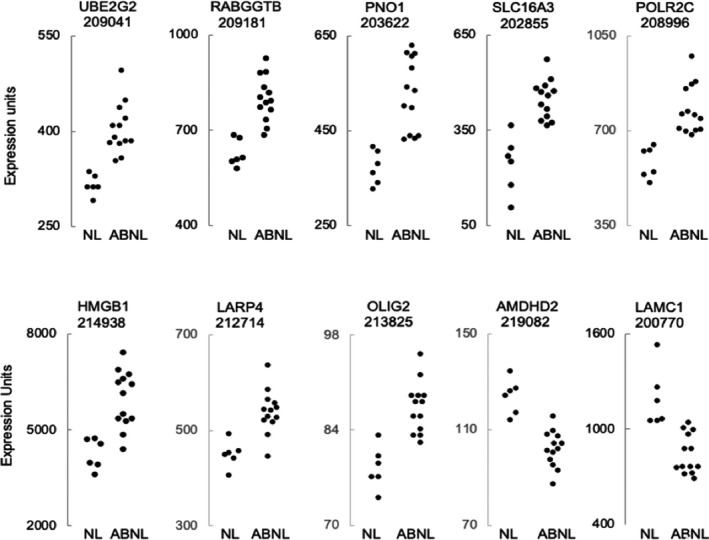
For individual normal and abnormal study subjects, expression values are plotted for the 9 transcripts (identified by gene name and Affymetrix probe set number) exhibiting differential expression at false discovery rate <0.1%, and also for LAMC1 (laminin gamma 1). For the abnormal group, note the suggestion of an outlying cluster of 7 subjects with highest‐HMGB1 (high mobility group box 1) and an outlying cluster of 7 subjects with lowest‐LAMC1. Gene names are expanded in Table 2.
The 43 transcripts exhibiting differential expression at the threshold of Welch P<0.001 are provided in Table S2.
Focus on HMGB1
Of the identified genes, we herein focus on the increased expression of HMGB1 by abnormals (1.4‐fold, FDR <0.1%, P=5.7×10−5) because of this protein's known and prominent role in arterial disease pathogenesis, specifically including the biology of atherosclerosis, per Discussion. 20 Arguing that high‐HMGB1 expression amongst abnormal subjects is not reflective of any proinflammatory in vivo milieu, HMGB1 elevation was not accompanied by differential expression by any of 40 other inflammatory genes (Table S3).
Inverse Expression Linkage
Inspection of individual subject expression values suggested a sub‐cluster of 7 abnormal with highest HMGB1 expression and a sub‐cluster of 7 abnormal with lowest LAMC1 expression (Figure 1). These 2 sub‐clusters were composed, nearly perfectly, of the same subjects, and there was a strong inverse correlation between HMGB1 and LAMC1 expression for the 19 study subjects (Figure 2A)
Figure 2.
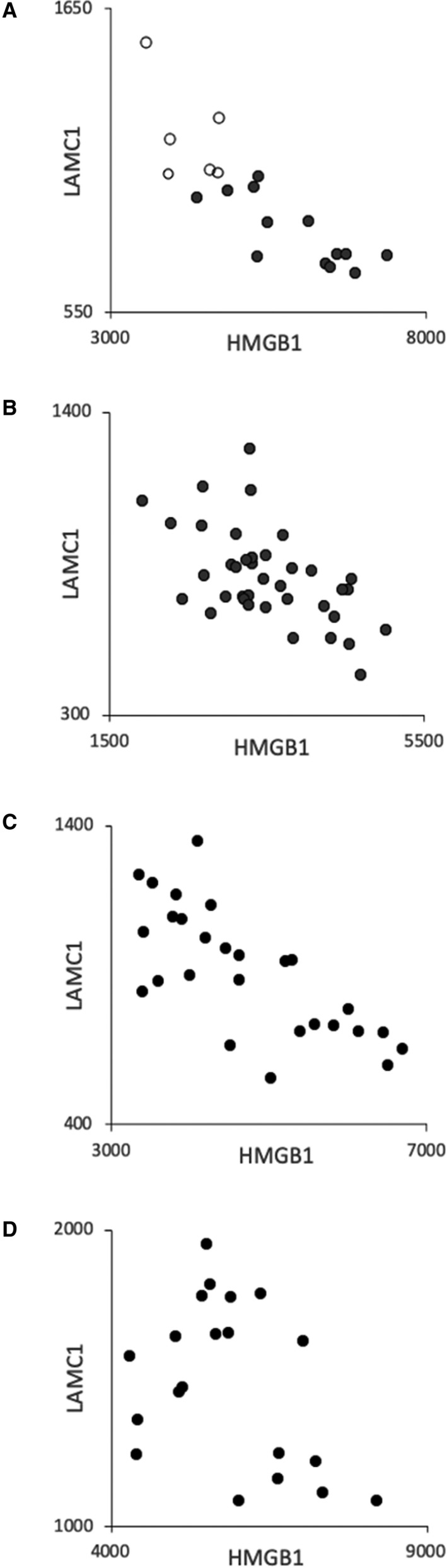
Inverse correlation between high‐HMGB1 (high mobility group box 1) and low‐LAMC1 (laminin gamma 1) expression. Scales display expression units which differ amongst panels because each study was done in a different time frame. A, The present study in which normal are in open symbols and abnormal in closed symbols, r=−0.844. B, Healthy 20‐ to 29‐year‐olds, r=−0.569. 5 C, Random 23‐ to 69‐year‐olds, r=−0.728. 4 D, Children with sickle cell anemia, r=−0.383. 4 Gene expression data are deposited at Gene Expression Omnibus, series GSE132651, GSE22688, GSE9877, GSE9877, respectively.
Providing indirect corroboration of this unexpected observation, we uncovered the same high‐HMGB1/low‐LAMC1 relationship in our past studies of BOEC gene expression for separate groups of: 38 healthy 20‐ to 29‐year‐olds (Figure 2B), 5 27 random normal subjects (Figure 2C), 4 and 20 children with sickle cell anemia (Figure 2D). 4 There was no apparent effect of sex on this high‐HMGB1/low‐LAMC1 relationship.
Hierarchical Clustering
Hierarchical clustering analysis using the universe of 43 transcripts exhibiting differential expression at P<0.001 generated 2 primary clusters, one containing all normals and the other containing all abnormals (Figure S1A). The latter cluster had 2 secondary sub‐clusters that separate the abnormals having highest versus lowest HMGB1 expression. This suggests that HMGB1 expression is an important—but not sole—discriminator.
Indeed, a random forest analysis of the same transcript universe estimated the strongest discriminators to also include MCT4 (SLC16A3), RABGGTB, LAMC1, UBE2G2, POLR2C, PNO1, and HMGCS1 (Figure S2). That these help discriminate between lowest‐ versus highest‐HMGB1 expressers (regardless of subject group) was supported by another hierarchical clustering analysis using only the 9 transcripts having FDR <0.1% (Figure S1B). This accurately generated 2 primary clusters: 1 composed of those abnormal subjects having the highest HMGB1 expression, and the other containing those abnormals having the lowest HMGB1 expression plus all normals.
Bio‐Functional Validation
To bio‐functionally test for elevated HMGB1 protein, we measured its release from BOEC incubated±lipopolysaccharide. Consistent with their abnormally high HMGB1 gene expression, BOEC from abnormal released 13 ±3‐fold greater HMGB1 than normal BOEC released; P=8.5×10−5 (Figure 3, left). This provides bio‐functional confirmation that higher HMGB1 gene expression by abnormal subjects is real.
Figure 3.
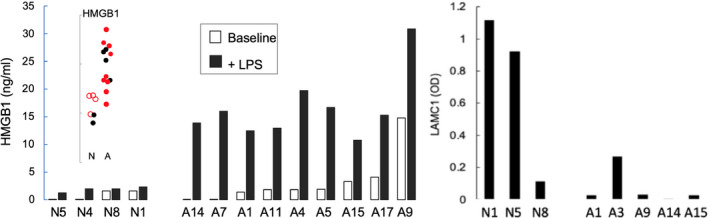
Bio‐functional testing corroborates high‐HMGB1 (high mobility group box 1) and low‐LAMC1 (laminin gamma chain 1) expression in abnormal subjects (A) vs normal subjects (N). Left, HMGB1 content of culture medium over blood outgrowth endothelial cells measured ± lipopolysaccharide stimulation for 24 hours. Baseline release (white bars) tended to be higher for abnormal vs normal subjects (P=0.158). In response to lipopolysaccharide (black bars), abnormal blood outgrowth endothelial cells released 13.3‐fold greater HMGB1 (P=8.5×10−5). The specific subjects studied are indicated in red in the embedded HMGB1 expression plot. Right, LAMC1 deposition into collagen subendothelial matrix by abnormal blood outgrowth endothelial cells over 14 days incubation was, on average, one tenth that by normal blood outgrowth endothelial cells. LPS indicates lipopolysaccharide.
To bio‐functionally test for lowered LAMC1 protein, we incubated BOEC on plates originally coated with collagen and measured LAMC1 deposition into the subendothelial matrix after 14 days. Consistent with their abnormally depressed LAMC1 expression, BOEC from abnormals deposited, on average, one tenth as much LAMC1 as identically‐incubated normal BOEC (Figure 3, right). Despite the discernable difference, the result from this test was not statistically significant, likely because of small sample size.
Clinical Follow‐Up
Since this clinical study was performed in 2005 to 2007, follow‐up data are available but incomplete (institutional review board rules prohibit contacting non‐Mayo subjects from this closed study) (Table 3). For 4 normals followed for 13.4±0.1 years, 1 experienced a major adverse cardiovascular event. For 12 abnormals followed for 9.1±4.5 years, 4 of 12 developed major adverse cardiovascular events. Interestingly, of these 4 abnormals who developed major adverse cardiovascular events, 3 were among the top‐4 highest HMGB1 expressors.
Table 3.
Available Subject Data at Subsequent Clinical Follow‐Up
| Coronary Endothelial Function | ||
|---|---|---|
| Normal | Abnormal | |
| Number available | 4/6 | 12/13 |
| Age at coronary study, y | 38.0±10.4 (24–49) | 40.8±1.6 (38–42) |
| Follow‐up duration, y | 13.4±0.1 (13.3–13.5) | 9.1±4.5 (1.2–14.0) |
| Experienced MACE, n | 1/4 | 4/12 |
| Age at MACE, y | 55 | 50±6 (39–54) |
| Study‐to‐MACE interval, y | 13 | 1, 12, 12, 12 |
For the normal subject this was a myocardial infarction. For the abnormal subjects it was: 1 new diagnosis of congestive heart failure; 1 stroke; 1 myocardial infarction; 1 peripheral artery disease and carotid endarterectomy needed. MACE indicates major adverse cardiovascular events.
DISCUSSION
In addressing risk for early coronary endothelial dysfunction, the earliest clinical form of atherosclerosis, 8 , 9 , 10 we surveyed gene expression by Blood Outgrowth Endothelial Cells as an approach to bridging the troublesome gap between genetics and clinical phenotype. Our operational definition of “early” was subject age <50 years, and coronary reactivity testing identified subjects as having normal or abnormal coronary endothelial function. Our results indicate that the approach is, indeed, feasible and possibly can shed light upon underlying risk. Comparing abnormals versus normals we identified differential expression of 29 transcripts at FDR <10%, of which 9 were significant at the stringent threshold of FDR <0.1%.
Of the latter group, we here focus on the abnormals having elevated expression of HMGB1 in apparent linkage with lowered expression of LAMC1. We first review the most relevant aspects of HMGB1 and LAMC1 proteins, as each has unambiguous implications for atherogenesis. Then we focus on their expected gene‐gene interactions that predict a remarkable, pathobiological convergence that would jeopardize endothelial cell responsiveness to shear stress.
HMGB1
HMGB1 expression was greater for abnormal subjects (1.4‐fold, FDR <0.1%, P=5.7×10−5), and the ranges for normal versus abnormal subjects barely overlapped. HMGB1, an “alarmin”, is one of the damage‐associated molecular pattern molecules. Its roles are diverse, remarkable, and highly relevant to atherosclerosis. 20 , 21 , 22
Nucleus
HMGB1 is the most abundant non‐histone nuclear protein, and it regulates multiple nuclear functions, among them gene expression. For example, HMGB1 enhances binding of nuclear factor‐κB p50/p50 and p65/p50 to DNA, and it is reportedly required for p50 to be functional. 23 Although it traffics bidirectionally across the nuclear membrane, HMGB1 is normally highly skewed towards the nucleus. In monocytes, however, the skew is heavily towards cytoplasm. 24
Cytoplasm
In cytoplasm, HMGB1 is part of many regulatory protein complexes, and it promotes translocation of nuclear factor‐κB1, RelA and SP1 into the nucleus. Notably, HMGB1 associates with cytoplasmic Src, exerting an inhibitory influence that is discussed in the Gene‐Gene Interactome section below.
Export
During necrosis or in response to injury or stimuli, HMGB1 is passively or actively exported from various cell types. 20 , 21 , 22 For example, endothelial cells release it in response to tumor necrosis factor, 25 abnormal shear stress, 26 or hypoxia. 20 Smooth muscle cells release it in response to cholesterol. 27 Neutrophils disgorge it into neutrophil extracellular traps, 28 and platelets contribute it to thrombi. 29 Monocytes/macrophages release HMGB1 in response to inflammatory stimuli and when manifesting their inflammatory reprogramming.
Extracellular
Upon release, HMGB1 can act as a cytokine or chemokine, able to induce and amplify inflammation by engaging multiple receptors, especially toll‐like receptor 4 and the receptor for advanced glycation end‐products. Indeed, HMGB1 is a dominant driver of sterile inflammation. 20 , 21 , 22 It activates monocytes/macrophages and platelets, as well as endothelial and smooth muscle cells that then adopt a proliferative phenotype. It causes endothelial barrier hyperpermeability, facilitating egress of WBC and atherogenic lipid, and it activates endothelial NADPH oxidase. 22
Atherogenesis
Each of the above HMGB1 effects is atheropromotive in nature and would plausibly be exaggerated in those with inherently high HMGB1 expression. Indeed, HMGB1 already is believed to promote all stages of atherosclerosis from inception to plaque rupture, and it is specifically implicated in coronary, peripheral and cerebral arterial disease. 20 For example, antibody‐mediated neutralization of HMGB1 attenuated atherosclerosis by >50% in apolipoprotein E deficient mice, 30 and genetic deletion or neutralization of HMGB1 prevented intimal hyperplasia in response to carotid wire injury. 31
SNP Insight
We did not conduct SNP testing here, but extant data link increased HMGB1 expression to arterial disease. The HMGB1 3814 polymorphism (rs2249825) is predicted to create a strong enhancer effect on HMGB1 expression, 32 and leukocytes from such individuals act ually do exhibit exaggerated HMGB1 release when challenged with lipopolysaccharide 33 —just as we saw here for BOEC from abnormals. Studied in Chinese, this SNP is associated with hypertension, 34 increased cerebral ischemia size, 35 and exaggerated inflammation in sepsis. 33
LAMC1
LAMC1 expression was lower for abnormal subjects (0.71‐fold, FDR=8.64%, P=4.2×10−4), and ranges for normal versus abnormal subjects barely overlapped. Laminins are important to all vessel wall cells and engage them with functional reciprocity. 36
Endothelial cells produce the 2 laminin heterotrimers (α4β1γ1 and α5β1γ1) of their basement membrane contact layer, and assembly of these heterotrimers specifically requires involvement of LAMC1, the laminin γ1 chain. 36 Indeed, experimental knockout of LAMC1 resulted in embryonic lethality with failure to make basement membranes. 36 , 37 Remarkably, laminin α5β1γ1 was experimentally found to be necessary for endothelial response to shear stress. 38
Thus, even if LAMC1 production is just lowered, harmful consequences may be predictable. One is enhanced endothelial barrier hyperpermeability. 39 Another consequence is that in endothelial basement membranes if laminin is missing it is maladaptively replaced with fibronectin, replacing laminin‐enforced endothelial quiescence with fibronectin‐mediated inflammatory signaling. 40 Particularly notable is that endothelial cells on laminin are shear responsive, but those on fibronectin are not. 41 Thus, depressed LAMC1 production could replicate several key features of atherogenesis. 42
Atherogenesis
We find nothing in the literature that directly links LAMC1 to atherosclerosis, but there are some suggestive data. First, a study of quantitative trait loci identified the Ath44 region of chromosome 1 as being associated with aortic root lesion size in murine atherosclerosis; LAMC1 is a gene in this region. 43 Second, an analysis of bio‐functional pathways enriched in advanced versus early coronary atherosclerosis identified focal adhesion (critical in shear responsiveness) to be an implicated functional module, within which LAMC1 was one of the abnormally downregulated genes associated with arteriopathy severity. 44
Gene‐Gene Interactome Impacting Endothelial Function
For all these reasons, we predict that the combination of high‐HMGB1 plus low‐LAMC1 expression in endothelium would converge biologically and detrimentally at the focal adhesion complex that is required for endothelial cell mechanosensing of shear stress. 36 , 41 , 45 Specifically, focal adhesion complex function would be impaired by HMGB1 because it inhibits the reciprocal phosphorylations 46 between Src and FAK (focal adhesion kinase) that enable focal adhesion complex to participate in normal shear sensing. As for LAMC1, it is necessary for the proper basement membrane engagement with endothelial abluminal integrins 36 , 37 , 38 , 41 that is required for their clustering, an on‐switch for focal adhesion complex function. That could be jeopardized if LAMC1 production is low.
We suggest that these known effects of high‐HMGB1 and of low‐LAMC1 would synergistically undermine the endothelial cell's ability to respond adaptively and optimally to shear stress. Thus, we hypothesize that impairment of shear responsiveness in this manner would maladaptively foster endothelial dysfunction and, thereby, earlier development of detectable atherogenesis.
Concordance of High‐HMGB1 and Low‐LAMC1 Expression
The unexpected linkage between high‐HMGB1 and low‐LAMC1 expression revealed by our data is corroborated via archived data sets from our 3 previous studies of BOEC gene expression (Figure 2). This suggests an underlying regulatory relationship, although our data cannot inform as to mechanism. The primary aberrancy could be high‐HMGB1, or low‐LAMC1, or something else affecting both. So, to illustrate relevance but simplify discussion, we here arbitrarily assume that elevated HMGB1 expression is the primary aberrancy that drives lower LAMC1 expression.
Suppression of LAMC1 is possible via microRNA (miR) species already implicated in atherosclerosis. 47 , 48 Perhaps most intriguing is miR‐21 that is induced by both HMGB1 and oscillatory flow regimes, 49 , 50 and miR‐21 is known to target LAMC1. 48 , 49 Interestingly, miR‐21 is the most abundant miR in normal BOEC (Hebbel and Steer, unpublished observation, 2012), and it is upregulated in human atherosclerotic tissue. 47 , 48 Other LAMC1‐targeting miRNAs are identified, as well as some that target LAMC1 transcription factors. One of these is ESR1 (estrogen receptor alpha) that not only is hypermethylated in atherosclerosis but also is a target of miR‐206, another miR that can be induced by HMGB1. 51
Caveats and Limitations
This study probes the early onset of atherosclerosis, but we recognize that “early” here can mean truly earlier onset and/or accelerated progression and/or earlier symptom awareness. The gene expression changes we have highlighted are perhaps consistent with each. Regardless, we surmise that such changes would not only nudge an individual's homeostatic balance in a maladaptive direction but also enhance susceptibility to risk factors. For example, elevated constitutive HMGB1 expression could result in its exaggerated release from endothelium residing in anatomically atheroprone areas. 26
We emphasize that all of our subjects were studied by coronary catheterization because they had an episode of chest pain. Thus, our normal subjects were not truly normal individuals. Rather, our labels “ normal” and “abnormal” refer specifically to their coronary endothelial function status. Consistent with this, our abnormal subjects did exhibit somewhat greater frequency of some risk factors, most notably hyperlipidemia and elevated C‐reactive protein. Experimentally, HMGB1 can induce C‐reactive protein, 52 but hyperlipidemia can stimulate HMGB1 release of HMGB1. 27 We find no evidence that C‐reactive protein or lipid increase HMGB1 expression.
Cultured cells always raise concerns about laboratory‐induced variations. However, our prior standardization, reproducibility, and validation studies, 2 , 3 , 4 , 5 plus our extensive experience with BOECs, justify confidence that our data identify true differences between endothelial cells from abnormal versus normal. We emphasize that a critical factor in achieving this is the fastidious application of all our extraordinary culture precautions described in Data S1.
Further, measured BOEC gene expression is not influenced by in vivo signaling exposures 2 , 3 , 4 , 5 as the BOEC themselves have never been exposed to inflammation or tissue‐specific signaling. Their 106‐fold expansion is logs beyond what is needed for inflammatory signaling effects to wash out, and it is logs before any phenotypic or genotypic drift appears. 3 , 4 BOEC are far more stable in culture than other endothelial types. Thus, we believe that here, as in our previous studies of this nature, 4 , 5 results likely reflect heritable differences between abnormal and normal subjects.
Of course, it is impossible to absolutely exclude the possibility that, in the abnormal subjects, an atherogenic environment created durable epigenetic changes within the circulating endothelial progenitors, changes then passed to their outgrowth progeny, the BOEC. Somewhat mitigating this concern, elevated HMGB1 expression by the BOEC from abnormal subjects was not accompanied by differential expression of any of 40 other inflammation‐responsive genes.
Finally, we do not know if these expression changes are actually endothelial specific. If not, perhaps they could be identified in a more easily accessible cell type. For other cell types, however, there would be magnified concern about acquired influences or artifacts. And, of course, if interest lies in the functional biology of endothelial cells, BOEC uniquely offer the opportunity to examine this.
Conclusions
Our data reveal an association between high‐HMGB1 plus low‐LAMC1 expression with coronary endothelial dysfunction at age <50 years. This pathobiologically‐relevant, probably‐heritable combination could create risk via a detrimental biological convergence that maladaptively impairs endothelial mechanosensing. Our results may have practical clinical implications since the approach can be applied even in children, 4 perhaps enabling identification of those who would most benefit from earlier, more aggressive medical intervention.
We recognize, of course, that this feasibility study is too limited to be definitive. Nonetheless, it does support the notion that BOEC can be used to bridge the information gap between genomics and clinical phenotype in understanding atherosclerosis risk. Indeed, BOEC comprise a unique platform that enables matching donor characteristics with endothelial functional assessment with various “omics.” In that regard, sufficient BOEC can easily be produced to enable studying all “omics” simultaneously for each endothelial cell culture. We suggest that this would be uniquely useful for achieving the truly integrative endothelial “omics” that may be key in understanding the heritable component of atherosclerosis risk.
Sources of Funding
This study was funded by a State of Minnesota Biotechnology Initiative grant to Hebbel and Lerman; also supported by the National Institutes of Health (P01‐HL55552 and P01‐HL76540).
Disclosures
None.
Supporting information
Data S1
Tables S1–S3
Figures S1–S2
(J Am Heart Assoc. 2020;9:e016134 DOI: 10.1161/JAHA.120.016134.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016134
For Sources of Funding and Disclosures, see page 9.
REFERENCES
- 1. Lusis AJ. Genetics of atherosclerosis. Trends Genet. 2012;28:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebbel RP. Blood endothelial cells: utility from ambiguity. J Clin Invest. 2017;127:1613–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Milbauer L, Wei P, Enenstein J, Jiang A, Hillery CA, Scott JP, Nelson SC, Bodempudi V, Topper JN, Yang RB, et al. Genetic endothelial systems biology of sickle stroke risk. Blood. 2008;111:3872–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei P, Milbauer LC, Enenstein J, Nguyen J, Pan W, Hebbel RP. Differential endothelial cell gene expression by African Americans versus Caucasian Americans: a possible contribution to health disparity in vascular disease and cancer. BMC Med. 2011;9:2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gutierrez E, Flammer AJ, Lerman LO, Elizaga J, Lerman A, Fernandez‐Aviles F. Endothelial dysfunction over the course of coronary artery disease. Eur Heart J. 2013;34:3175–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111:363–368. [DOI] [PubMed] [Google Scholar]
- 8. Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long‐term follow‐up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948–954. [DOI] [PubMed] [Google Scholar]
- 9. Targonski PV, Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Lerman A. Coronary endothelial dysfunction is associated with an increased risk of cerebrovascular events. Circulation. 2003;107:2805–2809. [DOI] [PubMed] [Google Scholar]
- 10. Siasos G, Sara JD, Zaromytidou M, Park KH, Coskun AU, Lerman LO, Oikonomou E, Maynard CC, Fotiadis D, Stefanou K, et al. Local low shear stress and endothelial dysfunction in patients with nonobstructive coronary atherosclerosis. J Am Coll Cardiol. 2018;71:2092–2102. [DOI] [PubMed] [Google Scholar]
- 11. Al Suwaidi J, Higano ST, Holmes DR Jr, Rihal CS, Lerman A. Measuring maximal percent area stenosis poststent placement with intracoronary Doppler and the continuity equation and correlation with intracoronary ultrasound and angiography. Am J Cardiol. 1999;84:650–654. [DOI] [PubMed] [Google Scholar]
- 12. Doucette JW, Corl PD, Payne HM, , Flynn AE, Goto M, Nassi M, Segal J. Validation of a Doppler guide wire for intravascular measurement of coronary artery flow velocity. Circulation. 1992;85:1899–1911. [DOI] [PubMed] [Google Scholar]
- 13. Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051. [DOI] [PubMed] [Google Scholar]
- 14. Egashira K, Inou T, Hirooka Y, Yamada A, Urabe Y, Takeshita A. Evidence of impaired endothelium‐dependent coronary vasodilatation in patients with angina pectoris and normal coronary angiograms. N Engl J Med. 1993;328:1659–1664. [DOI] [PubMed] [Google Scholar]
- 15. Hasdai D, Cannan CR, Mathew V, Holmes DR Jr, Lerman A. Evaluation of patients with minimally obstructive coronary artery disease and angina. Int J Cardiol. 1996;53:203–208. [DOI] [PubMed] [Google Scholar]
- 16. Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. [DOI] [PubMed] [Google Scholar]
- 17. Core Team R . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 18. Pan W. A comparative review of statistical methods for discovering differentially expressed genes in replicated microarray experiments. Bioinformatics. 2002;18:546–554. [DOI] [PubMed] [Google Scholar]
- 19. Gospodarowicz D, Gonzalez R, Fujii DK. Are factors originating from serum, plasma, or cultured cells involved in the growth‐promoting effect of the extracellular matrix produced by cultured bovine corneal endothelial cells? J Cell Physiol. 1983;114:191–202. [DOI] [PubMed] [Google Scholar]
- 20. Cai J, Wen J, Bauer E, Zhong H, Yuan H, Chen AF. The role of HMGB1 in cardiovascular biology: danger signals. Antioxid Redox Signal. 2015;23:1351–1369. [DOI] [PubMed] [Google Scholar]
- 21. Lotze MT, Tracey KJ. High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. [DOI] [PubMed] [Google Scholar]
- 22. Tang D, Kang R, Zeh HJ III, Lotze MT. High‐mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Agresti A, Lupo R, Bianchi ME, Muller S. HMGB1 interacts differentially with members of the Rel family of transcription factors. Biochem Biophys Res Commun. 2003;302:421–426. [DOI] [PubMed] [Google Scholar]
- 24. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mullins GE, Sunden‐Cullberg J, Johansson AS, Rouhiainen A, Erlandsson‐Harris H, Yang H, Tracey KJ, Rauvala H, Palmblad J, Andersson J, et al. Activation of human umbilical vein endothelial cells leads to relocation and release of high‐mobility group box chromosomal protein 1. Scand J Immunol. 2004;60:566–573. [DOI] [PubMed] [Google Scholar]
- 26. Qin WD, Mi SH, Li C, Wang GX, Zhang JH, Wang H, Zhang F, Ma Y, Wu DW, Zhang M. Low shear stress induced HMGB1 translocation and release via PECAM‐1/PARP‐1 pathway to induce inflammation response. PLoS One. 2015;10:e0120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haraba R, Suica VI, Uyy E, Ivan L, Antohe F. Hyperlipidemia stimulates the extracellular release of the nuclear high mobility group box 1 protein. Cell Tissue Res. 2011;346:361–368. [DOI] [PubMed] [Google Scholar]
- 28. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, Ritis K. Neutrophil extracellular trap formation is associated with IL‐1β and autophagy‐related signaling in gout. PLoS One. 2011;6:e29318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Bruhl ML, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, Takahashi HK, Liu K, Peter K, Nishibori M, et al. High‐mobility group box protein 1 neutralization reduces development of diet‐induced atherosclerosis in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:313–319. [DOI] [PubMed] [Google Scholar]
- 31. Cai J, Yuan H, Wang Q, Yang H, Al Abed Y, Hua Z, Wang J, Chen D, Wu J, Lu B, et al. HMGB1‐driven inflammation and intimal hyperplasia after arterial injury involves cell‐specific actions mediated by TLR4. Arterioscler Thromb Vasc Biol. 2015;35:2579–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kornblit B, Munthe‐Fog L, Petersen SL, Madsen HO, Vindelov L, Garred P. The genetic variation of the human HMGB1 gene. Tissue Antigens. 2007;70:151–156. [DOI] [PubMed] [Google Scholar]
- 33. Zeng L, Zhang AQ, Gu W, Chen KH, Jiang DP, Zhang LY, Du DY, Hu P, Huang SN, Wang HY, et al. Clinical relevance of single nucleotide polymorphisms of the high mobility group box 1 protein gene in patients with major trauma in southwest China. Surgery. 2012;151:427–436. [DOI] [PubMed] [Google Scholar]
- 34. Yao Y, Guo D, Yang S, Jin Y, He L, Chen J, Zhao X, Chen Y, Zhou W, Shen C. HMGB1 gene polymorphism is associated with hypertension in Han Chinese population. Clin Exp Hypertens. 2015;37:166–171. [DOI] [PubMed] [Google Scholar]
- 35. Hendrix P, Foreman PM, Harrigan MR, Fisher WS, Vyas NA, Lipsky RH, Lin M, Walters BC, Tubbs RS, Shoja MM, et al. Impact of high‐mobility group box 1 polymorphism on delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. World Neurosurg. 2017;101:325–330. [DOI] [PubMed] [Google Scholar]
- 36. Berrier AL, Yamada KM. Cell‐matrix adhesion. J Cell Physiol. 2007;213:565–573. [DOI] [PubMed] [Google Scholar]
- 37. Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di Russo J, Luik AL, Yousif L, Budny S, Oberleithner H, Hofschroer V, Klingauf J, van Bavel E, Bakker EN, Hellstrand P, et al. Endothelial basement membrane laminin 511 is essential for shear stress response. EMBO J. 2017;36:183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song J, Zhang X, Zhao Y, Zhou X, Sun L, Zeng S, Zuo M, Zhang X. Endothelial basement membrane laminin 511 contributes to endothelial junctional tightness and thereby inhibits leukocyte transmigration. Cell Rep. 2017;18:1256–1269. [DOI] [PubMed] [Google Scholar]
- 40. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF‐kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nature Rev Mol Cell Biol. 2009;10:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kayashima Y, Tomita H, Zhilicheva S, Kim S, Kim HS, Bennett BJ, Maeda N. Quantitative trait loci affecting atherosclerosis atgthe aortic root identified in an intercross between DBA2J and 129S6 apoplipoprotein E‐null mice. PLoS One. 2014;9:e88274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan X, Zhang X, Pan L, Tian X, Dong P. Identification of key pathways and genes in advanced coronary atherosclerosis using bioinformatics analysis. Biomed Res Int. 2017;2017:4323496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zebda N, Dubrovskyi O, Birukov KG. Focal adhesion kinase regulation of mechanotransduction and its impact on endothelial cell functions. Microvasc Res. 2012;83:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol. 2011;187:4686–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feinberg MW, Moore KJ. MicroRNA regulation of atherosclerosis. Circ Res. 2016;118:703–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun X, Belkin N, Feinberg MW. Endothelial microRNAs and atherosclerosis. Curr Atheroscler Rep. 2013;15:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheedy FJ. Turning 21: induction of miR‐21 as a key switch in the inflammatory response. Front Immunol. 2015;6:19, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Neth P, Nazari‐Jahantigh M, Schober A, Weber C. MicroRNAs in flow‐dependent vascular remodelling. Cardiovasc Res. 2013;99:294–303. [DOI] [PubMed] [Google Scholar]
- 51. Adams BD, Furneaux H, White BA. The micro‐ribonucleic acid (miRNA) miR‐206 targets the human estrogen receptor‐alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007;21:1132–1147. [DOI] [PubMed] [Google Scholar]
- 52. Inoue K, Kawahara K, Biswas KK, Ando K, Mitsudo K, Nobuyoshi M, Maruyama I. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc Pathol. 2007;16:136–143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S3
Figures S1–S2