

Abstract
Background
Cardiac remodeling predisposes individuals to heart failure if the burden is not solved, and heart failure is a growing cause of morbidity and mortality worldwide. The cardiac extracellular matrix not only provides structural support, but also is a core aspect of the myocardial response to various biomechanical stresses and heart failure. MFAP4 (microfibrillar‐associated protein 4) is an integrin ligand located in the extracellular matrix, whose biological functions in the heart remain poorly understood. In the current study we aimed to test the role of MFAP4 in cardiac remodeling.
Methods and Results
MFAP4‐deficient (MFAP4−/−) and wild‐type mice were subjected to aortic banding surgery and isoproterenol to establish models of cardiac remodeling. We also evaluated the functional effects of MFAP4 on cardiac hypertrophy, fibrosis, and cardiac electrical remodeling. The expression of MFAP4 was increased in the animal cardiac remodeling models induced by pressure overload and isoproterenol. After challenge of 8 weeks of aortic banding or 2 weeks of intraperitoneal isoproterenol, MFAP4−/− mice exhibited lower levels of cardiac fibrosis and fewer ventricular arrhythmias than wild‐type mice. However, there was no significant effect on cardiomyocyte hypertrophy. In addition, there was no significant difference in cardiac fibrosis severity, hypertrophy, or ventricular arrhythmia incidence between wild‐type‐sham and knockout‐sham mice.
Conclusions
These findings are the first to demonstrate that MFAP4 deficiency inhibits cardiac fibrosis and ventricular arrhythmias after challenge with 8 weeks of aortic banding or 2 weeks of intraperitoneal isoproterenol but does not significantly affect the hypertrophy response. In addition, MFAP4 deficiency had no significant effect on cardiac fibrosis, hypertrophy, or ventricular arrhythmia in the sham group in this study.
Keywords: cardiac remodeling, extracellular matrix proteins, heart failure, microfibrillar-associated protein 4, pressure overload
Subject Categories: Cardiomyopathy, Heart Failure, Remodeling
Nonstandard Abbreviations and Acronyms
- AB
aortic banding
- Ad‐shMFAP4
pDKD‐CMV‐eGFP‐U6‐shRNA‐MFAP4
- Ad‐shRNA
pDKD‐CMV‐eGFP‐U6‐shRNA
- PCL
pacing cycle length
- MFAP4
microfibrillar‐associated protein 4
- VA
ventricular arrhythmias
- Ad‐MFAP4
Adenovirus vector pAdeno‐EF1A(S)‐mNeonGreen‐CMVMfap4‐3FLAG
Clinical Perspective
What Is New?
For the first time we have defined the expression of MFAP4 (microfibrillar‐associated protein 4) was increased in the animal cardiac remodeling models induced by pressure overload and isoproterenol.
MFAP4 deficiency inhibits cardiac fibrosis and ventricular arrhythmias after challenge with 8 weeks of aortic banding or 2 weeks of intraperitoneal isoproterenol but does not significantly affect the hypertrophy response.
What Are the Clinical Implications?
MFAP4 may act as a novel therapeutic target for the upstream prevention of cardiac remodeling and heart failure.
Heart failure (HF) is the common ultimate outcome of different types of cardiovascular diseases, and is a growing public health problem associated with high hospitalization and mortality. 1 , 2 , 3 , 4 Cardiac remodeling is an adaptive cellular response to various kinds of biomechanical stresses or hemodynamic overload, that involves a series of structural and functional alterations, including increase in the size and/or thickness of cardiomyocytes and the left ventricle, perivascular and interstitial fibrosis, various types of ventricular and atrial arrhythmias and so on. 5 , 6 Cardiac remodeling predisposes a patient to HF if the burden is not solved, and can be divided into structural remodeling and electrical remodeling. Structural remodeling includes cardiac fibrosis and cardiac hypertrophy, and electrical remodeling mainly includes ventricular arrhythmias (VA) and atrial arrhythmias. 7 Considerable progress has been made in understanding the pathogenesis of HF and treating HF, but there are few options for efficacious treatment of end‐stage HF. 8
HF is often accompanied by progressive perivascular and interstitial fibrosis, which reduces myocardial compliance and function and is an independent predictor of overall mortality. 8 Cardiac fibrosis is characterized by exacerbated accumulation of extracellular matrix (ECM) components in the perivascular and interstitial regions of the myocardium. The cardiac ECM serves many functions: it not only provides support to cardiac myocytes, but also acts as a signal transducer for cell–cell and cell‐ECM interactions, facilitating force transmission and so on. 9 , 10
MFAP4 (microfibrillar‐associated protein 4) is a 36‐kDa ECM glycoprotein of the fibrinogen‐related protein superfamily that is widely expressed in elastin‐rich tissues, such as the lungs and heart. 11 , 12 It has an N‐terminal arginine‐glycine‐aspartic acid integrin‐binding sequence. 13 MFAP4 is also often referred to as MAGP36 (36‐kDa microfibril‐associated glycoprotein) in other species. 14
MFAP4 is closely associated with remodeling‐related diseases, including liver fibrosis, atherosclerosis, arterial injury stimulated remodeling, and asthma. 12 , 13 , 15 , 16 , 17 However, there have been no related studies to clarify the role of MFAP4 in cardiac remodeling. In our previous research, we downloaded 4 microarray profile data sets from the Gene Expression Omnibus database with a bioinformatics method and screened the differentially expressed genes implicated in cardiac remodeling. Twenty‐four differentially expressed genes were screened, of which MFAP4 exhibited the greatest fold difference. 18
To study the role of MFAP4 in cardiac remodeling, we generated MFAP4‐deficient (MFAP4−/−) mice and investigated the potential role of MFAP4 in cardiac fibrosis, hypertrophy, and VA..
METHODS
Data, Materials, and Code Disclosure Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reagents
The following main antibodies were obtained from Abcam (Cambridge, UK): inducible MFAP4 (#ab 80319), transforming growth factor‐β (TGF‐β, #ab64715), α‐smooth muscle actin (α‐SMA, #ab5694), Connexin 43 (Cx43, #ab11370); The antibodies against GAPDH (#2118), focal adhesion kinase (FAK, #3285), P‐FAK (#3283), phosphorylated‐Smad2 (P‐Smad2, #3108s), P‐Smad3 (#8769), total‐Smad2 (T‐Smad2, #3103s), T‐Smad3 (# 9513s), total—extracellular regulated protein kinase 1/2 (T‐ERK1/2, #4695), P‐ERK1/2 (#4370P), T‐AKT (4691#), P‐AKT (4060#), T‐PI3K (#4257), P‐PI3K (#4228S), T‐MEK1/2 (#9122s), P‐MEK1/2 (#9154s) were purchased from Cell Signaling Technology (Danvers, MA, USA); The antibodies against ANP (atrial natriuretic peptide) (# sc‐20158), β‐MHC (β‐myosin heavy chain) (sc‐53090), Col2a1 (collagen type III alpha 1) (sc‐8781) were obtained from Santa Cruz Inc. (TX, USA). The secondary antibody for immunofluorescence staining was purchased from LI‐COR Biosciences (Lincoln, USA); The BCA protein assay kit was purchased from Thermo Scientific (23227, MIT, USA); TRIzol and 6‐Diamidino‐2‐phenylindole (DAPI; S36939) were purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Transcriptor First Strand cDNA Synthesis kit (No. 04896866001) and LightCycler 480 SYBR Green I Master mix (No. 04707516001) were purchased from Roche Diagnostics (Basel, Switzerland). TGF‐β, isoproterenol, pentobarbital sodium were obtained from Sigma‐Aldrich (St. Louis, MO, United States). Adenovirus vector pAdeno‐EF1A(S)‐mNeonGreen‐CMVMfap4‐3FLAG (Ad‐MFAP4), pAdeno‐EF1A(S)‐NeonGreen‐CMV‐MCS‐3FLAG, pDKD‐CMV‐eGFP‐U6‐shRNA (Ad‐shRNA), pDKD‐CMV‐eGFP‐U6‐shRNA‐MFAP4 (Ad‐shMFAP4) were obtained from Obio Technology (Shanghai, China).
Mice
All animal studies adhered to the G uidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication, revised 2011) and the guidelines of the Animal Care and Use Committee of Renmin Hospital of Wuhan University. 19 MFAP4‐deficient (knockout, KO) mice originally obtained from Cyagen Biosciences Inc (Guangzhou, China); Wild‐type C57/B6 mice (8 weeks old; 23–27 g) were purchased from the Institute of Laboratory Animal Science at the Chinese Academy of Medical Sciences (Beijing, China). All mice were housed and maintained in a specific pathogen‐free barrier environment in a 12‐hour light‐dark cycle, and observed daily to ensure free access to water and food. Randomization and blinding was used for animal grouping in all experiments to ensure the reliability of the data.
Aortic Banding Models
After 7 days of adaptive feeding, mice were subjected to aortic banding (AB) surgery to generate pressure overload‐induced cardiac remodeling, as previously described. 20 Briefly, the mice were anesthetized with 3% pentobarbital sodium (by intraperitoneal injection [IP]) and the thoracic aorta was then identified. Under a microscope, a 27G blunt needle was placed next to the aortic segment and tightly tied to it with 7‐0 silk thread. After ligation, the blunt needle was pulled out, resulting in ≈70% contraction. Sham‐operated mice were subjected to a similar procedure without constriction of the aorta. Eight weeks after surgery, the mice were euthanized by an overdose of sodium pentobarbital (200 mg/kg, IP).
Neurohormonal Stimulation Models
Cardiac remodeling was also induced by repetitive IP of 50 mg/kg per day isoproterenol for 14 days. Control mice were treated in parallel with saline solution.
Echocardiography and Hemodynamic Evaluation
After anesthetization with 1.5% isoflurane, echocardiography was performed to evaluate the structure and function of the left ventricle with a MyLab 30CV ultrasound system (Esaote SpA, Genoa, Italy) equipped with a 10‐MHz phased array transducer as previously described. 6 The left ventricular (LV) end systolic diameter, LV end diastolic diameter, and posterior wall thickness were recorded. Based on these data, the ejection fraction and fractional shortening were calculated.
Invasive hemodynamic monitoring was performed with an aria pressure–volume conductance system (MPVS‐300 Signal Conditioner, Millar Instruments, Houston, TX, USA). Briefly, mice were anesthetized by 2% isoflurane inhalation, and a 1.4‐French catheter transducer (SPR‐839; Millar Instruments, Houston, TX, USA) was inserted into the LV via the right carotid artery. All data were recorded and analyzed via pressure–volume analysis data analysis software (Millar, Houston, TX, United States).
Histological Analysis and Immunohistochemical Staining
The hearts were washed in 10% potassium chloride solution to ensure that they were stopped in diastole and fixed with 4% paraformaldehyde for >24 hours. The hearts were then embedded in paraffin and cut transversely into 5‐μm slices to visualize the left and right ventricles. Several heart sections of the heart were stained with 0.1% Picrosirius red for evaluation of collagen deposition and with hematoxylin and eosin and wheat germ agglutinin for evaluation of cross‐sectional areas (CSAs). The images were then visualized by light microscopy (ECLIPSE 80i; Nikon, Japan). The data were analyzed with Image‐Pro Plus 6.0 software (Bethesda, MD, USA). We took the average of all photos of each heart (10 photos of each heart) and then conducted statistical analysis between groups.
Immunohistochemical staining was used to evaluate the expression levels of α‐SMA and MFAP4. After rehydration, the sections were heated using the pressure cooker method for antigen retrieval and then incubated anti‐MFAP4 (1:50) and anti‐α‐SMA (1:100) antibodies overnight at 4°C. Subsequently, the sections were incubated with goat anti‐rabbit EnVisionTM+/horseradish peroxidase reagent and stained using a 3,3N‐diaminobenzidine tertrahydrochloride detection kit.
Cell Culture
Neonatal rat cardiac fibroblasts (CFs) and neonatal rat cardiomyocytes were obtained from Sprague‐Dawley rats born within 3 days of each other according to the previous literature. 5 , 21 , 22 Depending on the experiment, CFs and cardiomyocytes were seeded in culture dishes, 6‐well plates, or 24‐well plates. The cardiomyocytes were cultured in DMEM/F12 (Gibco, C11995) containing 15% fetal bovine serum (FBS, Gibco, 10099) and CF medium containing 10% FBS at 37°C in a humidified incubator with 5% CO2. Bromodeoxyuridine (BrdU, 100 μmol/L) was used to prevent CF growth in cardiomyocyte cultures. After they were cultured to 70%–80% confluency and deprived of serum for 16 hours to synchronize, the CFs and cardiomyocytes were assigned randomly to groups, and different kinds of adenoviruses or reagents were added depending on the experiment.
Immunofluorescence Staining
CFs were analyzed for cardiac α‑SMA expression by immunofluorescence to assess the transformation of CFs into myofibroblasts. The cells on the glass coverslips were washed 3 times with PBS (for 5 minutes each), fixed with 4% paraformaldehyde (10 minutes), permeabilized in 0.2% Triton X‐100 in PBS (10 minutes), and blocked with 10% goat serum for 60 minutes at room temperature before being stained with an anti‑α‑SMA antibody at a dilution of 1:100 with 1% goat serum overnight at 4°C. The cells were washed 5 times with PBS and then incubated with an Alexa Fluor 568‐conjugated goat anti‑rabbit immunoglobulin G (Invitrogen, USA) secondary antibody for 1 hour at 37°C. Finally, the coverslips were washed 5 times and then mounted onto glass slides with DAPI. Immunofluorescence images were obtained with a fluorescence microscope (Olympus DX51, Japan).
Quantitative Real‐Time Reverse Transcription‑Polymerase Chain Reaction
For reverse transcription‐polymerase chain reaction, total RNA was extracted from pulverized LV myocardium tissue using TRIzol. The purity and concentration of the extracted total RNA were spectrophotometrically estimated with an ultraviolet spectrophotometer (NanoDrop2000; Thermo Fisher Scientific). The RNA (2 μg/sample) was then reverse‐transcribed into cDNA with an Advantage RT‐for‐PCR Kit. Reverse transcription‐polymerase chain reaction amplifications and analyses were performed with LightCycler 480 SYBR Green 1 Master Mix. All of the primer information is provided in Table. The mRNA levels of each gene were analyzed with the 2−ΔΔCt method and normalized against GAPDH gene expression.
Table.
The Primers Sequences for RT‐PCR
| Targets | Species | Forward | Reverse |
|---|---|---|---|
| GAPDH | Mice | TCATCAACGGGAAGCCCATC | CTCGTGGTTCACACCCATCA |
| MFAP4 | Mice | GCAACCCCTGGACTGTGATG | TTGTCATGTCGCAGAAGACGG |
| ANP | Mice | ACCTGCTAGACCACCTGGAG | CCTTGGCTGTTATCTTCGGTACCGG |
| BNP | Mice | GAGGTCACTCCTATCCTCTGG | GCCATTTCCTCCGACTTTTCTC |
| α‐MHC | Mice | GTCCAAGTTCCGCAAGGT | AGGGTCTGCTGGAGAGGTTA |
| β‐MHC | Mice | CCGAGTCCCAGGTCAACAA | CTTCACGGGCACCCTTGGA |
| Fibronectin | Mice | CCG GTG GCT GTC AGT CAG A | CCG TTC CCA CTG CTG ATT TATC |
| CTGF | Mice | TGT GTG ATG AGC CCA AGG AC | AGT TGG CTC GCA TCA TAG TTG |
| TGF‐β | Mice | GGTGGTATACTGAGACACCTTG | CCCAAGGAAAGGTAGGTGATAG |
| Collagen1a | Mice | AGGCTTCAGTGGTTTGGATG | CACCAACAGCACCATCGTTA |
| Collagen III | Mice | AAGGCTGCAAGATGGATGCT | GTGCTTACGTGGGACAGTCA |
| αSMA | Mice | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| GAPDH | Rat | GACATGCCGCCTGGAGAAAC | AGCCCAGGATGCCCTTTAGT |
| MFAP4 | Rat | AGATTCAACGGCTCAGTGAGT | TGCTTCAGTGTCAGGAGGTG |
| ANP | Rat | AAAGCAAACTGAGGGCTCTGCTCG | TTCGGTACCGGAAGCTGTTGCA |
| BNP | Rat | CAGCAGCTTCTGCATCGTGGAT | TTCCTTAATCTGTCGCCGCTGG |
| β‐MHC | Rat | TCTGGACAGCTCCCCATTCT | CAAGGCTAACCTGGAGAAGATG |
| αSMA | Rat | CATCCGACCTTGCTAACGGA | GTCCAGAGCGACATAGCACA |
| Fibronectin | Rat | GGATCCCCTCCCAGAGAAGT | GGGTGTGGAAGGGTAACCAG |
| Collagen1a | Rat | GAGAGAGCATGACCGATGGATT | TGGACATTAGGCGCAGGAA |
| Collagen III | Rat | AAGGGCAGGGAACAACTGAT | GTGAAGCAGGGTGAGAAGAAAC |
ANP indicates atrial natriuretic peptide; BNP, B‐type natriuretic peptide; CTGF, connective tissue growth factor; MFAP4, microfibrillar‐associated protein 4; RT‐PCR, reverse transcriptase polymerase chain reaction; TGF‐β, transforming growth factor‐β; α‐MHC, α‐myosin heavy chain; αSMA, alpha‐smooth muscle actin; and β‐MHC, β‐myosin heavy polypeptide.
Western Blot
For western blotting, heart samples or cultured cells were lysed in RIPA assay lysis buffer, and the total protein concentration was assessed using a BCA Protein Assay Kit (23227, Thermo Scientific, MIT, USA). The total protein concentration in all samples was normalized before western blot analysis was performed. Protein lysates (50 μg) were subjected to 8% to 12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (IPFL00010, EMD Millipore, Billerica, MA, USA). After blocking with 5% non‐fat milk (in Tris‐buffered saline with 0.1% Tween 20) for 1 hour at room temperature, the membranes were incubated with primary antibodies overnight at 4°C. The next day, after washing 3 times with Tris‐buffered saline with 0.1% Tween 20, the blots were incubated with a 1:1000 dilution of anti‐mouse or anti‐rabbit immunoglobulin G for 1 hour at room temperature while protected from light. After washing 3 times with Tris‐buffered saline with 0.1% Tween 20, the membranes were subsequently incubated with enhanced chemiluminescence reagents (170‐5061, BioRad, Hercules, CA, USA) and then visualized using Bio‐Rad ChemiDoc XRS+ according to the manufacturer's instructions. For quantification, the protein expression levels were normalized to GAPDH levels.
Electrocardiography Analysis
Mice were lightly anesthetized with 1.5% isoflurane. After the mice were calm and stable, the concentration of isoflurane was maintained at 0.8% to 1%. According to the instructions, the subcutaneous leads were connected, and ECG recordings were obtained on limb lead II. Lead II was analyzed for heart rate (HR; RR interval) and PR, QRS, and QTc were analyzed off‐line by LabChart 7 Pro (ADInstruments, Sydney, Australia). ECG waveforms averaged over 4 consecutive beats were used for the analyses. 23
Preparation of Langendorff‐Perfused Hearts
After anticoagulation with heparin sodium (100 U, IP), the mice were anesthetized with pentobarbital sodium (50 mg/kg, IP). Then, thoracotomy was performed and the heart was quickly isolated and transferred to ice‐cold (4°C) 100% O2 oxygenated cold Tyrode solution (mmol/L: NaCl 130, KCl 5.4, CaCl2 1.8, MgCl2 1,Na2HPO4 0.3, HEPES 10, glucose 10, pH adjusted to 7.4 with NaOH). Excess tissue such as tracheal, pericardial, and esophageal tissue was removed to expose the ascending aorta under a microscope, a small section of aorta was identified and then rapidly cannulated with a tailor‐made 21‐gauge cannula that had been prefilled with cold Tyrode buffer. The aorta was secured onto the cannula with a micro‐aneurysm clip and the isolated heart was then fixed to the Langendorff‐perfusion system (AD Instruments, Australia). The heart was perfused with 100% O2 saturated Tyrode solution through the aorta by a peristaltic pump (AD Instruments, Australia). The perfusion temperature was maintained at 37°C, the perfusion pressure was maintained at 80 to 100 mm Hg and the perfusion rate was 2 to 3 mL. Five minutes usually passed from the removal of the heart to the resumption of beating of the isolated heart. ECG electrodes were placed in the posterior wall of the right atrium and the apex of the heart to simulate normal electrocardiography, and the readings stabilized by 10 minutes. Heart specimens with obvious arrhythmia or irreversible myocardial ischemia after reperfusion for 10 minutes were excluded.
Monophasic Action Potential Recording and Electrical Stimulation Protocol
The epicardial monophasic action potential (MAP) of the adventitia of the LV anterior free wall was recorded with a self‐made MAP electrode. The MAP electrode was composed of 2 Teflon‐coated Ag‐AgCl electrodes (99.99% purity) with diameters of 0.25 mm that were twisted together and electroplated to eliminate direct current offset. A pair of platinum stimulation electrodes was placed on the basal surface of the right ventricle and delivered regular pacing stimulation (pulse width 1 ms, voltage 3 mV). Epicardial bipolar electrograms were recorded with an AgCl Teflon‐coated bipolar electrode with a diameter of 0.25 mm and coated with Teflon. All electrical signals were amplified by the ECG amplifier module with filtering between 0.3 Hz and 1 kHz. The MAP waveforms were analyzed with LabChart 7 Pro software (AD Instruments).
S1‐S1 pacing was used to determine the alternating threshold of the 90% action potential duration (APD) (APD90) and APD. The pacing cycle length (PCL) ranged from 150 to 30 ms, with successive 10 ms decreases. The pacing at each PCL last for 30 seconds, and was then interrupted for at least 30 seconds to eliminate the pacing memory effect. The APD90 at a PCL of 150 ms was defined as the average 90% repolarization time for 6 to 8 successive MAPs. APD alternans (ALT) was determined by 2 consecutive beats whose APD90 differed by ≥5% over 10 beats. The ALT threshold was defined as the maximal PCL (PCLmax) that induced ALT. VAs were induced by burst pacing (2 ms pulses at 50 Hz, 2 seconds burst duration) repeated 20 times at an interval of 2 seconds.
Statistical Analysis
The data are presented as mean±SEM and were analyzed by GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA, USA) and SPSS 24.0. The Kolmogorov‐Smirnov test was used to validate the normal distribution of the data (n>4). Multiple groups were tested by 1‐way ANOVA followed by Tukey posthoc test, whereas differences between 2 groups were evaluated using unpaired Student t‐tests. Categorical data are presented as percentages and were tested using the Fisher exact test. A value of P<0.05 was considered to indicate statistical significance.
RESULTS
MFAP4 Expression in a Pressure Overload‐Induced Cardiac Remodeling Model
We found that MFAP4 levels were slightly increased at 1 week but significantly upregulated at 2, 4, 6, and 8 weeks after AB (Figure 1A and 1B, 2 weeks: 1.592 times; 4 weeks: 1.921 times; 6 weeks: 2.108 times; 8 weeks: 2.471 times; P<0.05). In addition, immunohistochemical staining showed that the expression of MFAP4 at 8 weeks after AB was significantly higher than that in the sham group, (Figure 1C). Together, these findings indicate that MFAP4 is indeed expressed in hearts and that its protein levels are compensatorily increased during the development of cardiac remodeling.
Figure 1. MFAP4 is overexpressed in response to pressure overload.
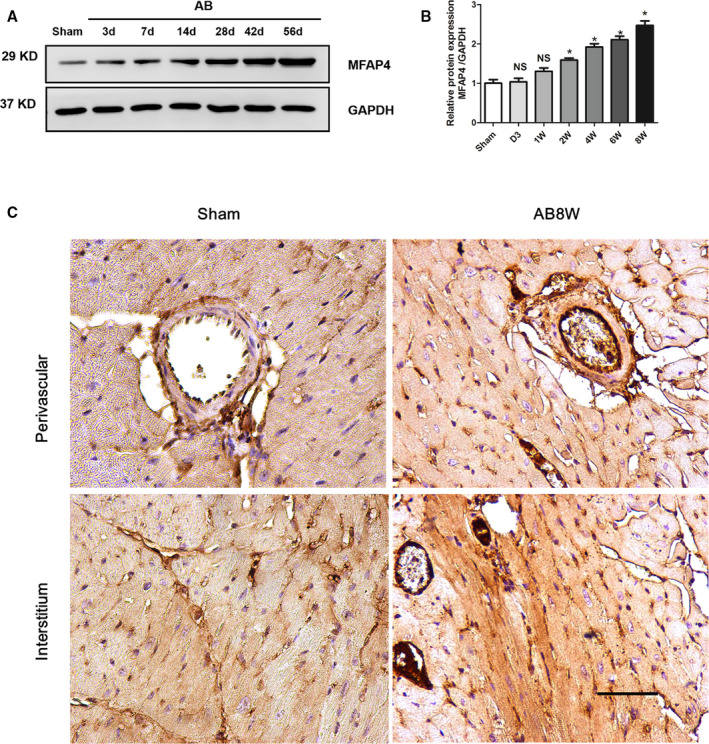
A and B, Representative western blot of MFAP4 (microfibrillar‐associated protein 4) in heart tissues after aortic banding at the indicated time points (3, 7, 14, 28, 42, and 56 days) (n=5). C, Immunohistochemical detection of MFAP4 expression in the sham group and the 8‐week aortic banding group. Scale bar: 50 μm (n=3). AB indicates aortic banding; AB 8W, 8‐week aortic banding group; and MFAP4, microfibrillar‐associated protein 4. *P<0.05 compared with Sham group.
MFAP4 Ablation Improves Cardiac Function
To confirm the role of MFAP4 in cardiac remodeling, we reproduced and identified homozygous MFAP4 knockout (KO) mice (Figure S1). We performed AB surgery or sham surgery in MFAP4‐KO mice and their wild‐type (WT) littermates and evaluated the effects of MFAP4 on cardiac function. After 8 weeks, echocardiographic analysis was performed to assess the function of the left ventricle. There were no significant changes in sham group (WT‐sham versus KO‐sham, P>0.05). However, pressure overload induced significantly better cardiac systolic function in KO mice compared with WT mice (WT‐AB VS KO‐AB, P<0.05), as measured by ejection fraction and fractional shortening (Figure 2A through 2C). However, MFAP4‐KO and AB surgery had no effect on heart rate (HR, Figure 2D, P>0.05).
Figure 2. MFAP4 deficiency alleviates cardiac dysfunction after pressure overload.
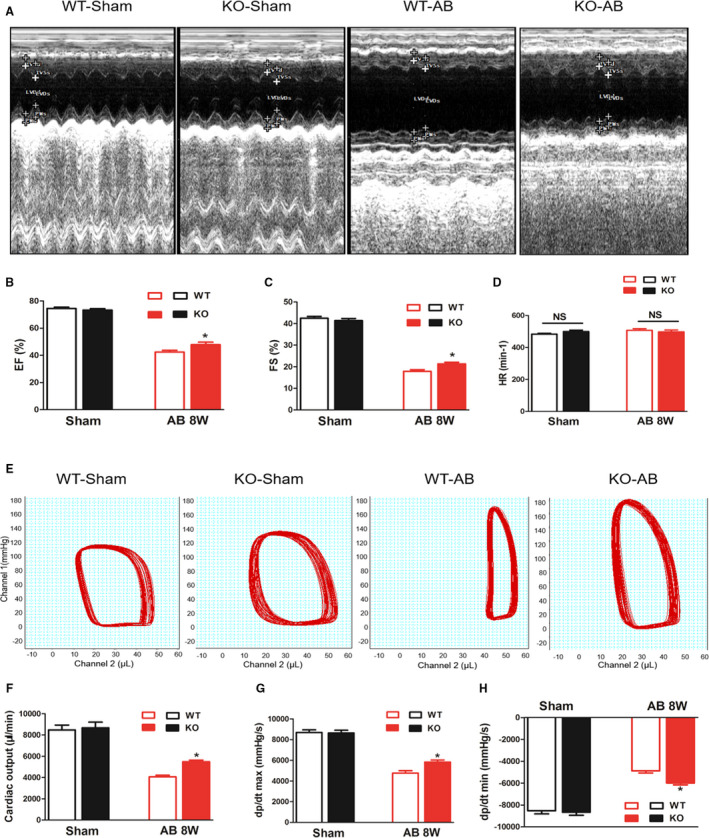
A, Representative echocardiographic images for each group. B through D, Statistical results for the ejection fraction, left ventricular fractional shortening, and heart rate (n=15 for the wild‐type‐sham and knockout‐sham groups; n=12 for the wild‐type‐aortic banding [AB] and knockout‐AB groups). E, Representative pressure‐volume loop images for each group. F through H, Statistical results for the cardiac output, maximal rate of pressure development (dP/dtmax), and maximal rate of pressure decay (dP/dtmin) (n=12 for the WT‐sham and knockout‐sham groups n=11 for the WT‐AB and knockout‐AB groups). AB indicates aortic banding; CO, cardiac output; dp/dtmax, maximal rate of left ventricle pressure development; dp/dtmin, minimal rate of left ventricle pressure development; EF, ejection fraction; FS, fractional shortening; and HR, heart rate. *P<0.05 represents the comparison between WT‐AB and KO‐AB group.
Pressure‐volume loop analysis further illustrated the improvements in LV hemodynamic dysfunction in MFAP4‐KO mice, as measured by parameters that reflect systolic function (assessed by +dP/dt), diastolic function (assessed by +dP/dt), and cardiac output (Figure 2E through 2H). These results suggest that MFAP4 deficiency could improve cardiac function after AB surgery.
Effect of MFAP4 on Cardiac Hypertrophy Induced by Pressure Overload
As shown in Figure 3, pressure‐overloaded WT mice showed significantly greater heart weight (HW)/body weight and HW/tibia length ratios and cardiomyocyte CSAs than WT‐sham mice (HW/body weight, 8.680±0.361 versus 4.383±0.090; HW/tibia length, 13.72±0.573 versus 6.982±0.086, CSA, 502.0±21.81 versus 203.5±7.818, P<0.05). However, no significant differences were observed between the KO‐AB and WT‐AB groups (P>0.05). Moreover, the mRNA expression levels of several hypertrophic markers, including ANP, BNP (Brain natriuretic peptide), α‐MHC (α‐myosin heavy chain), and β‐MHC (β‐myosin heavy chain), were not different between the KO‐AB and WT‐AB groups (P>0.05). Western blotting was used to detect the protein levels of ANP and β‐MHC, and the results were consistent with the above results (P>0.05).
Figure 3. MFAP4 deficiency does not improve aortic banding‐induced cardiac hypertrophy.
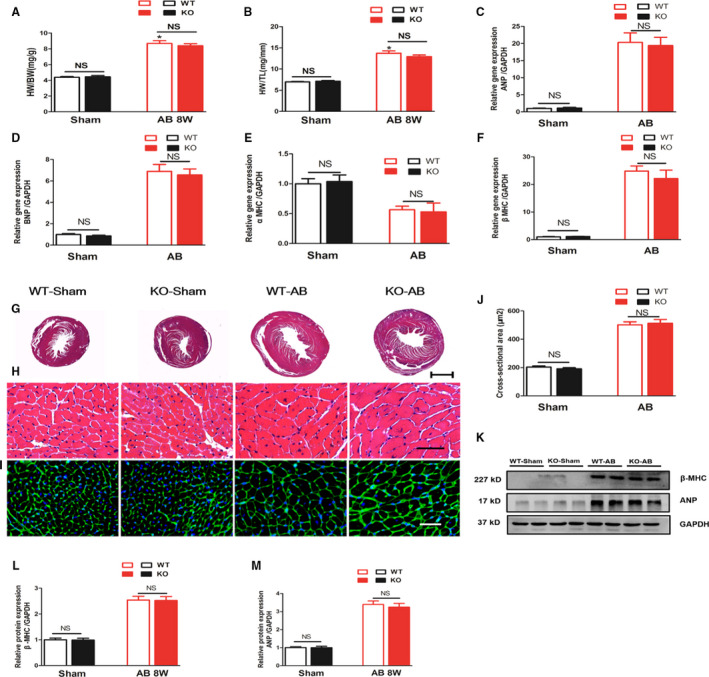
A and B, heart weight/body weight and heart weight/tibia length ratios of wild‐type (WT) and MFAP4‐knockout mice (KO) after sham or aortic banding (AB) operation (n=15 for the WT‐sham and KO‐sham groups; n=16 for the WT‐AB and KO‐AB groups). C through F, The effects of AB on the mRNA expression of ANP, BNP, α‐MHC, and β‐MHC were determined by reverse transcription‐polymerase chain reaction analysis (n=6). G and H, Representative hematoxylin and eosin‐stained heart sections of WT or MFAP4‐KO mice after sham or AB operation. G, 10× (scale bar: 400 μm), (H) 400× (scale bar: 50 μm). n=6. I, Representative wheat germ agglutinin‐fluorescein isothiocyanate isomer staining of sham and AB mice at 8 weeks post‐surgery, Scale bar: 50 μm. J, Quantification of the CSA in each group (n=6). K through M, Representative blots and quantitative results for ANP and β‐MHC protein expression in myocardium in each group (n=6). AB indicates aortic banding; AB 8W, 8‐week aortic banding group; ANP indicates atrial natriuretic peptide; BNP, B‐type natriuretic peptide; HW/BW, heart weight/body weight; HW/TL, heart weight/tibia length; KO, knockout mice; NS, no statistically difference; WGA, wheat germ agglutinin; and β‐MHC, β‐myosin heavy polypeptide. *P<0.05 compared with the corresponding sham group.
Effect of MFAP4 on Cardiac Fibrosis Induced by Pressure Overload
Increased cardiac fibrosis was observed in WT mice in response to AB operation (10.74±0.879% versus 2.598±0.243%, P<0.05), but was markedly limited in MFAP4‐KO mouse hearts (Figure 4A and 4B, 8.271±0.758% versus 10.74±0.879%, P<0.05). The mRNA levels of the fibrotic markers α‐SMA, collagen Iα (Col1α), collagen III (Col3), connective tissue growth factor, fibronectin, TGF‐β, and the protein levels of α‐SMA, Col3, P‐Smad2/ T‐Smad2, P‐Smad3/ T‐Smad3 were measured (Figure 4C through 4M); these molecules are known mediators of fibrosis. Decreased expression levels of connective tissue growth factor, TGF‐β, Col1α, Col3, fibronectin, α‐SMA, P‐Smad2/T‐Smad2, P‐Smad3/T‐Smad3 were detected in the AB‐induced KO mice than that of in the AB‐induced WT mice (Figure 4C through 4M, Compared with WT‐sham, the multiples increased by 3.054 times, 3.836 times, 5.046 times, 9.272 times, 4.072 times, 2.149 times, 2.004 times, 1.716 times, 2.009 times, respectively, P<0.05). All these data indicate that deletion of MFAP4 blocks pathological cardiac fibrosis induced by chronic pressure overload.
Figure 4. MFAP4 deficiency attenuates aortic banding‐induced cardiac fibrosis.
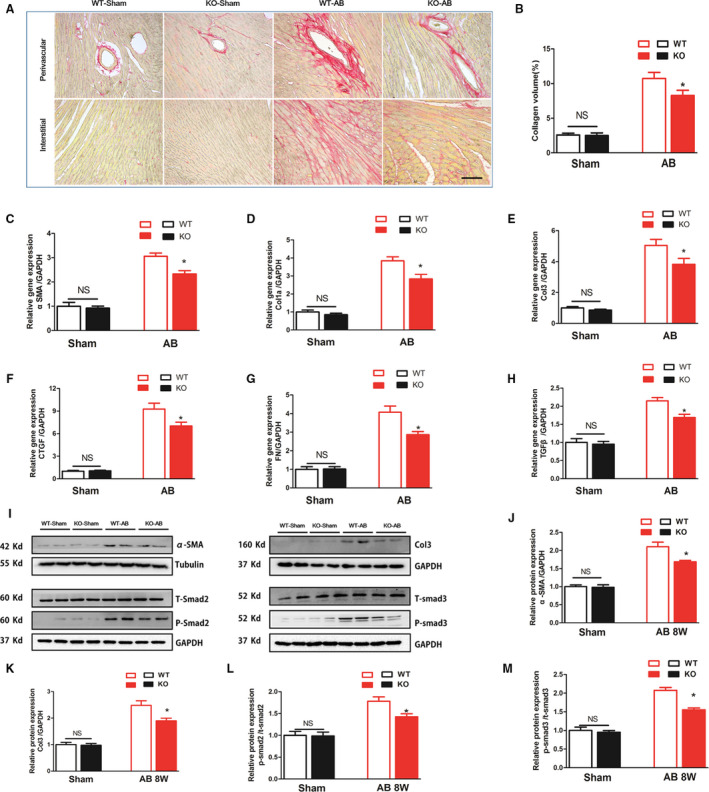
A, Representative image of the heart with Picrosirius staining. scale bar: 50 μm. B, Quantification of the total collagen volume in the indicated group (n=6). C through H, PCR analysis of fibrotic markers (αSMA, collagen Iα, collagen III, CTGF, fibronectin, TGF‐β) (n=6). I through M, Representative blots and quantitative results for αSMA, collagen III and P‐Smad2/T‐Smad2, and P‐Smad3/T‐Smad3 protein expression in the myocardium in each group (n=6). AB indicates aortic banding; Col1α, collagen Iα; Col3, collagen type 3; CTGF, connective tissue growth factor; KO, knockout mice; MFAP4, microfibrillar‐associated protein 4; PSR, Picrosirius red; TGF‐β, transforming growth factor‐β; WT, wild‐type; and αSMA, alpha‐smooth muscle actin. *P<0.05 compared with the corresponding wild‐type‐aortic banding group.
MFAP4 Deficiency Significantly Inhibits FAK and Downstream Signaling
We found that pressure overload–induced FAK activation was strongly downregulated in MFAP4‐KO mice after 8 weeks of AB (Figure 5A and 5B, P<0.05). Consistent with the observed increase in FAK activity, pressure overload caused decreases the levels of phosphorylated PI3K, AKT, MEK1/2, and ERK1/2 (Figure 5A, 5C through 5F, P<0.05). Therefore, our study demonstrates that the FAK and its downstream signaling molecules forma critical pathway by which MFAP4 influences cardiac fibrosis induced by pressure overload.
Figure 5. MFAP4 deficiency significantly inhibits FAK and downstream signaling.
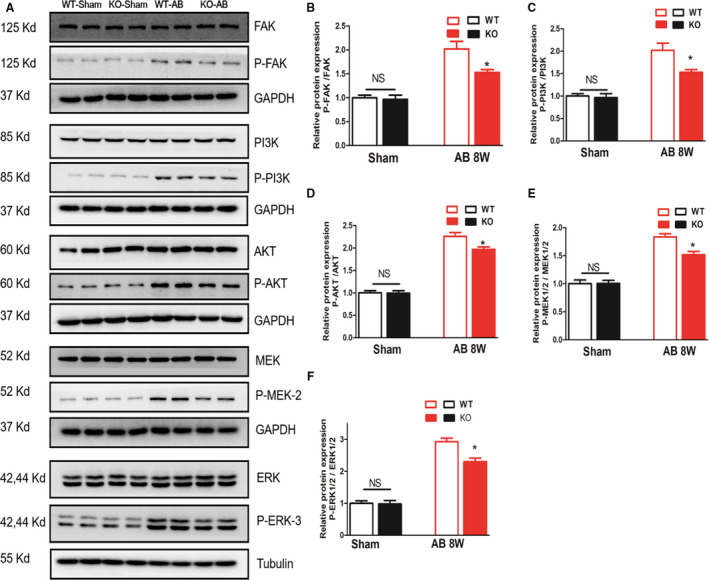
A through F, Representative blots and quantitative results for P‐FAK/T‐FAK, P‐PI3K/T‐PI3K, P‐AKT/T‐AKT, P‐MEK1/2/ T‐MEK1/2, P‐ERK1/2/T‐ERK1/2 protein expression in the myocardium in each group (n=6). *P<0.05 compared with the corresponding WT‐AB group. AB indicates aortic banding; ERK, extracellular signal regulated kinase; FAK, focal adhesion kinase; KO, knockout mice; MEK, mitogen‐activated protein kinase (MAPK)/ERK kinase; PI3K, phosphatidylinositol 3 kinase; and WT, wild‐type.
Effect of MFAP4 on Hypertrophy and Dysfunctional Responses Induced by Isoproterenol
We found that IP of isoproterenol significantly increased the expression of the MFAP4 gene in the myocardium, but MFAP4 expression in MFAP4‐KO mice was almost negligible (Figure 6A, P<0.05). Two weeks post‐commencement of isoproterenol injection, the mice exhibited obviously worse cardiac function (P<0.05). The changes in ejection fraction and fractional shortening in mice challenged with isoproterenol were significantly reversed by MFAP4 deficiency (Figure 6B and 6C, P<0.05). To explore the effect of MFAP4 deletion on isoproterenol‐mediated cardiac hypertrophy, we measured HW/body weight and HW/tibia length ratios, CSAs, and the levels of hypertrophic markers. As shown in Figure 6, WT‐isoproterenol mice showed significantly higher HW/body weight, HW/tibia length ratios, and CSAs than WT‐Control mice (P<0.05). However, no significant differences were observed between the KO‐isoproterenol and WT‐isoproterenol groups (P>0.05). Moreover, the mRNA expression levels of several hypertrophic markers (ANP, BNP, α‐MHC, and β‐MHC) and the protein expression levels of ANP and β‐MHC were also not different between the KO‐isoproterenol and WT‐isoproterenol groups (P>0.05). The results were consistent with those obtained for the pressure overload induced cardiac hypertrophy model.
Figure 6. MFAP4 deficiency does not improve isoproterenol‐induced cardiac hypertrophy.
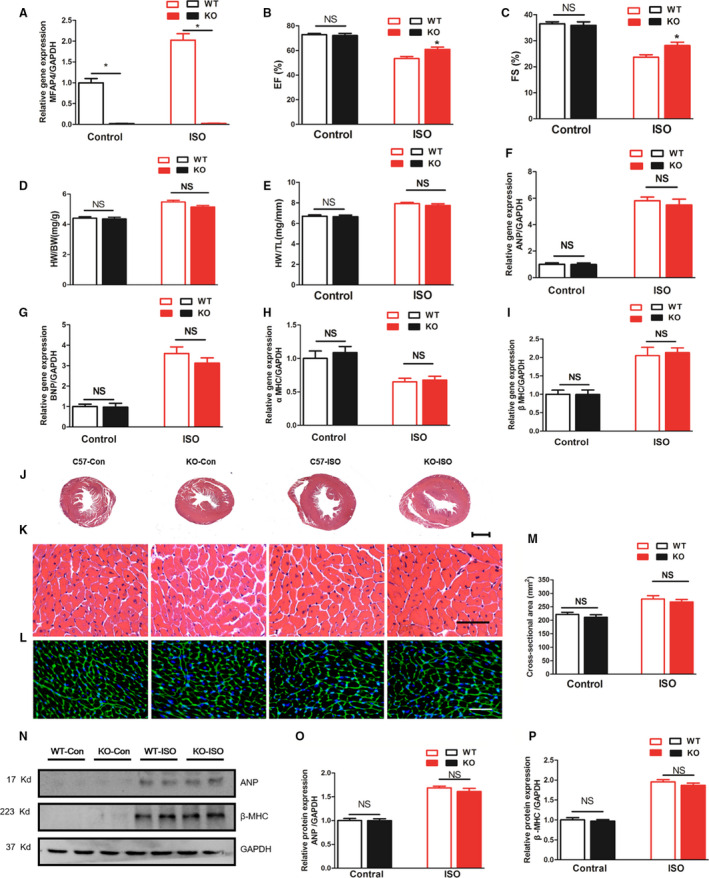
A, Polymerase chain reaction analysis of MFAP4 mRNA expression in the myocardium in each group (n=6). B and C, Statistical results for the ejection fraction and fractional shortening in the indicated group (n=10 for the WT‐Control and knockout mice (KO)‐Control groups; n=13 for the WT‐isoproterenol group; n=12 for the KO‐isoproterenol group). D and E, HW/BW and HW/tibia length ratios of WT and MFAP4‐KO mice after vehicle (control) or isoproterenol IP injection (n=12 for the WT‐Control and KO‐Con groups; n=23 for the WT‐isoproterenol group; n=13 for the KO‐isoproterenol group). F through I, The effects of isoproterenol on the mRNA expression of ANP, BNP, α‐MHC, and β‐MHC were determined by reverse transcription‐polymerase chain reaction analysis (n=6). J through L, Hematoxylin and eosin staining and wheat germ agglutinin‐fluorescein isothiocyanate isomer staining of the control or isoproterenol mice. J, 10× (scale bar: 400 μm), (K) 400× (scale bar: 50 μm). M, Quantification of the cross‐sectional areas in each group (n=6). N through P, Representative blots and quantitative results for ANP and β‐MHC protein expression in the myocardium in each group (n=6). ANP indicates atrial natriuretic peptide; HW/BW, heart weight/body weight; HW/TL, heart weight/tibia length; KO, knockout mice; NS, no statistically difference; WGA, wheat germ agglutinin; WT, wild‐type; and β‐MHC, β‐myosin heavy polypeptide. *P<0.05 compared with the corresponding control group.
Effect of MFAP4 on Cardiac Fibrosis Induced by Isoproterenol
As shown in Figure 7, we found markedly increased myocardial collagen volumes and fibrosis markers in the isoproterenol‐induced KO and WT hearts compared with the controls after 2 weeks (P<0.05). However, it should be noted that considerably decreased cardiac collagen volumes and fibrosis markers were also present in the KO mice (P<0.05). The results were consistent with those obtained for the pressure overload induced cardiac fibrosis model.
Figure 7. MFAP4 deficiency attenuated isoprotenerol‐induces cardiac fibrosis.
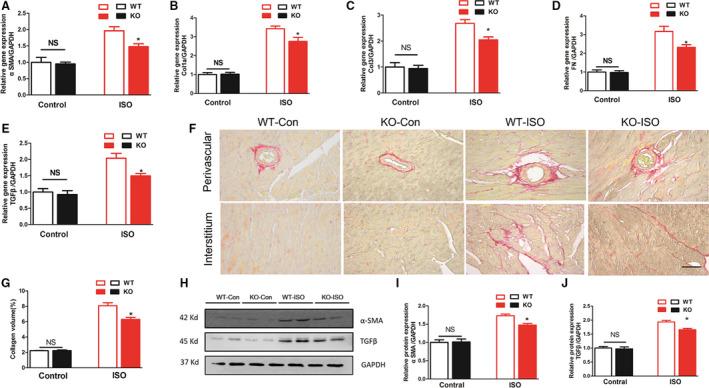
A through E, Polymerase chain reaction analysis of fibrotic markers (αSMA, collagen Iα, collagen III, fibronectin, transforming growth factor‐β) in the myocardium in each group (n=6). F, Representative image of the heart with Picrosirius staining. G, Quantification of the total collagen volume in the indicated group (n=6). H and I, Representative blots and quantitative results for ANP and β‐MHC protein expression in the myocardium in each group (n=6). Col1α indicates collagen Iα; CTGF, connective tissue growth factor; KO, knockout mice; MFAP4, microfibrillar‐associated protein 4; PSR, Picrosirius red; TGF‐β, transforming growth factor‐β; WT, wild‐type; and αSMA, alpha‐smooth muscle actin. *P<0.05 compared with the corresponding wild‐type‐isoproterenol group.
Expression of MFAP4 in Cardiomyocytes and CFs
To elucidate the expression of MFAP4 in cardiomyocytes and CFs, we extracted cardiomyocytes and CFs from neonatal rats and detected the basal MFAP4 expression in these 2 kinds of cells by polymerase chain reaction. We found that the basal expression of MFAP4 in CFs was >40 times that in cardiomyocytes. MFAP4, as an ECM protein, may be produced mainly by CFs (Figure 8A). In addition, we also stimulated these 2 kinds of cells with angiotensin II (Ang II), phenylephrine, and TGF‐β and measured the expression of MFAP4 and markers of hypertrophy and fibrosis. We found that Ang II and TGF‐β increased the expression of α‐SMA, Col3, and MFAP4 in CFs (P<0.05), while phenylephrine only slightly increased the expression of MFAP4 (P<0.05). Among the factors, TGF‐β increased MFAP4 expression most significantly in CFs (2.895 times, P<0.001). In cardiomyocytes, Ang II, phenylephrine, and TGF‐β increased the expression of ANP and β‐MHC (P<0.05), but only Ang II and TGF‐β increased the expression of MFAP4 (Figure 8B and 8C, P<0.05). Given the basal expression of MFAP4, we believe that the expression of MFAP4 in CFs is much higher than that in cardiomyocytes, and that its expression significantly increases upon stimulation with TGF‐β.
Figure 8. Role and mechanism of MFAP4 in cardiac fibroblasts.
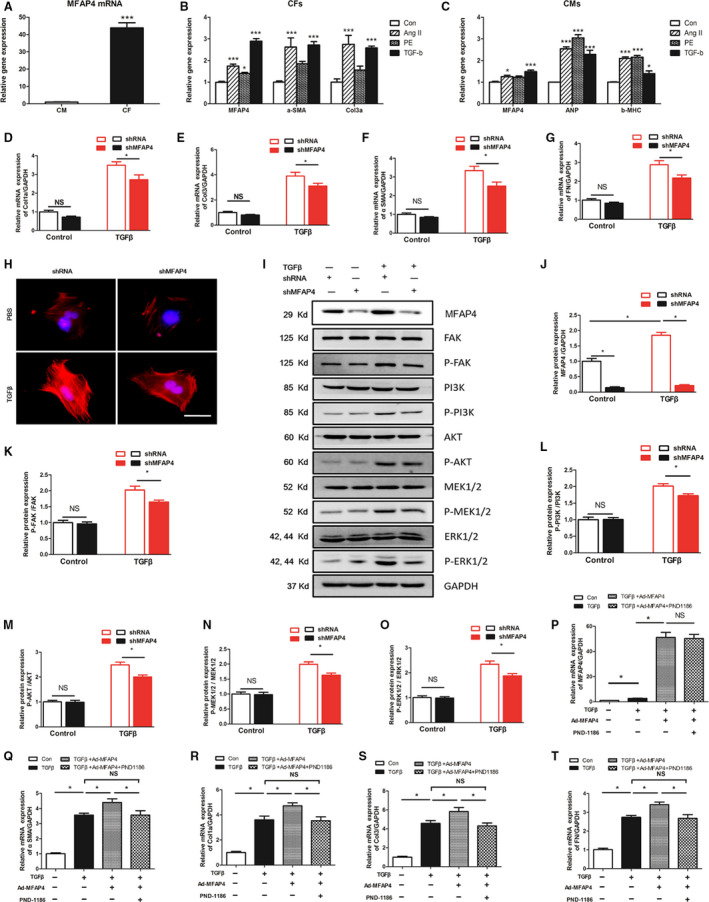
A, Polymerase chain reaction analysis of mRNA level of MFAP4 (microfibrillar‐associated protein 4) between cardiomyocytes and cardiac fibroblasts (n=6). The data were compared by unpaired Student t‐test. B and C, Polymerase chain reaction analysis of the mRNA expression level of MFAP4 and markers of, hypertrophic and fibrosis markers when in cardiomyocytes and cardiac fibroblasts were stimulated by angiotensin II (Ang II), phenylephrine, and or TGF‐β, respectively (n=6). (D through G) PCR analysis of the mRNA expression levels of αSMA, Col1α, Col3, and fibronectin (n=6). H, Immunofluorescence staining of αSMA (n=6). I through O, Representative blots and quantitative results for P‐FAK/T‐FAK, P‐PI3K/T‐PI3K, P‐AKT/T‐AKT, P‐MEK1/2/T‐MEK1/2, P‐ERK1/2/T‐ERK1/2 protein expression in cardiac fibroblasts each group (n=4). P through T, Polymerase chain reaction analysis of the mRNA expression levels of MFAP4 and fibrosis markers in cardiac fibroblasts treated with Ad‐MFAP4 and an inhibitor of FAK (PND‐1186) (n=6). Ang II indicates angiotensin II; CF, cardiac fibroblasts; CMs, Cardiomyocyte; Col1α, collagen Iα; Col3, collagen 3; CTGF, connective tissue growth factor; ERK, extracellular signal regulated kinase; FAK, focal adhesion kinase; ; MEK, mitogen‐activated protein kinase (MAPK)/ERK kinase; MFAP4, microfibrillar‐associated protein 4; PI3K, phosphatidylinositol 3kinase; PSR, Picrosirius red; TGF‐β, transforming growth factor‐β; and αSMA, alpha‐smooth muscle actin. *P<0.05; **P<0.01; *P<0.001.
MFAP4 Silencing Prevents the Accumulation of Collagen in CFs Stimulated by TGF‐β
Next, we used adenoviral shRNA (Figure S2), to silence the MFAP4 gene (Ad‐shMFAP4) in CFs and compared its effects with those of a control adenovirus (Ad‐shRNA) by observing the expression of fibrosis markers in CFs. Ad‐shMFAP4‐treated CFs cultured with TGF‐β1 exhibited significantly less fibrosis than Ad‐shRNA‐treated CFs cultured with TGF‐β1, which showing lower levels of α‐SMA, Col1a, Col3, and fibronectin mRNA expression (Figure 8D through 8G, P<0.05) and lower α‐SMA fluorescence intensity (Figure 8H). Given the results of the in vivo experiments, we detected the activation of FAK and its downstream signaling pathway in CFsby TGF‐β stimulation and Ad‐shMFAP4‐mediated silencing. Consistent with the in vivo results, the phosphorylation activation of FAK and downstream PI3K‐AKT and MEK1/2‐ERK1/2 in the Ad‐shMFAP4+TGF‐β group was significantly lower than that in the Ad‐shRNA+TGF‐β group (Figure 8I through 8N, P<0.05).
Overexpression of MFAP4 Aggravates the Fibrotic Response of CFs Stimulated by TGF‐β
Subsequently, we used MFAP4‐overexpressing adenoviruses (Ad‐MFAP4) and control adenoviruses (pAdeno‐EF1A(S)‐NeonGreen‐CMV‐MCS‐3FLAG) (Figure S3). Ad‐MFAP4 significantly increased the MFAP4 expression in CFs (Figure 8O, P<0.05). We found that overexpression of MFAP4 alone had no significant effect on fibrosis markers through Ad‐MFAP4 (Figure S4, P>0.05). However, overexpressing MFAP4 showed higher levels of fibrosis markers after TGF‐β stimulation (TGF‐β+Ad‐MFAP4), than pAdeno‐EF1A(S)‐NeonGreen‐CMV‐MCS‐3FLAG‐treated cells+TGF‐β (Figure 8P through 8S, P<0.05). To investigate whether FAK is essential for the deteriorative effects of MFAP4 in vivo, we used PND‐1186 (VS‐4718), an inhibitor of FAK, to inhibit the activation of FAK in CFs stimulated by TGF‐β. After combined treatment with PND‐1186 (TGF‐β+Ad‐MFAP4+PND‐1186), the profibrotic effect of Ad‐MFAP4 in the heart induced by TGF‐β was significantly inhibited (TGF‐β+Ad‐MFAP4+PND‐1186 group VS TGF‐β+Ad‐MFAP4 group, P<0.05), as evidenced by downregulation of the mRNA expression of α‐SMA, Col1α, Col3, and fibronectin compared with TGF‐β+Ad‐MFAP4 group. Therefore, FAK‐related signaling pathways play an important role in MFAP4 aggravating the fibrotic response of CFs stimulated by TGF‐β.
Effect of MFAP4 on Ventricular Arrhythmia
To explore the effect of MFAP4 on cardiac electrical remodeling, we first monitored electric signals using surface ECG (lead II) recordings obtained in mice under light anesthesia (Figure 9A). No statistically significant differences in PR intervals or RR intervals were observed between the WT‐AB and KO‐AB groups (Figure 9B and 9C, P>0.05). However, the KO‐AB group exhibited significantly lower QRS intervals and QTc values than the WT‐AB group (Figure 9D and 9E, P<0.05).
Figure 9. MFAP4 deficiency alleviates aortic banding‐induced ventricular arrhythmia.
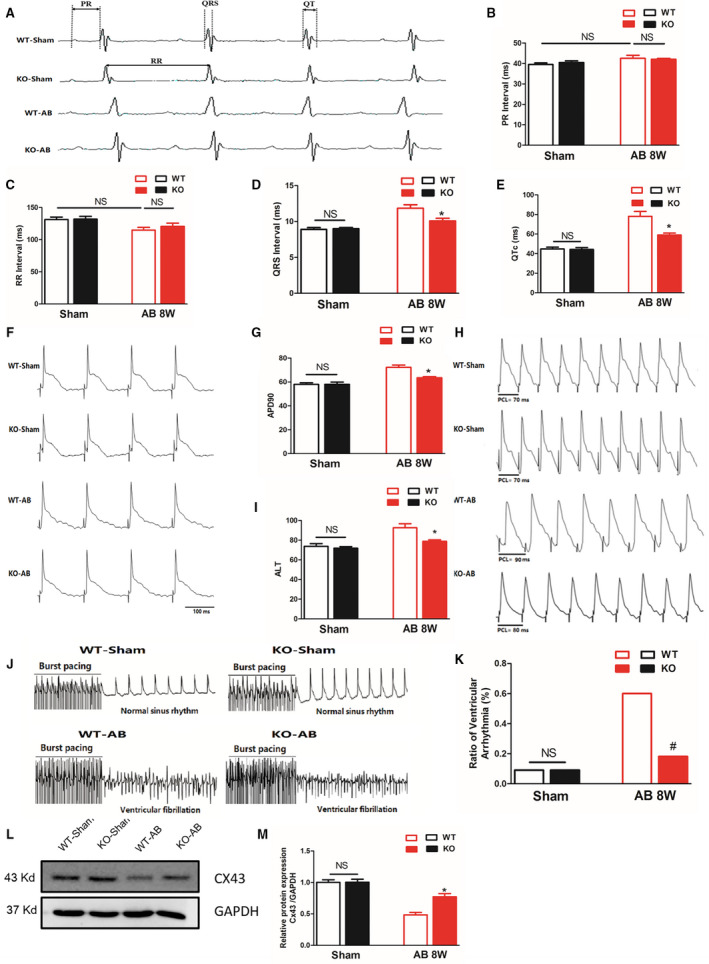
A, Representative ECG recordings and a comparison of ECG parameters. B through E, Statistical analysis of the PR, RR, QRS, and QTc intervals in MFAP4 (microfibrillar‐associated protein 4)‐knockout (KO) and wild‐type (WT) mice 8 weeks after the sham operation or aortic banding (AB) surgery (n=8 for the WT‐sham and KO‐sham groups; n=13 for the WT‐AB group; n=7 for the knockout [KO]‐AB group). F and G, Representative action potential figures and statistical analysis of the APD90 values in MFAP4‐KO and WT mice 8 weeks after the sham operation or AB surgery (n=8). H and I, Representative electric alternans figures and statistical analysis of the ALT thresholds in MFAP4‐KO and WT mice 8 weeks after the sham operation or AB surgery (n=8). J and K, Representative arrhythmia induced by burst‐pacing stimulation and statistical analysis of MFAP4‐KO and WT mice 8 weeks after the sham operation or AB surgery (n=11 for the WT‐sham, KO‐sham, and KO‐AB groups; n=10 for the WT‐AB group). L and M, Representative western blots, and statistical analysis of electroconduction‐related protein (Connexin 43) levels in MFAP4‐KO and WT mouse heart tissues (n=4). AB indicate aortic banding; AB 8W, 8‐week aortic banding group; ALT, action potential duration alternans; APD90, 90% action potential duration; Cx43, Connexin 43; HW/BW, heart weight/body weight; KO, knockout mice; MFAP4, microfibrillar‐associated protein 4; and WT, wild‐type. *P<0.05 represents the comparison between WT‐AB and KO‐AB group.
Then Langendorff‐perfused hearts were used to characterize changes in electrophysiological parameters (APD90, ALT, and VA incidence). APD90 was measured under programmed electrical stimulation with different pacing cycle length (PCLs) in Langendorff‐perfused hearts. In AB‐induced hearts (the WT‐AB group), APD90 and ALT were significantly prolonged, but they were much shorter in the KO‐AB group (Figure 9F through 9I). There was no significant difference in the induction of VA between the WT‐sham group and the KO‐sham group. In contrast, the VA induction rate was remarkably increased in the WT‐AB group (P<0.05), but MFAP4vKO attenuated these changes (Figure 9J and 9K, P<0.05). Connexin 43 is the major component of gap junctions, anatomical structures that regulate the function of ion channels responsible for action potential transmission. 24 , 25 We found that AB surgery significantly reduced the expression of Cx43 in the heart and that the levels of Cx43 after AB were significantly higher in the MFAP4‐KO group than in WT group (Figure 9L and 9M, P<0.05).
Discussion
Our data provide evidence that MFAP4 deficiency inhibits cardiac fibrosis and VA induced by pressure overload and isoproterenol, but has no significant effect on the hypertrophy response. We have demonstrated that pressure overload and isoproterenol cause accumulation of MFAP4 protein in the heart. We have further provided evidence of a loss of MFAP4 function in MFAP4‐KO mice and have shown that MFAP4 deficiency attenuates cardiac fibrosis, VA and preserves cardiac function following pressure overload and/or isoproterenol, strongly suggesting that MFAP4 is an essential regulator of cardiac structural and electrical remodeling. Based on the in vivo and in vitro data, we concluded that the activation of downstream signal pathway of FAK is involved in the promotion of cardiac fibrosis by MFAP4. The process of HF is always accompanied by the accumulation of ECM. There is robust clinical evidence of a strong correlation among increases in cardiac ECM, deterioration of cardiac function, and adverse outcomes in patients with HF. In addition, it is well known that CFs are the main cell types that mediated cardiac fibrosis during cardiac remodeling. When the heart is stimulated by pressure overload, activated CFs begin to proliferate and transdifferentiate into myofibroblasts, which then begin to secrete elevated levels of collagens and other ECM structural and matricellular proteins.
MFAP4 has been associated with various remodeling‐related disorders, including liver fibrosis, atherosclerosis, arterial injury stimulated remodeling, and asthma. 11 However, the expression levels and physiological functions of MFAP4 in heart disease are largely unknown. In this study, we found that MFAP4 mRNA and protein expression was upregulated in hearts with HF mediated by overload/isoproterenol, or in TGF‐β stimulated CFs. This result is consistent with the predicted expression of MFAP4 revealed in our previously published bioinformatics study on cardiac pathological remodeling. 16
Compared with WT mice, KO mice showed better cardiac function and less cardiac fibrosis after pressure overload. We also established a model of isoproterenol‐mediated cardiac remodeling. We found that, MFAP4 deficiency improved cardiac function and inhibited cardiac fibrosis. Cardiac hypertrophy is another important type of cardiac remodeling, but MFAP4 deficiency had no significant effects on hypertrophy indexes. Therefore, MFAP4 may mainly affect CFs and have little effect on cardiomyocytes.
The mechanisms underlying the antifibrotic effects of MFAP4 remain largely unclear. Some previous studies have proposed a novel mechanism linking MFAP4 to the activation of FAK by integrin signaling. Integrin is a transmembrane receptor that mediates cell–ECM and cell–cell connections. Among the 24 known human integrin superfamilies, 8 integrin dimers (αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α5β1, α8β1, αIIbβ3) recognize tripeptide N‐terminal arginine‐glycine‐aspartic acid motifs in ECM proteins. 26 During signal transduction, integrins transmit information about the chemical composition and mechanical state of the ECM into cells. 27 , 28 , 29 It is well established that FAK, a cytoplasmic non‐receptor protein tyrosine kinase, plays a critical role in cell migration and activated by cell binding to ECM proteins, 6 such as MFAP4. 13 Our experiments have confirmed that the process of cardiac pathological remodeling is accompanied by increased FAK phosphorylation. Our in vitro experiments have also shown that TGF‐β stimulation can enhance the phosphorylation of FAK, while MFAP4 deletion can inhibit the phosphorylation of FAK to some extent. We used PND‐1186 (VS‐4718), an inhibitor of FAK, to inhibit the activation of FAK in CFs stimulated by TGF‐β and found that the profibrotic effect of Ad‐MFAP4 in the heart induced by TGF‐β was significantly inhibited. Therefore, the fibrogenic effect of MFAP4 may be achieved by activation of FAK.
A series of studies have shown that when FAK‐linked integrin binds to the ECM, it contributes to the activation of the Ras–extracellular signal‐regulated kinase (ERK) mitogen‐activated protein kinase (MAPK) cascade. 30 ERK1/2 is a key transmitter of signals from surface receptors to the nucleus. Activation of ERK1/2 is triggered by MAPK/ERK kinase‐1/2 (MEK1/2) via phosphorylation of serine/threonine residues. 31 Our previous studies and a large number of other studies have confirmed that ERK1/2 activation can aggravate cardiac fibrosis, and inhibiting the activation of ERK1/2 can improve cardiac function and inhibit the cardiac pathological remodeling. 6 , 32 , 33 Our experiments have also proved that MFAP4 can further activate the MEK1/2‐ERK1/2 signaling pathway through activation of FAK. On the other hand, deletion of MFAP4 leads to decreases in the phosphorylation levels of MEK1/2 and ERK1/2.
Phosphorylation of FAK can activate not only MEK1/2‐ERK1/2, but also PI3K/AKT signaling pathway. 21 , 28 PI3K is the key upstream target for AKT activation. 20 Our previous studies and numerous other studies have confirmed that over activation of AKT is an important pathogenetic mechanism of cardiac remodeling. 20 , 34 , 35 In addition, MFAP4 deletion has been found to inhibit overactivation of PI3K/AKT during cardiac remodeling and to play a protective role in the myocardium.
Various types of cardiovascular diseases, especially HF, are often accompanied by structural and electrical remodeling of the heart. Among the key components of cardiac remodeling, fibrosis, which can occur as interstitial fibrosis, dense scarring or patchy fibrosis, constitutes a key histological substrate for atrial arrhythmia and VA. 36 VA also includes ventricular tachycardia, which accounts for ≈80% of sudden cardiac death and is usually a lethal arrhythmia. 37 Cardiac remodeling, whether it is mediated by AB (pressure overload) or isoproterenol injection, can lead to diffuse myocardial interstitial fibrosis. Diffuse fibrosis, in which too much ECM is deposited among myocardial fibers, increases the possibility of arrhythmia by selectively reducing side‐to‐side gap junction connections between cardiomyocytes. 38 This reduction in gap junctions between cells slows down the transverse propagation of waves and increases anisotropy, thus predisposing the fibrotic myocardium to wave break and anisotropic reentry. 38 In addition to reentry, many studies have shown that fibrosis interferes with all the basic electrophysiological mechanisms that lead to arrhythmias. 35 , 36 , 38 , 39
In our study, we found that MFAP4 deficiency not only reduced cardiac fibrosis caused by pressure overload, but also inhibited prolongation of the QRS interval and QTc recorded by body surface ECG. In addition, prolonged APD90 and ALT were observed in the AB‐induced cardiac remodeling model, and MFAP4 deficiency attenuated the prolongations ofAPD90 and ALT. Burst stimuli are often used in experiments to induce VA, reflecting the possibility of VA in the Langendorff‐perfused hearts. In our study, the incidence of VA induced by burst stimuli in MFAP4‐KO group was significantly lower than that in WT group. Cx43 is the major component of gap junctions, anatomical structures that regulate the function of ion channels responsible for the action potential transmission, and it is critical for maintaining normal cardiac electrical conduction. We found that the expression of Cx43 decreased significantly after cardiac fibrosis, while MFAP4 deletion increased the expression of Cx43, which may be one of the mechanisms by which MFAP4 deletion inhibits VA. Therefore, MFAP4 deficiency not only improves cardiac fibrosis after pressure overload, but also reduces the incidence of VA.
There are at least 2 limitations of our investigation. First, the study did not collect samples from clinical patients, so the expression of MFAP4 in patients with HF was not studied. Second, we used an MFAP4‐KO mouse model with reduced total body expression of MFAP4. In the next study, we will collect clinical patient samples, and construct CFs specific KO and overexpression mice to further clarify the role of MFAP4 in cardiac remodeling in more detail.
Conclusions
In summary, our study shows that MFAP4 promoted pressure overload‐induced cardiac fibrosis and VA, and the FAK and downstream signaling may play an important role in this process. These findings are the first to demonstrate that MFAP4 deficiency inhibits cardiac fibrosis and VA after challenge with 8 weeks of AB or 2 weeks of IP isoproterenol without significantly affecting the hypertrophy response. In addition, we found that MFAP4 deficiency had no significant effects on cardiac fibrosis, hypertrophy, or VA in the sham group. These observations advance our understanding of the mechanisms of pathological cardiac remodeling and HF.
Sources of Funding
This work was supported by grants from the Key Project of the National Natural Science Foundation (No. 81530012); National Key R&D Program of China (2018YFC1311300); National Natural Science Foundation of China (81770360), Development Center for Medical Science and Technology National Health and Family Planning Commission of the People's Republic of China (The prevention and control project of cardiovascular disease, 2016ZX‐008‐01); the Fundamental Research Funds for the Central Universities (2042018kf1032); Hubei Province Health and Family Planning Scientific Research Project (WJ2019Z004), and Hubei Provincial Natural Science Foundation of China (2018CFA044), and Hubei Province’s Outstanding Medical Academic Leader program.
Disclosures
None.
Supporting information
Figures S1–S4
Acknowledgments
Author contributions: Hui‐bo Wang and Qi‐Zhu Tang contributed to conception and designed experiments; Hui‐bo Wang, Wei Shuai performed out experiments; Li‐bo Liu, Jun Yang, and Jian Yang analyzed experimental results.
(J Am Heart Assoc. 2020;9:e015307 DOI: 10.1161/JAHA.119.015307.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.119.015307
For Sources of Funding and Disclosures, see page 18.
Contributor Information
Man Xu, Email: xuman987@whu.edu.cn.
Qi-zhu Tang, Email: qztang@whu.edu.cn.
References
- 1. Schuttler D, Clauss S, Weckbach LT, Brunner S. Molecular mechanisms of cardiac remodeling and regeneration in physical exercise. Cells. 2019;8:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569–582. [DOI] [PubMed] [Google Scholar]
- 3. Misra A, Mann DL. Treatment of heart failure beyond practice guidelines. Role of cardiac remodeling. Circ J. 2008;72(suppl A):A1–A7. [DOI] [PubMed] [Google Scholar]
- 4. Aimo A, Gaggin HK, Barison A, Emdin M, Januzzi JL Jr. Imaging, biomarker, and clinical predictors of cardiac remodeling in heart failure with reduced ejection fraction. JACC Heart Fail. 2019;7:782–794. [DOI] [PubMed] [Google Scholar]
- 5. Wang HB, Duan MX, Xu M, Huang SH, Yang J, Yang J, Liu LB, Huang R, Wan CX, Ma ZG, et al. Cordycepin ameliorates cardiac hypertrophy via activating the ampkalpha pathway. J Cell Mol Med. 2019;23:5715–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang HB, Huang SH, Xu M, Yang J, Yang J, Liu MX, Wan CX, Liao HH, Fan D, Tang QZ. Galangin ameliorates cardiac remodeling via the MEK1/2-ERK1/2 and PI3K-AKT pathways. J Cell Physiol. 2019;234:15654–15667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeong MH, Kim HJ, Pyun JH, Choi KS, Lee DI, Solhjoo S, O'Rourke B, Tomaselli GF, Jeong DS, Cho H, et al. Cdon deficiency causes cardiac remodeling through hyperactivation of WNT/beta-catenin signaling. Proc Natl Acad Sci USA. 2017;114:E1345–E1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F, Gibson AM, Nieman ML, Perkins C, Sargent MA, et al. Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation. 2018;138:1236–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frangogiannis NG. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. 2019;125:117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ Res. 2016;118:1906–1917. [DOI] [PubMed] [Google Scholar]
- 11. Pilecki B, Schlosser A, Wulf-Johansson H, Trian T, Moeller JB, Marcussen N, Aguilar-Pimentel JA, de Angelis MH, Vestbo J, Berger P, et al. Microfibrillar-associated protein 4 modulates airway smooth muscle cell phenotype in experimental asthma. Thorax. 2015;70:862–872. [DOI] [PubMed] [Google Scholar]
- 12. Wulf-Johansson H, Lock Johansson S, Schlosser A, Trommelholt Holm A, Rasmussen LM, Mickley H, Diederichsen AC, Munkholm H, Poulsen TS, Tornoe I, et al. Localization of microfibrillar-associated protein 4 (MFAP4) in human tissues: clinical evaluation of serum MFAP4 and its association with various cardiovascular conditions. PLoS One. 2013;8:e82243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saekmose SG, Mossner B, Christensen PB, Lindvig K, Schlosser A, Holst R, Barington T, Holmskov U, Sorensen GL. Microfibrillar-associated protein 4: a potential biomarker for screening for liver fibrosis in a mixed patient cohort. PLoS One. 2015;10:e0140418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi R, Tashima Y, Masuda H, Shozawa T, Numata Y, Miyauchi K, Hayakawa T. Isolation and characterization of a new 36-kDa microfibril-associated glycoprotein from porcine aorta. J Biol Chem. 1989;264:17437–17444. [PubMed] [Google Scholar]
- 15. Schlosser A, Pilecki B, Hemstra LE, Kejling K, Kristmannsdottir GB, Wulf-Johansson H, Moeller JB, Fuchtbauer EM, Nielsen O, Kirketerp-Moller K, et al. MFAP4 promotes vascular smooth muscle migration, proliferation and accelerates neointima formation. Arterioscler Thromb Vasc Biol. 2016;36:122–133. [DOI] [PubMed] [Google Scholar]
- 16. Molleken C, Sitek B, Henkel C, Poschmann G, Sipos B, Wiese S, Warscheid B, Broelsch C, Reiser M, Friedman SL, et al. Detection of novel biomarkers of liver cirrhosis by proteomic analysis. Hepatology. 2009;49:1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holm AT, Wulf-Johansson H, Hvidsten S, Jorgensen PT, Schlosser A, Pilecki B, Ormhoj M, Moeller JB, Johannsen C, Baun C, et al. Characterization of spontaneous air space enlargement in mice lacking microfibrillar-associated protein 4. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1114–L1124. [DOI] [PubMed] [Google Scholar]
- 18. Wang HB, Huang R, Yang K, Xu M, Fan D, Liu MX, Huang SH, Liu LB, Wu HM, Tang QZ. Identification of differentially expressed genes and preliminary validations in cardiac pathological remodeling induced by transverse aortic constriction. Int J Mol Med. 2019;44:1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ma ZG, Yuan YP, Zhang X, Xu SC, Kong CY, Song P, Li N, Tang QZ. C1q-tumour necrosis factor-related protein-3 exacerbates cardiac hypertrophy in mice. Cardiovasc Res. 2019;115:1067–1077. [DOI] [PubMed] [Google Scholar]
- 20. Ma ZG, Zhang X, Yuan YP, Jin YG, Li N, Kong CY, Song P, Tang QZ. A77 1726 (leflunomide) blocks and reverses cardiac hypertrophy and fibrosis in mice. Clin Sci (Lond). 2018;132:685–699. [DOI] [PubMed] [Google Scholar]
- 21. Schorb W, Booz GW, Dostal DE, Conrad KM, Chang KC, Baker KM. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ Res. 1993;72:1245–1254. [DOI] [PubMed] [Google Scholar]
- 22. Toth A, Jeffers JR, Nickson P, Min JY, Morgan JP, Zambetti GP, Erhardt P. Targeted deletion of puma attenuates cardiomyocyte death and improves cardiac function during ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;291:H52–60. [DOI] [PubMed] [Google Scholar]
- 23. Shuai W, Kong B, Fu H, Shen C, Jiang X, Huang H. MD1 deficiency promotes inflammatory atrial remodelling induced by high-fat diets. Can J Cardiol. 2019;35:208–216. [DOI] [PubMed] [Google Scholar]
- 24. Leo-Macias A, Agullo-Pascual E, Delmar M. The cardiac connexome: non-canonical functions of connexin43 and their role in cardiac arrhythmias. Semin Cell Dev Biol. 2016;50:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, Makita N, Kowase S, Boonmee N, Vitayakritsirikul V, et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol. 2015;66:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nieberler M, Reuning U, Reichart F, Notni J, Wester HJ, Schwaiger M, Weinmuller M, Rader A, Steiger K, Kessler H. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel). 2017;9:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun Z, Costell M, Fassler R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol. 2019;21:25–31. [DOI] [PubMed] [Google Scholar]
- 28. Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. [DOI] [PubMed] [Google Scholar]
- 30. Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. [DOI] [PubMed] [Google Scholar]
- 31. Cai J, Yi FF, Bian ZY, Shen DF, Yang L, Yan L, Tang QZ, Yang XC, Li H. Crocetin protects against cardiac hypertrophy by blocking MEK-ERK1/2 signalling pathway. J Cell Mol Med. 2009;13:909–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gruhle S, Sauter M, Szalay G, Ettischer N, Kandolf R, Klingel K. The prostacyclin agonist iloprost aggravates fibrosis and enhances viral replication in enteroviral myocarditis by modulation of ERK signaling and increase of iNOS expression. Basic Res Cardiol. 2012;107:287. [DOI] [PubMed] [Google Scholar]
- 33. Liu R, van Berlo JH, York AJ, Vagnozzi RJ, Maillet M, Molkentin JD. DUSP8 regulates cardiac ventricular remodeling by altering ERK1/2 signaling. Circ Res. 2016;119:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine attenuates pathological cardiac fibrosis via PPAR-gamma/AKT pathways. EBioMedicine. 2017;18:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation. 2015;131:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen MN, Kiriazis H, Gao XM, Du XJ. Cardiac fibrosis and arrhythmogenesis. Compr Physiol. 2017;7:1009–1049. [DOI] [PubMed] [Google Scholar]
- 37. Albert CM, Chae CU, Grodstein F, Rose LM, Rexrode KM, Ruskin JN, Stampfer MJ, Manson JE. Prospective study of sudden cardiac death among women in the United States. Circulation. 2003;107:2096–2101. [DOI] [PubMed] [Google Scholar]
- 38. Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. 2014;70:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shenasa M. Fibrosis and ventricular arrhythmogenesis: role of cardiac MRI. Card Electrophysiol Clin. 2019;11:551–562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4