

Abstract
Background
APOL1 high‐risk genotypes are associated with increased risk for hypertension‐attributed kidney disease among Black adults in the United States. Biopsy studies show differences in kidney vasculature by APOL1 status; less is known about the variants' associations with systemic vascular and endothelial function. Whether APOL1 risk variants are associated with blood pressure (BP) is also uncertain.
Methods and Results
Using linear regression, we examined cross‐sectional associations of APOL1 risk genotypes (high=2 risk alleles, low=0 or 1 risk allele) with subclinical measures of vascular function (small arterial elasticity, n=1586; large arterial elasticity, n=1586; ascending aortic distensibility, n=985) and endothelial function (flow‐mediated dilation, n=777). Using linear mixed‐effects models, we studied longitudinal associations of APOL1 risk genotypes with BP (n=1619), adjusting for age, sex, and African ancestry. Among 1619 (12% APOL1 high‐risk) Black participants in MESA (Multi‐Ethnic Study of Atherosclerosis), mean age was 62 years old, 58% had hypertension, and mean systolic BP was 131 mm Hg at baseline. At examination 1 (2000–2002), there was no significant difference in small arterial elasticity, large arterial elasticity, ascending aortic distensibility, or flow‐mediated dilation in participants with APOL1 high‐ versus low‐risk genotypes (P>0.05 for all). Over a mean follow‐up of 7.8 years, relative annual changes in systolic and diastolic BP and pulse pressure did not differ significantly by APOL1 risk status (between‐group differences of −0.20, −0.14, and −0.25, respectively; P>0.05 for all).
Conclusions
Among Black participants in MESA, APOL1 high‐risk genotypes were not associated with subclinical vascular and endothelial function or BP trajectories. The relationship of APOL1 with kidney disease may be intrinsic to the kidney rather than through peripheral effects on systemic vasculature or BP.
Keywords: APOL1, apolipoprotein L1, arterial stiffness, blood pressure, hypertension
Subject Categories: Genetics, Hypertension, Race and Ethnicity, Epidemiology, Blood Pressure
Nonstandard Abbreviations and Acronyms
- AAD
ascending aortic distensibility
- APOL1
apolipoprotein L1
- BP
blood pressure
- C1
large arterial elasticity
- C2
small arterial elasticity
- FMD
flow‐mediated dilation
- MESA
Multi‐Ethnic Study of Atherosclerosis
Clinical Perspective
What Is New?
APOL1 high‐risk genotypes are not associated with subclinical measures of vascular and endothelial function or blood pressure trajectories in Black adults without baseline clinical cardiovascular disease.
What Are the Clinical Implications?
The results of the current study argue against a role of APOL1 risk variants in the development of hypertension.
Black adults are more likely to have hypertension and difficult‐to‐control blood pressure (BP) compared with White adults. 1 , 2 , 3 , 4 Although environmental factors account for some of this excess risk, genetic factors likely also contribute. 5 , 6 The APOL1 high‐risk genotypes, present in 12% to 14% of Black Americans, have been associated with an increased risk for various types of kidney disease, including hypertension‐attributed chronic kidney disease. 7 , 8 , 9 , 10 , 11 Expression of APOL1 (Apolipoprotein L1) protein and mRNA has been shown in vascular endothelial and smooth muscle cells of both healthy and diseased (eg, focal segmental glomerulosclerosis and HIV‐associated nephropathy) human kidneys. 12 , 13 Some histopathologic studies have also reported an association between the APOL1 high‐risk genotypes and arteriosclerosis, whereas others have not. 14 , 15 , 16 , 17 These observations have led to an interest in understanding whether the APOL1 risk variants confer an increased risk of hypertension and, if so, whether the association is mediated by kidney damage or mediates the association of APOL1 with kidney disease.
We previously demonstrated in CARDIA (Coronary Artery Risk Development in Young Adults), a cohort of healthy young adults, that the APOL1 risk variants were not associated with BP trajectories over the life course. 18 In contrast, others have reported a positive association using data from electronic health records. 19 Differences in study design and population may explain the discrepant findings; however, the question remains as to whether an association exists between APOL1 and BP. Moreover, the mechanisms by which the APOL1 risk variants relate to progressive chronic kidney disease and possibly also hypertension warrant further investigation. APOL1 risk variants may be associated with microvascular damage. To our knowledge, no study to date has examined the association of the APOL1 risk variants with subclinical measures of vascular function (eg, small arterial elasticity [C2], large arterial elasticity [C1], ascending aortic distensibility [AAD]) and endothelial function (eg, flow‐mediated dilation [FMD]). This question is important because we previously demonstrated that decreased C2 and C1 were associated with an increased risk of incident hypertension and faster decline of estimated glomerular filtration rate. 20 , 21
In a cohort of adults without baseline clinical cardiovascular disease, we aimed to determine whether the APOL1 risk variants were associated with subclinical measures of vascular and endothelial function and BP trajectories. We hypothesized that individuals with the APOL1 high‐risk genotypes would have lower C2, C1, AAD, and FMD at baseline and greater increases in BP over time compared with individuals with low‐risk genotypes.
METHODS
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Study Population
MESA (Multi‐Ethnic Study of Atherosclerosis) is a prospective cohort study that began in July 2000 with the overall goal of better understanding factors related to the progression of subclinical to clinical cardiovascular disease. Details on the study design of MESA have been reported previously. 22 Briefly, 6814 men and women between the ages of 45 and 84 years old without baseline clinical cardiovascular disease were enrolled. Four racial/ethnic groups (White, Black, Hispanic, and Asian) were represented, and study participants were recruited from 6 communities in the United States (Baltimore, Maryland; Chicago, Illinois; Forsyth County, North Carolina; Los Angeles County, California; New York, New York; and St. Paul, Minnesota). 22 Institutional review board approval was obtained at each study site, and all study participants provided informed consent at the initial examination. 22 , 23 We utilized data from the first 5 examinations: exam 1 (July 2000 to August 2002), exam 2 (September 2002 to February 2004), exam 3 (March 2004 to September 2005), exam 4 (September 2005 to May 2007), and exam 5 (April 2010 to December 2011). 24 Our study population was derived from the 1746 participants who self‐identified as Black and who underwent APOL1 genotyping, of whom 1586 had C2, 1586 had C1, 985 had AAD, and 777 had FMD measured at exam 1 and 1619 had at least 1 systolic BP measurement during follow‐up (at exam 2, 3, 4, or 5; Figure S1).
Outcomes
Measurements of vascular and endothelial function were obtained at exam 1 and have been described previously. 20 , 21 , 25 , 26 , 27 , 28 , 29 , 30 , 31 Briefly, C2 and C1 were measured using the PulseWave CR‐2000 Research CardioVascular Profiling Instrument (Hypertension Diagnostics). A tonometer was placed over the right radial artery, which was supported by a wrist stabilizer. Measurements of the radial artery waveforms were collected for 30 seconds with accompanying systolic and diastolic BP measurements using an automated, oscillometric BP cuff in the left arm. Elasticities of the small and large arteries, or change in arterial volume per change in arterial pressure, were estimated by analysis of the diastolic pulse contour and a computer‐based, third‐order, 4‐element Windkessel modified model. 20 , 21 , 27 In a random subset of MESA participants, 2 measurements taken on the same day were shown to have between‐measure correlations of 0.84 for C2 and 0.74 for C1. 29 AAD was measured using magnetic resonance imaging (1.5‐T whole‐body magnetic resonance imaging systems Signa CV/I or Signa LX; General Electric Medical Systems). Images of the aorta were captured using gradient echo phase‐contrast cine magnetic resonance imaging with electrocardiographic gating, and cross‐sectional areas of the ascending aorta were determined using FLOW software (Medis Medical Imaging Systems). Aortic distensibility was then calculated as follows: (maximum cross‐sectional area−minimum aortic cross‐sectional area)/(minimum cross‐sectional area×pulse pressure). 20 , 27 , 28 , 31
FMD was measured using high‐resolution ultrasonography of the brachial artery. After at least 6 hours of fasting and smoking, a standard BP cuff was placed on the right arm of each participant at 2 inches below the antecubital fossa. A 9‐MHz linear array transducer (M12L transducer; General Electric Healthcare) was then used to image the brachial artery at 5 to 9 cm above the antecubital fossa. After images were recorded at rest, reactive hyperemia was induced by occluding the brachial artery for 5 minutes at a cuff pressure ≥50 mm Hg above the participant's systolic BP. Digitalized images were collected for 30 seconds before inflation of the cuff and for 2 minutes beginning immediately before deflation of the cuff. FMD was defined as the percentage of increase in the brachial artery with reactive hyperemia and calculated as follows: [(peak brachial artery diameter after cuff deflation−diameter at rest)/diameter at rest]×100%. Intraclass correlation coefficients were 0.90 for baseline diameter, 0.90 for maximum diameter, and 0.54 for percentage of FMD. 21 , 26 , 30
Systolic and diastolic BPs were measured using a Dinamap BP device (Dinamap Monitor Pro 100; Critikon). At each visit, 3 resting BPs were obtained with the participant in a seated position following a 5‐minute rest period. The average of the second and third BP readings was used. 22 , 32 , 33 , 34 Pulse pressure was calculated as seated systolic BP minus seated diastolic BP. 21
Exposure
The primary exposure was APOL1 risk status, which we defined using a recessive genetic model consistent with prior studies on APOL1 and kidney disease. 7 , 8 , 10 , 35 , 36 DNA was extracted from buffy coat samples collected at exam 1 and genotyped for the APOL1 risk variants (rs73885319 and rs71785313) via TaqMan assays (Applied Biosystems 7900). 22 , 37 The G1 risk allele (rs73885319 and rs60910145) consists of 2 missense mutations, whereas the G2 risk allele (rs71785313) consists of a 6‐bp deletion. 7 , 8 The APOL1 high‐risk genotypes were specified by the presence of 2 risk alleles (G1/G1, G1/G2, or G2/G2), whereas the low‐risk genotypes were specified by the presence of 0 or 1 risk allele (G0/G0, G1/G0, or G2/G0). As previously described, 406 ancestry‐informative markers from the Affymetrix 6.0 array and 4 ancestry populations in the ADMIXMAP software were used to estimate proportion of global African ancestry. 37
Additional Variables
Data on sociodemographic factors, personal and family medical histories, and medication use were obtained through questionnaires. 22 The MESA Typical Week Physical Activity Survey, adapted from the Cross‐Cultural Activity Participation Survey, was used to quantify baseline physical activity. 22 , 33 Body mass index was calculated by dividing weight in kilograms by height in meters squared. Hypertension was defined by a systolic BP ≥140 mm Hg, a diastolic BP ≥90 mm Hg, 38 or use of BP‐lowering medications, whereas diabetes mellitus was defined by a fasting glucose ≥126 mg/dL (measured using the Vitros analyzer; Johnson & Johnson Clinical Diagnostics) or use of glucose‐lowering medications. 34 , 37 , 39 Fasting lipids were determined using the cholesterol oxidase method (for total cholesterol and high‐density lipoprotein; Roche Diagnostics), the triglyceride GB reagent (for triglycerides; Roche Diagnostics), and the Friedewald equation (for low‐density lipoprotein). 32 , 34 , 40 Glomerular filtration rate was estimated based on cystatin C using the Chronic Kidney Disease‐Epidemiology Collaboration equation. 41 A particle‐enhanced immunonephelometric assay (N Latex Cystatin C; Siemens) on a nephelometer (BNII; Siemens) and corrected for assay drift was used to measure cystatin C from serum specimens stored at −70°C. 42 , 43 Urine albumin was measured by nephelometry (Array 360 CE Protein Analyzer; Beckman Instruments), and creatinine was measured by the Jaffe method (Vitros 950IRC instrument; Johnson & Johnson Clinical Diagnostics) from a single morning urine sample. 43 , 44
Statistical Analysis
We compared baseline characteristics, overall and by APOL1 risk status, using count (percentage), mean (SD), and median (interquartile range). To assess the cross‐sectional associations of APOL1 risk status with C2, C1, AAD, and FMD at exam 1, we constructed linear regression models adjusting for age, sex, and African ancestry. Utilizing linear mixed‐effects models, we then examined the longitudinal associations of APOL1 risk status with systolic BP, diastolic BP, and pulse pressure, also adjusting for age, sex, and African ancestry. To model the dependence between repeated‐outcome measures, we used the autoregressive order 1 covariance structure of the residuals, which assumed that BP values measured at consecutive visits were correlated more strongly than those separated by longer time intervals. In sensitivity analyses, we further adjusted for antihypertensive medication use as a time‐updated binary variable. Statistical analyses were performed using SPSS v24 (IBM Corp) and Stata release 13 (StataCorp). We considered P<0.05 to be statistically significant.
RESULTS
Baseline Characteristics
Based on a recessive genetic model, 12% of the study population had an APOL1 high‐risk genotype (2 risk alleles), whereas 88% had an APOL1 low‐risk genotype (0 or 1 risk allele). At baseline, mean age was 62 years old, 54% were female, 58% had hypertension, and mean systolic BP was 131±21 mm Hg. Mean estimated glomerular filtration rate (based on cystatin C) was 90±20 mL/min per 1.73 m2, and 11% had albuminuria, which we defined by a urine albumin‐to‐creatinine ratio ≥30 mg/g. Although hypertension and use of anti‐hypertensive medications were more common in the APOL1 high‐risk group, BP control appeared fairly similar between the 2 APOL1 risk groups (Table 1).
Table 1.
Baseline Characteristics of Study Population at MESA Exam 1 by APOL1 Risk Status
| Characteristic | All (n=1619) | APOL1 high‐risk (n=190) | APOL1 low‐risk (n=1429) |
|---|---|---|---|
| Age, y | 62±10 | 62±9 | 62±10 |
| Female | 876 (54) | 96 (51) | 780 (55) |
| Education | |||
| Less than high school | 180 (11) | 25 (13) | 155 (11) |
| High school graduate | 312 (19) | 32 (17) | 280 (20) |
| Postsecondary | 1116 (69) | 132 (70) | 984 (69) |
| Employment | |||
| Employed | 658 (41) | 76 (40) | 582 (41) |
| Unemployed/employed part‐time | 125 (8) | 18 (10) | 107 (8) |
| Retired/homemaker | 824 (51) | 95 (50) | 729 (51) |
| Annual family income | |||
| <$25 000 | 437 (29) | 49 (28) | 388 (29) |
| $25 000–$49 999 | 486 (33) | 57 (33) | 429 (32) |
| $50 000–$74 999 | 310 (21) | 35 (20) | 275 (21) |
| ≥$75 000 | 263 (18) | 32 (19) | 231 (18) |
| Smoking status | |||
| Never | 738 (46) | 84 (44) | 654 (46) |
| Former | 579 (36) | 67 (35) | 512 (36) |
| Current | 291 (18) | 38 (20) | 253 (18) |
| Diabetes mellitus | 274 (17) | 40 (21) | 234 (16) |
| Fasting glucose, mg/dL | 100±32 | 102±32 | 100±32 |
| Hypertension | 946 (58) | 119 (63) | 827 (58) |
| Systolic BP, mm Hg | 131±21 | 132±21 | 131±21 |
| Diastolic BP, mm Hg | 75±10 | 74±10 | 75±10 |
| Pulse pressure, mm Hg | 57±17 | 58±18 | 57±17 |
| Antihypertensive medication use | 802 (50) | 104 (55) | 698 (49) |
| Total cholesterol, mg/dL | 190±36 | 190±37 | 190±36 |
| Triglycerides, mg/dL | 89 [66–122] | 88 [65–122] | 89 [66–122] |
| HDL, mg/dL | 52±15 | 54±16 | 52±15 |
| LDL, mg/dL | 117±33 | 116±35 | 117±33 |
| Lipid‐lowering medication use | 253 (16) | 33 (17) | 220 (15) |
| Body mass index, kg/m2 | 30.1±5.8 | 30.1±6.0 | 30.1±5.7 |
| Moderate‐vigorous PA, MET‐min/wk | 4613 [2160–8588] | 4455 [2135–8370] | 4636 [2160–8678] |
| Family history of heart disease | 634 (42) | 63 (35) | 571 (43) |
| eGFRCysC, mL/min/1.73 m2 | 90±20 | 90±19 | 90±20 |
| UACR, mg/g | 5.3 [3.1–12.0] | 5.5 [3.3–14.5] | 5.2 [3.0–11.9] |
| UACR ≥30 | 170 (11) | 27 (14) | 143 (10) |
Values presented as mean±SD, median [interquartile range], or number (percentage). BP, blood pressure; eGFRCysC, estimated glomerular filtration rate based on cystatin C; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MESA, Multi‐Ethnic Study of Atherosclerosis; MET, metabolic equivalent; PA, physical activity; and UACR, urine albumin‐to‐creatinine ratio.
Subclinical Measures of Vascular Function
Mean baseline measures of C2 and C1 did not differ significantly between the 2 APOL1 risk groups (C2, 4.01 versus 4.20 mL/mm Hg×100; C1, 13.41 versus 13.49 mL/mm Hg×10 comparing APOL1 high‐ versus low‐risk, respectively). Mean baseline AAD was also comparable among APOL1 high‐ versus low‐risk individuals (1.55 versus 1.69 mm Hg−1×103, respectively). In unadjusted models and models adjusted for age, sex, and African ancestry, there were no significant differences in these subclinical measures of vascular function by APOL1 risk status (Table 2).
Table 2.
Association of APOL1 Risk Status With Subclinical Measures of Vascular and Endothelial Function at MESA Exam 1
| C2 | ||||
|---|---|---|---|---|
| n |
Mean (SD) mL/mm Hg×100 |
Model 1 β (95% CI) |
Model 2 β (95% CI) |
|
| APOL1 low‐risk | 1393 | 4.20 (2.55) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high‐risk | 193 | 4.01 (2.30) | −0.15 (−0.55, 0.25) | −0.11 (−0.47, 0.24) |
| P value | 0.33 | 0.47 | 0.54 | |
| C1 | ||||
|---|---|---|---|---|
| n |
Mean (SD) mL/mm Hg×10 |
Model 1 β (95% CI) |
Model 2 β (95% CI) |
|
| APOL1 low‐risk | 1393 | 13.49 (5.88) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high‐risk | 193 | 13.41 (2.10) | −0.06 (−0.98, 0.86) | −0.05 (−0.88, 0.78) |
| P value | 0.85 | 0.90 | 0.91 | |
| AAD | ||||
|---|---|---|---|---|
| n |
Mean (SD) mm Hg−1×103 |
Model 1 β (95% CI) |
Model 2 β (95% CI) |
|
| APOL1 low‐risk | 863 | 1.69 (1.31) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high‐risk | 122 | 1.55 (0.75) | −0.22 (−0.48, 0.04) | −0.21 (−0.46, 0.03) |
| P value | 0.09 | 0.10 | 0.09 | |
| FMD | ||||
|---|---|---|---|---|
| N |
Mean (SD) mm |
Model 1 β (95% CI) |
Model 2 β (95% CI) |
|
| APOL1 low‐risk | 692 | 0.15 (0.10) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high‐risk | 85 | 0.16 (0.09) | 0.01 (−0.02, 0.03) | 0.01 (−0.02, 0.03) |
| P‐value | 0.58 | 0.50 | 0.56 | |
Model 1 unadjusted. Model 2 adjusted for age, sex, and African ancestry. AAD indicates ascending aortic distensibility; C1, large arterial elasticity; C2, small arterial elasticity; FMD, flow‐mediated dilation; and MESA, Multi‐Ethnic Study of Atherosclerosis.
Subclinical Measure of Endothelial Function
Mean baseline FMD was similar for the 2 APOL1 risk groups (0.16 versus 0.15 mm comparing APOL1 high versus low risk, respectively). In the unadjusted model and the model adjusted for age, sex, and African ancestry, FMD did not differ significantly by APOL1 risk status (Table 2).
Blood Pressure
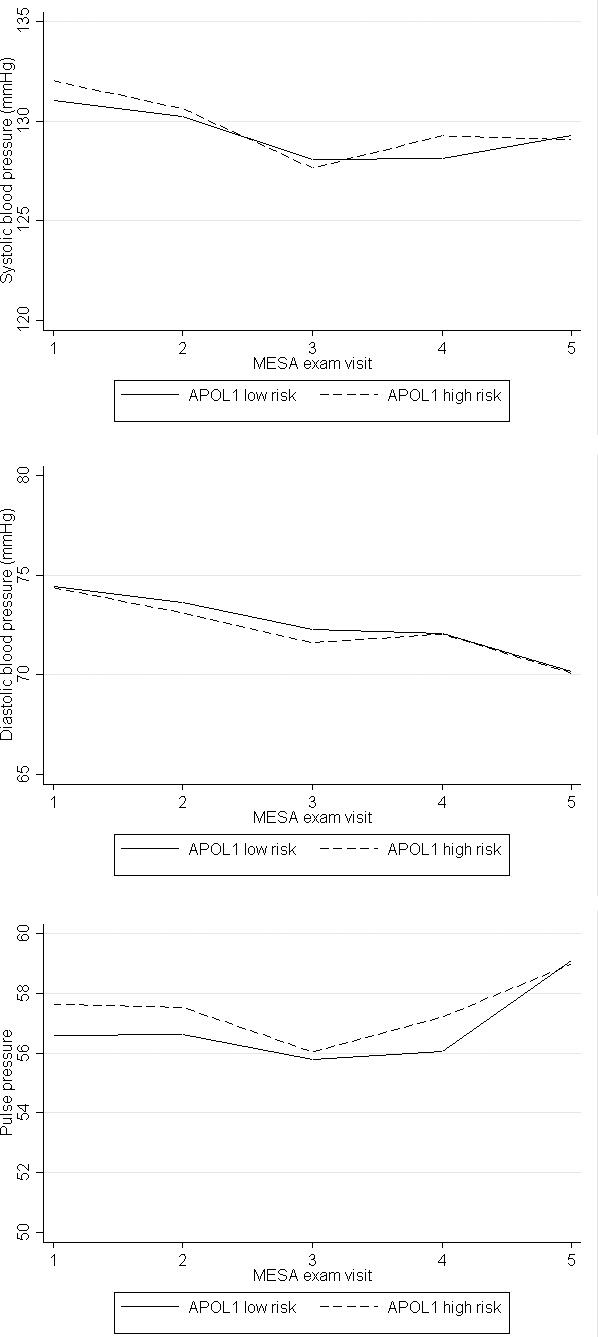
The mean number of BP visits was 4.5±0.85 for each APOL1 risk group over a mean follow‐up of 7.8 years. In longitudinal analyses, relative annual change in systolic BP did not differ significantly between APOL1 high‐ versus low‐risk individuals (between‐group difference, −0.20; 95% CI, −0.48 to 0.09; P=0.17). Similar conclusions were obtained when considering diastolic BP or pulse pressure as the outcome of interest (Table 3; Figure).
Table 3.
Association of APOL1 Risk Status With Longitudinal BP Change From MESA Exams 1 to 5
| n |
Relative Annual Change % Change/Year (95% CI) |
Model 1 Between‐Group Difference |
Model 2 Between‐Group Difference |
|
|---|---|---|---|---|
| Systolic BP | ||||
| APOL1 low risk | 1429 | −0.10 (−0.20, −0.01) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high risk | 190 | −0.21 (−0.46, 0.04) | −0.11 (−0.38, 0.16) | −0.20 (−0.48, 0.09) |
| P value | 0.42 | 0.17 | ||
| Diastolic BP | ||||
| APOL1 low risk | 1429 | −0.63 (−0.71, −0.55) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high risk | 190 | −0.69 (−0.90, −0.49) | −0.06 (−0.28, 0.16) | −0.14 (−0.37, 0.09) |
| P value | 0.57 | 0.24 | ||
| Pulse pressure | ||||
| APOL1 low risk | 1429 | 0.56 (0.41, 0.71) | 0.00 (ref) | 0.00 (ref) |
| APOL1 high risk | 190 | 0.41 (0.01, 0.82) | −0.15 (−0.58, 0.28) | −0.25 (−0.71, 0.21) |
| P value | 0.50 | 0.28 | ||
Model 1 unadjusted. Model 2 adjusted for age, sex, and African ancestry. BP, blood pressure; MESA, Multi‐Ethnic Study of Atherosclerosis; and ref, referent.
Figure 1. Longitudinal changes in systolic blood pressure, diastolic blood pressure, and pulse pressure by APOL1 risk status from MESA (Multi‐Ethnic Study of Atherosclerosis) exams 1 to 5.
From exam 1 to exam 5, the number of individuals receiving pharmacologic therapy for hypertension increased from 50% to 68%, with the number of individuals on ≥3 antihypertensive medications increasing from 7% to 15% (Table S1). Given this intensification in treatment over time, we further adjusted for time‐updated antihypertensive medication use and still found a lack of association between APOL1 risk status and longitudinal systolic BP, diastolic BP, or pulse pressure (between‐group differences in relative annual change for APOL1 high versus low risk of −0.18 [95% CI, −0.46 to 0.10; P=0.22], −0.13 [95% CI, −0.36 to 0.10; P=0.28], and −0.32 [95% CI, −0.69 to 0.23; P=0.32], respectively).
DISCUSSION
In this study, among Black individuals without baseline clinical cardiovascular disease, we found that APOL1 risk status was not associated with subclinical measures of vascular or endothelial function. In addition, there was no association between APOL1 risk status and change in BP levels over time. Taken together, our results provide further evidence against a role of the APOL1 risk variants in the development of hypertension.
To our knowledge, the associations between APOL1 and subclinical measures of vascular and/or endothelial function have not been examined previously. In MESA, we previously reported that lower C2, C1, and AAD were each associated with an increased risk of incident hypertension, 20 whereas Shimbo et al reported that lower FMD levels were associated with prevalent but not incident hypertension. 26 Given these findings, we hypothesized that if the APOL1 risk variants were to confer an increased risk of hypertension, this might begin with subclinical changes in vascular and/or endothelial function. In the present study, however, we did not observe a significant association between APOL1 high‐risk status and baseline levels of C2, C1, AAD, or FMD. These findings warrant replication in other cohorts of individuals with clinical cardiovascular disease or hypertension. Still, 58% of our study population already had hypertension at baseline, with 50% on antihypertensive medications.
We also reported that APOL1 high‐risk status was not associated with longitudinal change in systolic BP, diastolic BP, or pulse pressure. These findings are in line with our prior findings in CARDIA in which we also observed a lack of association between the APOL1 risk variants and longitudinal BP change and trajectory among healthy young adults. 18 Also consistent with our findings, previous studies of admixture mapping for hypertension did not identify the chromosomal region of APOL1. 6 , 45 In contrast, utilizing data from 3 electronic medical record–linked biobanks (as part of the Electronic Medical Records and Genomics [eMERGE] Network), others have reported that the APOL1 risk variants were associated with higher systolic BP within age strata of 20 to 23, 24 to 27, and 28 to 31 years old but not in older age strata. 19 Potential explanations for these incongruent findings include differences in study population, analytical approach, and sample size. In MESA and CARDIA, we utilized data that were collected prospectively and that examined change in BP over time but had smaller sample sizes (n=1619 for MESA and n=1330 for CARDIA). 18 In eMERGE, data were extracted from electronic medical records, and differences in BP within age strata were compared, but sample sizes were much larger (n=9203). 19 Although null, the results of the current study contribute to our understanding of the potential role of APOL1 risk variants in extrarenal disease. In clinical practice, APOL1 screening is already being offered to some patients. Intervention for those with high‐risk genotypes may include aggressive BP monitoring and control to help prevent development or progression of chronic kidney disease. Our results provide further support for the notion that having an APOL1 high‐risk genotype without other risk factors for kidney disease may not necessarily warrant more intensive BP management beyond standard care. 36
Interestingly, systolic BP appeared to decrease over time, particularly between MESA exams 1 and 3. On further investigation, we found that this finding was likely due to an intensification of antihypertensive treatment. In additional analyses adjusting for antihypertensive medication use, the lack of association between APOL1 high‐risk status and longitudinal systolic BP persisted, suggesting that our null findings were not due to differences in antihypertensive medication use between the 2 risk groups.
Our study has several notable strengths. First, our outcomes of interest included not only BP but also potential subclinical precursors of hypertension. Second, we leveraged prospectively collected data from a well‐described cohort of individuals without baseline clinical cardiovascular disease. In particular, C2, C1, AAD, FMD, and BP were each measured in a standardized manner. Third, follow‐up was relatively long at nearly 8 years, with 88% of study participants having ≥4 BP measurements over the course of 5 visits. Limitations include the cross‐sectional analyses of C2, C1, AAD, and FMD at baseline; limited generalizability to other populations (eg, those with clinical cardiovascular disease, more hypertension, or moderate to severe chronic kidney disease); and potential for residual confounding. Last, the CIs around our effect suggest that there may still be an association that we were unable to detect.
In conclusion, we found that among Black individuals without baseline clinical cardiovascular disease, APOL1 high‐risk status was not associated with subclinical measures of vascular and endothelial function at baseline or changes in BP during follow‐up.
Sources of Funding
Chen was supported by the Extramural Grant Program by Satellite Healthcare, a not‐for‐profit renal care provider and Clinician Scientist Career Development Award from Johns Hopkins University and is currently supported by a George M. O'Brien Center for Kidney Research Pilot and Feasibility Grant from Yale University (under Award Number NIH/NIDDK P30DK079310) and K08DK117068 from NIH/NIDDK. MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) and the National Center for Advancing Translational Sciences (NCATS) in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01‐HC‐95159, N01‐HC‐95160, N01‐HC‐95161, N01‐HC‐95162, N01‐HC‐95163, N01‐HC‐95164, N01‐HC‐95165, N01‐HC‐95166, N01‐HC‐95167, N01‐HC‐95168, N01‐HC‐95169 from NHLBI, UL1‐TR‐000040, UL1‐TR‐001079, UL1‐TR‐001420, UL1‐TR‐001881 from NCATS, and DK063491 from NIH/NIDDK. Funding for SHARe genotyping was provided by NHLBI contract N02‐HL‐64278. Genotyping was performed at Affymetrix (Santa Clara, California) and the Broad Institute of Harvard and MIT (Boston, Massachusetts) using the Affymetrix Genome‐Wide Human SNP Array 6.0.
Disclosures
Peralta is chief medical officer (CMO) for Cricket Health. The remaining authors have no disclosures to report.
Supporting information
Table S1
Figure S1
Acknowledgments
The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa‐nhlbi.org.
(J Am Heart Assoc. 2020;9:e017039 DOI: 10.1161/JAHA.120.017039.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017039
For Sources of Funding and Disclosures, see page 8.
Portions of this work were presented at the American Society of Nephrology Kidney Week, October 23 to 28, 2018, in San Diego, CA
REFERENCES
- 1. Kramer H, Han C, Post W, Goff D, Diez‐Roux A, Cooper R, Jinagouda S, Shea S. Racial/ethnic differences in hypertension and hypertension treatment and control in the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Hypertens. 2004;17:963–970. [DOI] [PubMed] [Google Scholar]
- 2. Allen NB, Siddique J, Wilkins JT, Shay C, Lewis CE, Goff DC, Jacobs DR Jr, Liu K, Lloyd‐Jones D. Blood pressure trajectories in early adulthood and subclinical atherosclerosis in middle age. JAMA. 2014;311:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu K, Ruth KJ, Flack JM, Jones‐Webb R, Burke G, Savage PJ, Hulley SB. Blood pressure in young blacks and whites: relevance of obesity and lifestyle factors in determining differences. The CARDIA Study. Coronary Artery Risk Development in Young Adults. Circulation. 1996;93:60–66. [DOI] [PubMed] [Google Scholar]
- 4. Grams ME, Chow EK, Segev DL, Coresh J. Lifetime incidence of CKD stages 3–5 in the United States. Am J Kidney Dis. 2013;62:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu X, Cooper RS. Admixture mapping provides evidence of association of the VNN1 gene with hypertension. PLoS One. 2007;2:e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu X, Luke A, Cooper RS, Quertermous T, Hanis C, Mosley T, Gu CC, Tang H, Rao DC, Risch N, et al. Admixture mapping for hypertension loci with genome‐scan markers. Nat Genet. 2005;37:177–181. [DOI] [PubMed] [Google Scholar]
- 7. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. 2010;128:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lipkowitz MS, Freedman BI, Langefeld CD, Comeau ME, Bowden DW, Kao WH, Astor BC, Bottinger EP, Iyengar SK, Klotman PE, et al. Apolipoprotein L1 gene variants associate with hypertension‐attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83:114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV‐associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus‐associated collapsing glomerulopathy. J Am Soc Nephrol. 2013;24:722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, et al. Localization of APOL1 protein and mRNA in the human kidney: nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol. 2015;26:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. 2011;22:2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larsen CP, Beggs ML, Saeed M, Ambruzs JM, Cossey LN, Messias NC, Walker PD, Freedman BI. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod Pathol. 2015;28:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kopp JB, Winkler CA, Zhao X, Radeva MK, Gassman JJ, D'Agati VD, Nast CC, Wei C, Reiser J, Guay‐Woodford LM, et al. Clinical features and histology of apolipoprotein L1‐associated nephropathy in the FSGS clinical trial. J Am Soc Nephrol. 2015;26:1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larsen CP, Beggs ML, Walker PD, Saeed M, Ambruzs JM, Messias NC. Histopathologic effect of APOL1 risk alleles in PLA2R‐associated membranous glomerulopathy. Am J Kidney Dis. 2014;64:161–163. [DOI] [PubMed] [Google Scholar]
- 17. Hughson MD, Hoy WE, Mott SA, Puelles VG, Bertram JF, Winkler CL, Kopp JB. APOL1 risk alleles are associated with more severe arteriosclerosis in renal resistance vessels with aging and hypertension. Kidney Int Rep. 2016;1:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen TK, Estrella MM, Vittinghoff E, Lin F, Gutierrez OM, Kramer H, Lewis CE, Kopp JB, Allen NB, Winkler CA, et al. APOL1 genetic variants are not associated with longitudinal blood pressure in young black adults. Kidney Int. 2017;92:964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nadkarni GN, Galarneau G, Ellis SB, Nadukuru R, Zhang J, Scott SA, Schurmann C, Li R, Rasmussen‐Torvik LJ, Kho AN, et al. Apolipoprotein L1 variants and blood pressure traits in African Americans. J Am Coll Cardiol. 2017;69:1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peralta CA, Adeney KL, Shlipak MG, Jacobs D Jr, Duprez D, Bluemke D, Polak J, Psaty B, Kestenbaum BR. Structural and functional vascular alterations and incident hypertension in normotensive adults: the Multi‐Ethnic Study of Atherosclerosis. Am J Epidemiol. 2010;171:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peralta CA, Jacobs DR Jr, Katz R, Ix JH, Madero M, Duprez DA, Sarnak MJ, Criqui MH, Kramer HJ, Palmas W, et al. Association of pulse pressure, arterial elasticity, and endothelial function with kidney function decline among adults with estimated GFR >60 mL/min/1.73 m (2): the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2012;59:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr, Kronmal R, Liu K, et al. Multi‐Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 23. Blaha MJ, Cainzos‐Achirica M, Greenland P, McEvoy JW, Blankstein R, Budoff MJ, Dardari Z, Sibley CT, Burke GL, Kronmal RA, et al. Role of coronary artery calcium score of zero and other negative risk markers for cardiovascular disease: the Multi‐Ethnic Study of Atherosclerosis (MESA). Circulation. 2016;133:849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olson JL, Bild DE, Kronmal RA, Burke GL. Legacy of MESA. Glob Heart. 2016;11:269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wassel CL, Jacobs DR Jr, Duprez DA, Bluemke DA, Sibley CT, Criqui MH, Peralta CA. Association of self‐reported race/ethnicity and genetic ancestry with arterial elasticity: the Multi‐Ethnic Study of Atherosclerosis (MESA). J Am Soc Hypertens. 2011;5:463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimbo D, Muntner P, Mann D, Viera AJ, Homma S, Polak JF, Barr RG, Herrington D, Shea S. Endothelial dysfunction and the risk of hypertension: the Multi‐Ethnic Study of Atherosclerosis. Hypertension. 2010;55:1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shimbo D, Shea S, McClelland RL, Viera AJ, Mann D, Newman J, Lima J, Polak JF, Psaty BM, Muntner P. Associations of aortic distensibility and arterial elasticity with long‐term visit‐to‐visit blood pressure variability: the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Hypertens. 2013;26:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheung N, Sharrett AR, Klein R, Criqui MH, Islam FM, Macura KJ, Cotch MF, Klein BE, Wong TY. Aortic distensibility and retinal arteriolar narrowing: the Multi‐Ethnic Study of Atherosclerosis. Hypertension. 2007;50:617–622. [DOI] [PubMed] [Google Scholar]
- 29. Brumback LC, Jacobs DR Jr, Dermond N, Chen H, Duprez DA. Reproducibility of arterial elasticity parameters derived from radial artery diastolic pulse contour analysis: the Multi‐Ethnic Study of Atherosclerosis. Blood Press Monit. 2010;15:312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, Lima JA, Crouse JR, Herrington DM. Predictive value of brachial flow‐mediated dilation for incident cardiovascular events in a population‐based study: the Multi‐Ethnic Study of Atherosclerosis. Circulation. 2009;120:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stacey RB, Bertoni AG, Eng J, Bluemke DA, Hundley WG, Herrington D. Modification of the effect of glycemic status on aortic distensibility by age in the multi‐ethnic study of atherosclerosis. Hypertension. 2010;55:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, Ouyang P, Jones SR, Blumenthal RS, Budoff MJ, et al. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the Multi‐Ethnic Study of Atherosclerosis. Circulation. 2014;129:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kershaw KN, Lane‐Cordova AD, Carnethon MR, Tindle HA, Liu K. Chronic stress and endothelial dysfunction: the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Hypertens. 2017;30:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goff DC Jr, Bertoni AG, Kramer H, Bonds D, Blumenthal RS, Tsai MY, Psaty BM. Dyslipidemia prevalence, treatment, and control in the Multi‐Ethnic Study of Atherosclerosis (MESA): gender, ethnicity, and coronary artery calcium. Circulation. 2006;113:647–656. [DOI] [PubMed] [Google Scholar]
- 35. Grams ME, Rebholz CM, Chen Y, Rawlings AM, Estrella MM, Selvin E, Appel LJ, Tin A, Coresh J. Race, APOL1 risk, and eGFR decline in the general population. J Am Soc Nephrol. 2016;27:2842–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parsa A, Kao WH, Xie D, Astor BC, Li M, Hsu CY, Feldman HI, Parekh RS, Kusek JW, Greene TH, et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med. 2013;369:2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen TK, Katz R, Estrella MM, Gutierrez OM, Kramer H, Post WS, Shlipak MG, Wassel CL, Peralta CA. Association between APOL1 genotypes and risk of cardiovascular disease in MESA (Multi‐Ethnic Study of Atherosclerosis). J Am Heart Assoc. 2017;6:e007199 DOI: 10.1161/JAHA.117.007199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med. 1997;157:2413–2446. [DOI] [PubMed] [Google Scholar]
- 39. Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, et al. Follow‐up report on the diagnosis of diabetes mellitus. Diabetes Care. 2003;26:3160–3167. [DOI] [PubMed] [Google Scholar]
- 40. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 41. Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, Van Lente F, Zhang YL, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Driver TH, Shlipak MG, Katz R, Goldenstein L, Sarnak MJ, Hoofnagle AN, Siscovick DS, Kestenbaum B, de Boer IH, Ix JH. Low serum bicarbonate and kidney function decline: the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2014;64:534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peralta CA, Katz R, Bonventre JV, Sabbisetti V, Siscovick D, Sarnak M, Shlipak MG. Associations of urinary levels of kidney injury molecule 1 (KIM‐1) and neutrophil gelatinase‐associated lipocalin (NGAL) with kidney function decline in the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2012;60:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carter CE, Katz R, Kramer H, de Boer IH, Kestenbaum BR, Peralta CA, Siscovick D, Sarnak MJ, Levey AS, Inker LA, et al. Influence of urine creatinine concentrations on the relation of albumin‐creatinine ratio with cardiovascular disease events: the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2013;62:722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu X, Young JH, Fox E, Keating BJ, Franceschini N, Kang S, Tayo B, Adeyemo A, Sun YV, Li Y, et al. Combined admixture mapping and association analysis identifies a novel blood pressure genetic locus on 5p13: contributions from the CARe consortium. Hum Mol Genet. 2011;20:2285–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figure S1