

Abstract
Background
Sorbs2b (sorbin and SH3 domain‐containing 2b) was recently identified as a cardiomyopathy gene from a zebrafish mutagenesis screen. However, cardiac functions of its mammalian ortholog remain elusive.
Methods and Results
We conducted a detailed expression and subcellular localization analysis of Sorbs2 ortholog in mice and a phenotypic characterization in Sorbs2 knockout mice. Sorbs2 is highly expressed in the mouse heart and encodes an adhesion junction/desmosome protein that is mainly localized to the intercalated disc. A mutation with near complete depletion of the Sorbs2 protein in mice results in phenotypes characteristic of human arrhythmogenic cardiomyopathy (ACM), including right ventricular dilation, right ventricular dysfunction, spontaneous ventricular tachycardia, and premature death. Sorbs2 is required to maintain the structural integrity of intercalated disc. Its absence resulted in profound cardiac electrical remodeling with impaired impulse conduction and action potential derangements. Targeted sequencing of human patients with ACM identified 2 rare splicing variants classified as likely pathogenic were in 2 unrelated individuals with ACM from a cohort of 59 patients with ACM.
Conclusions
The Sorbs2 knockout mouse manifests several key features reminiscent of human ACM. Although the candidacy of SORBS2 as a new ACM‐susceptibility gene is supported by preliminary human genetics study, future validation in larger cohorts with ACM is needed.
Keywords: arrhythmogenic cardiomyopathy, intercalated disc, sorbin and SH3 domain‐containing 2, susceptibility gene
Subject Categories: Arrhythmias, Animal Models of Human Disease, Genetics, Cardiomyopathy, Basic Science Research
Nonstandard Abbreviations and Acronyms
- ACM
arrhythmogenic cardiomyopathy
- DIC
doxorubicin‐induced cardiomyopathy
- GBT
gene‐breaking transposon
- ICD
intercalated disc
- LV
left ventricle
- MRI
magnetic resonance imaging
- RV
right ventricle
- SCD
sudden cardiac death
- Sorbs2
sorbin and SH3 domain‐containing 2
- VERP
ventricular effective refractory period
- VT
ventricular tachycardia
- WT
wild type
Clinical Perspective
What Is New?
Sorbs2 (sorbin and SH3 domain‐containing 2) encodes an adhesion junction/desmosome protein that is mainly localized to the intercalated disc.
Sorbs2 knockout mouse manifests phenotypes of right ventricular dilation, right ventricular dysfunction, spontaneous ventricular tachycardia, and premature deaths, which are key features of mouse arrhythmogenic cardiomyopathy models.
Two rare likely‐pathogenic splicing variants of SORBS2 gene were identified in patients with arrhythmogenic cardiomyopathy.
What Are the Clinical Implications?
SORBS2 could be a new candidate arrhythmogenic cardiomyopathy–susceptibility gene; however, more human genetic evidence is needed to confirm this statement.
Sorbs2 knockout mouse manifests several key features reminiscent of human arrhythmogenic cardiomyopathy.
Arrhythmogenic right ventricular (RV) cardiomyopathy, also referred to as arrhythmogenic right ventricular dysplasia, is a rare inherited cardiac muscle disorder that can lead to heart failure and sudden cardiac death (SCD). 1 The prevalence of arrhythmogenic RV cardiomyopathy in the general population is between 1:1000 and 1:5000, accounting for up to 20% of SCDs in people <35 years of age. 2 , 3 ARVC is characterized by structural abnormalities in the RV and ventricular arrhythmia at early stages. Left ventricular (LV) involvement becomes common with disease progression, and biventricular involvement prompted a recent recommendation of using a broader term, arrhythmogenic cardiomyopathy (ACM), for this disease. 4 Current management of ACM is mostly palliative and focuses on delaying disease progression and preventing SCD. No curative treatment is available for this life‐threatening disease. 1
Since the first disease‐causing variant was identified in the gene coding plakoglobin (JUP), 5 to date, at least 17 genes have been linked to ACM, mostly through human genetic studies, accounting for ≈50% to 60% ACM probands. 6 , 7 Among these 17 ACM‐associated genes, 5 of them (DSP, DSG2, DSC2, PKP2, and JUP) encode desmosomal proteins located in the intercalated disc (ICD). 5 , 8 , 9 , 10 , 11 Mutations in these desmosomal protein‐encoding genes account for most variants identified from the ACM probands. 1 A small percentage of ACM probands have sequence variants in nondesmosomal protein‐encoding genes, such as CTNNA3 and CDH2. 12 , 13 Mouse models for all these 5 desmosome/ICD protein‐coding genes have been generated through either targeted genetic deletion or transgenic overexpression of genes with disease‐causing mutations. 14 , 15 , 16 , 17 , 18 , 19 The key features of human ACM phenotypes, such as structural abnormalities in the ventricles, spontaneous ventricular tachycardia (VT), and SCD, can be recapitulated in most of these mouse models. However, certain aspects of human ACM, such as fibrofatty infiltration, were only noted in the mouse model involving DSP 20 , 21 but not in those involving DSG2, DSC2, and JUP. 19 , 22 Because of the convenience of embryonic morpholino knockdown and amenability to high‐throughput genetic and compound screening, zebrafish ACM models for DSC2 and JUP have also been developed. 23 , 24
Through an insertional mutagenesis screen in zebrafish, we recently identified the gene‐breaking transposon (GBT) 002 as a mutant that exerted deleterious modifying effects on doxorubicin‐induced cardiomyopathy (DIC). 25 GBT002 was later determined as a loss‐of‐function mutant that disrupts sorbs2b (sorbin and SH3 domain‐containing 2b), one of the SORBS2 orthologs in zebrafish. 25 SORBS2, also known as ArgBP2 (Arg kinase binding protein 2), encodes a member of the sorbin homology adaptor family protein that contains a Sorbin domain at the N terminus and 3 SH3 domains at the C terminus. 26 The encoded protein is highly abundant in cardiac muscle cells and manifests a restricted subcellular expression pattern, including association with stress fibers 27 and sarcomeric Z discs in cardiomyocytes. 28 , 29 In addition, several other recent studies have also suggested the cardiac involvement of Sorbs2. First, the knockdown of Sorbs2 in neonatal rat cardiomyocytes in culture resulted in cellular hypertrophy. 30 Second, SORBS2 was a candidate gene in regions of copy number variations linked to congenital heart disease in humans. 31 Third, the expression of SORBS2 protein was significantly increased in the sera of patients with acute myocardial infarction but depleted in the infarcted myocardia. 32 However, in vivo cardiac phenotypes have not been reported in the Sorbs2 knockout mouse model. 33
Herein, we report detailed cardiac studies of Sorbs2 in mice. We revealed Sorbs2 encodes an ICD protein that is required to maintain the ICD structural integrity. We showed Sorbs2 knockout mice manifest ACM‐like phenotypes, including RV cardiomyopathy, VT, and premature death. Scanning a cohort of 59 Chinese Han patients with ACM led to the identification of 2 likely pathogenic rare variants. Collectively, our results showed that the Sorbs2 knockout mouse manifests several key features reminiscent of human ACM and SORBS2 likely represents a previously unrecognized ACM‐susceptibility gene.
METHODS
All supporting data and materials within the article will be made available from the corresponding authors on reasonable request.
Experimental Animals
All procedures were performed in accordance with Mayo Clinic guidelines for animal use and care and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Sorbs2e8/e8 mice, also named B6N(Cg)‐Sorbs2 tm1.1(KOMP)Mbp/J, were purchased from the Jackson Laboratory (catalog No. 022780) with genotype characterization using polymerase chain reaction (PCR) primers: 5′‐ATTAGGACGGTAAGCCACGC‐3′ and 5′‐AGGCTTCACCTTGCTTTGATAG‐3′ for detecting the wild‐type (WT) allele (predicted size of 292 bp) and 5′‐TTTGGCTTCAACCACAACAC‐3′ and 5′‐CGGTCGCTACCATTACCAGT‐3′ for detecting the mutant allele (predicted size of 500 bp). The zebrafish (Danio rerio) WIK line was maintained under a 14‐hour light/10‐hour dark cycle at 28.5°C. WT zebrafish were bred to give embryos for morpholino injection and subsequent fractional shortening and heart rate analysis. The animal study protocols were approved by the Mayo Clinic Institutional Animal Care and Use Committee (A00002911 for mice and A3531 for zebrafish).
Human Studies
Human studies were approved from the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University and Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Institutional Review Board identification: TJ‐C20181101). All participating patients signed informed consent forms.
Tissue Collection
For tissue harvest, the mice were euthanized by the administration of 250 mg/kg pentobarbital according to the guidelines of the Mayo Clinic Institutional Animal Care and Use Committee. Heart tissues from different cardiac chambers were frozen in liquid nitrogen, stored at −80°C, and subsequently used for total RNA transcript and Western blot experiments. Some tissues were fixed in formalin and embedded in paraffin for histomorphometry. At least 3 heart samples were collected for each individual experiment.
Western Blot Analysis
Heart tissues were homogenized in radioimmunoprecipitation assay buffer with 0.5‐mm steel beads at speed 8 for 4 minutes, followed by speed 10 for 1 minute using a Bullet Blender (Next Advance). Lysate samples were denatured at 95°C for 10 minutes and loaded onto an SDS‐PAGE and Western blotting. Anti‐Sorbs2 (Sigma, catalog No. SAB4200183), anti–connexin 43 (Cx43) (Cell Signaling, catalog No. 3512), anti–α‐catenin (Thermofisher Scientific, catalog No. 71‐1200), anti–β‐catenin (Thermofisher Scientific, catalog No. 13‐8400), anti–desmoglein 1/2 (Progen, catalog No. 61002), anti‐plakoglobin (Progen, catalog No. 65105), anti–N‐cadherin (Sigma, catalog No. C1821), and anti‐GAPDH (Santa Cruz Biotechnology, catalog No. sc‐25778) antibodies were used.
Quantitative Reverse Transcription–PCR
Total RNA was extracted using an RNeasy extraction kit (Qiagen), according to the manufacturer's instructions. The extracted RNA was quantified, and 1 μg RNA was reverse transcribed to synthesize cDNA using a Super Script III First Strand Reverse Transcriptase Kit and random hexamer primers (Invitrogen). After initial denaturation with the LightCycler 480 Probes Master Mix (Roche), 50 ng of the cDNA underwent 50 rounds of amplification (LightCycler 480, Roche) with the following primers: 5′‐GTGCGGTGTCCAACACAGAT‐3′ and 5′‐TCCAATCCTGTCAATCCTACCC‐3′ for mouse Nppa gene; 5′‐GAGGTCACTCCTATCCTCTGG‐3′ and 5′‐GCCATTTCCTCCGACTTTTCTC‐3′ for mouse Nppb gene; 5′‐ACTGTCAACACTAAGAGGGTCA‐3′ and 5′‐TTGGATGATTTGATCTTCCAGGG‐3′ for mouse β‐Mhc gene; 5′‐GCCCAGTACCTCCGAAAGTC‐3′ and 5′‐GCCTTAACATACTCCTCCTTGTC‐3′ for mouse α‐Mhc gene; 5′‐CTGAATGGTATCGCACCGGAA‐3′ and 5′‐GGTCCACAAGCACTCCACAG‐3′ for mouse Cx40/Gja5 gene, 5′‐ACAGCGGTTGAGTCAGCTTG‐3′, 5′‐GAGAGATGGGGAAGGACTTGT‐3′ for mouse Cx43/Gja1 gene, 5′‐CAGAGCCAACCAAAACCTAAGC‐3′, 5′‐CTGCACACATAAAATGGGTGGA‐3′ for mouse Cx45/Gjc1 gene; and 5′‐CTATCAGAGGCCATTCTCCC‐3′ and 5′‐GAACATTGTCTTGTACCAGTCC‐3′ for mouse Sorbs2 gene. The data were analyzed, and relative expression was determined using the comparative cycle threshold (Ct) method (2−ΔΔCt), and expression was normalized using Gapdh (5′‐AGGTCGGTGTGAACGGATTTG‐3′ and 5′‐TGTAGACCATGTAGTTGAGGTCA‐3′) as the housekeeping internal control.
Immunohistochemistry
The heart samples harvested from mice were embedded in tissue freezing medium and stored at −80°C, followed by sectioning at 10 μm using a cryostat (Leica CM3050 S). The slides were subjected to immunostaining using a previously described protocol. 34 The following antibodies were used: anti‐Sorbs2 (Sigma, catalog No. SAB4200183) at 1:200, anti‐Cx43 (Cell Signaling, catalog No. 3512) at 1:200, anti–N‐cadherin (Sigma, catalog No. C1821) and anti–β‐catenin (Thermo Fisher Scientific, catalog No. 13‐8400) at 1:200, and anti‐plakoglobin (Progen, catalog No. 65105) at 1:200. A Zeiss LSM 780 confocal microscope was used to acquire the image. The tile scan function was used to acquire images covering a complete mouse heart. For each group, 5 to 10 slides were imaged and analyzed. The distribution of fluorescence density was analyzed by pseudoline scan using Zen software (Zeiss). The weighted colocalization of Sorbs2 with other ICD proteins was determined using a colocalization function in Zen software.
Magnetic Resonance Imaging
Mice were anesthetized by inhalational isoflurane (1.5%−2.5% in oxygen/air) via a nose cone during the imaging procedure. Respiration was continuously monitored during the scan, and the heart rate was maintained at 20 to 60 breaths per minute by adjusting the concentration of isoflurane. The animal might be maintained under anesthesia for >2 hours. The body temperature of the animals was maintained by a stream of air conditioned by a thermocouple‐based system at 32°C. Magnetic resonance imaging (MRI) was performed on a Bruker Avance 300‐MHz spectrometer (7‐T) vertical bore nuclear magnetic resonance spectrometer equipped with “mini‐imaging” accessories. The images were acquired with a field of view of 3.4 cm2, slice thickness of 1 mm, and an in‐plane resolution of 135 μm. The images were acquired from both long‐axial and short‐axial views and analyzed offline using custom‐designed code in MATLAB. 35 , 36 RV and LV endocardial and epicardia contours were drawn on each of the 10 short‐axial or long‐axial slices. The wall thickness of the LV was calculated directly from the contour data. The volume of the LV was calculated on the basis of a single‐plane ellipsoid model using the following equation 37 :
A is the area of the LV in the long‐axis view, and L is the length of the LV along the long‐axial view. RV volumes were calculated using a formula derived from the ellipsoidal shell model (difference‐of‐ellipsoids model): V=2/3Pd. The plain of area P was defined by 2 perpendicular axes of the ellipsoid, and distance d was the third axis perpendicular to area P. 37
Electrocardiogram
Anesthesia with isoflurane (0.5%–1.0% v/v) was administered via a nose cone. Mice were placed on an ECG‐heater board with 4 paws on individual electrode. The ECG‐heater board maintained the body temperature at 37°C and monitored the heart rates. The ECG signal was amplified through an amplifier (Axon CNS digital 1440 A) and recorded using Chart 5 software. For each mouse, 2 minutes of ECG signal was recorded.
Electrophysiology
Ex vivo cardiac electrophysiology study was performed in Langendorff‐perfused mouse hearts, as we have described previously. 38 Briefly, the excised heart was rapidly mounted on the cannulus of a modified Langendorff apparatus and perfused at 3.0 mL/min using a peristaltic pump (P720, Instech Laboratories) with Tyrode solution (in mmol/L): NaCl 119, KCl 4.8, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.0, NaHCO3 24.9, glucose 10.0, pyruvate 5.0, heparin 1200 U/L, pH 7.4, equilibrated with 95% O2/5% CO2 at 37°C. A bipolar electrode was placed on the RV epicardium for cardiac pacing. Programmed electrical stimulation was performed using a cardiac stimulator (Bloom‐Fisher Medical Technologies) with 2‐ms current pulses at a 2‐time diastolic threshold (100–200 μA). The cycle lengths of stimuli were 10 to 20 ms shorter than those of intrinsic heartbeat coupling intervals.
Action potential recordings of isolated mouse RV were performed using standard microelectrode techniques, as previously described. 39 The RV was rapidly excised, placed with the endocardial surface up in a temperature‐regulated 5‐mL chamber, and continuously superfused with oxygenated Tyrode solution at 4 mL/min at 37°C. The tissue was impaled with a micropipette with a tip resistance of 10 to 25 MΩ when filled with 3 mol/L KCl, and the output signals were sampled at 100 kHz without filter by a high‐input impedance preamplifier (Duo 773, World Precision Instruments, Sarasota, FL). The cardiac preparations were paced using a stimulator (Bloom Associates, Ltd, Reading, PA) with a stimulus output at twice threshold. The action potentials were recorded at a paced cycle length of 200 ms, and the resting potentials, action potential durations at 50% and 90%, were measured offline. Ventricular effective refractory period (VERP) was measured by a train of stimuli (10≈14 S1) at a fixed cycle length of 200 ms, coupled with an extrastimulus (S2) that was 10 ms shorter than the train and repeated at 10‐ms decrements until refractoriness was reached. Burst pacing was performed by trains of 20 pulses with coupling intervals 10 to 20 ms longer than VERP. VTs were induced by programmed electrical stimulation and burst pacing. Sustained VT was defined as wide QRS complex tachycardia with a rate >600 beats/min lasting >30 seconds. 40
Transmission Electron Microscopy
Tissues from mouse hearts were bathed in relaxation buffer (75 mmol/L KCl, 10 mmol/L imidazole, pH 7.2, 2 mmol/L MgCl2, 2 mmol/L EGTA, 1 mmol/L NaN3, and 4 mmol/L creatine phosphate) for 1 hour, fixed in Trump fixative (Electron Microscopy Sciences) overnight at 4°C, and then processed at the Mayo Clinic Electron Microscopy Core Facility. Images were obtained using a Philips CM10 transmission electron microscope (Philips, Amsterdam, the Netherlands).
Hematoxylin‐Eosin and Masson Trichrome Staining
Heart tissues were harvested from mice at 4 months of age after being euthanized by administration of 250 mg/kg pentobarbital. Dissected tissues were immediately fixed in 4% PBS‐buffered formaldehyde and sent to the Mayo Clinic Histology Core Laboratory for subsequent sample processing and hematoxylin‐eosin and Masson trichrome staining. Images were captured using the EVOS FL Auto Imaging System (Thermo Fisher Scientific). Fibrosis quantification was performed using Image J software (National Institutes of Health, Bethesda, MD), as described previously. 41 The blue color range was selected to represent fibrosis areas. Blue‐stained areas and nonstained myocyte areas from each section were determined using color‐based thresholding. The percentage of fibrosis area was calculated as the blue‐stained areas divided by total surface area from each section.
RNA‐Binding Protein Immunoprecipitation
The RNA‐binding protein immunoprecipitation assay was performed using an EZ‐Magna RNA immunoprecipitation kit (Sigma, 17‐704), according to the manufacturer's instructions. Briefly, 10 mg of RV tissue from C57 mice was homogenized using a Bullet Blender (Next Advance). One microgram of anti‐Sorbs2 antibody (Sigma, catalog No. SAB420183) was used for the immunoprecipitation. Primers for quantitative reverse transcription–PCR to detect the abundance of Cx43 mRNA are 5′‐ACAGCGGTTGAGTCAGCTTG‐3′ for Cx43/Gja1‐F and 5′‐GAGAGATGGGGAAGGACTTGT‐3′ for Cx43/Gja1‐R.
Participant Recruitment
From November 2010 to November 2017, 59 unrelated Han Chinese patients with ACM were recruited with approval from the Institutional Review Board of the local hospital (Institutional Review Board identification: TJ‐C20181101). Specifically, 46 patients were recruited in Nanjing from the First Affiliated Hospital of Nanjing Medical University and 13 patients were recruited in Wuhan from the Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. For controls, 402 sex‐ and ethnically matched subjects without cardiomyopathy were recruited from a healthy population undergoing health examinations during the same period. All recruited individuals have undergone ECG and echocardiography or computed tomography or MRI tests to evaluate the structure and function of their hearts. Patients with ACM were diagnosed on the basis of the 2019 Heart Rhythm Society (HRS) expert consensus statement. 42
Panel Construction and Targeted Sequencing
An ACM panel was designed that can be used for Ampliseq semiconductor next‐generation sequencing (Ion Torrent, Thermo Fisher). The panel included 159.11‐kb full coding regions with >5 bp flanking regulatory sequences of 13 known genes for hereditary ACM (Figure S7B). The coding sequences of CTNNA3 and SORBS2 genes were later sequenced separately using Sanger sequencing. Primer sequences are listed in the Table S5. Genomic DNA was extracted from peripheral leukocytes using a standard protocol. Barcode‐ligated libraries were constructed and mixed with the same molecular quantities. The pooled libraries were sequenced using an Ion Torrent sequencing platform.
Whole Exome Sequencing
In total, 200 ng of genomic DNA was fragmented into 250‐ to 350‐bp fragments using sonication followed by end repair. The DNA fragments were purified and ligated with adapters using a SureSelectXT Library Prep Kit (Agilent, Santa Clara, CA). Next, the adapter‐ligated DNA libraries were amplified and captured using a SureSelect capture library kit (Agilent). The captured sequences were further amplified for 350‐bp read‐length paired‐end sequencing using an Illumina X‐ten system (Illumina, San Diego, CA).
Mutation Identification and Bioinformatics Analysis
The targeted sequencing raw data were initially processed with the Ion Torrent platform‐specific software Torrent Suite v5.0. All reads were aligned to the GRCh37/hg19 human reference genome to analyze coverage status and to call variants. Extracted variants were annotated using Ion Reporter software 5.0 (Thermo Fisher). The whole exon sequencing data were processed according to the Genome Analysis Toolkit Best Practices recommendations. All potential pathogenic variants were identified with definite clinical significance, according to the recent American College of Medical Genetics and Genomics guidelines for the interpretation of sequence variants. 43 To identify potential pathogenic variants, we searched the Human Gene Mutation Database and the ClinVar database. All identified potential pathogenic variants that have been reported in the Human Gene Mutation Database or ClinVar database were reassessed by cosegregation data, identification of the variant in independent individuals, and functional analyses of the variant. The expected phenotypes of these potential pathogenic variants should be consistent with the clinical complications of the carrier. The frequencies of all variants were searched from the Genome Aggregation Database. Missense variants were predicted using Polyphen‐2, SIFT, and Mutation Taster. A variant was determined as deleterious only when the variant was predicted to be damaging and conserved by all computational software. After validation via Sanger sequencing by using an Applied Biosystems 3500xl capillary sequencer (Applied Biosystems, Foster City, CA) to eliminate false‐positive variants, each validated potential pathogenic variant was sequenced in 402 healthy controls using the same method. We then conducted Sanger sequencing of the SORBS2 gene in patients with ACM. Finally, the variants were categorized into 5 classes, including pathogenic, likely pathogenic, uncertain significance, likely benign, and benign.
Minigene Analysis
Because the human c.679+1G>T mutation is localized between intron 8 and intron 9 of the SORBS2 gene, a 2.4‐kb DNA fragment was cloned from the human SORBS2 gene (NM_021069) covering part of intron 9, whole exon 10, intron 10, exon 11, and part of intron 11. The amplified DNA fragment was cloned into an Nde I site of the pTBNde‐min vector (Addgene) to express a wide‐type SORBS2 fragment. To obtain the vector expressing a mutated SORBS2 fragment, mutagenesis was conducted using the site‐directed mutagenesis system (Invitrogen) with primer pairs 5′‐TCCTTATACATACAATGCAGTTAGGATGGGTTTCCTTGCTC‐3′ and 5′‐GAGCAAGGAAACCCATCCTAACTGCATTGTATGTATAAGGA‐3′. The vectors were transfected into AD‐293 cells. Forty‐eight hours later, RNA was extracted from the transfected cells to check alternative splicing via reverse transcription–PCR, with 5′‐CAACTTCAAGCTCCTAAGCCACTGC‐3′ as the forward primer and 5′‐TAGGATCCGGTCACCAGGAAGTTGGTTAAATCA‐3′ as the reverse primer.
Zebrafish Morpholino Injection and Cardiac Function Quantification
Zebrafish embryos were injected with sorbs2a‐e6 morpholino (5′‐TTTATTTGAAAGAACTCACCGTTGT‐3′; 0.25 μmol/L) and/or sorbs2b‐e10 morpholino (5′‐ATGTGAAACAATAGGAAACCTTGTT‐3′; 0.25 μmol/L) at 1‐cell stage. Heart fractional shortening and heart rate were measured at 3 days post‐fertilization, according to our previously reported methods. 44 Reverse transcription–PCR was conducted to assess the knockdown efficiency of morpholinos. The exon skipping event in zebrafish injected with the sorbs2a morpholino was detected with the forward primer 5′‐CCCAAGGACTGGTACAAGAC‐3′ and the reverse primer 5′‐GAGGATTTTCCAGGCTCATACTC‐3′. The intron retention event in zebrafish injected with the sorbs2b morpholino was detected with the forward primer 5′‐GATTGGTACAAGACCATGTTCAAAC‐3′ and the reverse primer 5′‐GTCTGCACCCTTCTGCCAACAC‐3′.
Statistical Analysis
Kaplan‐Meier survival curve, echocardiography, and MRI analyses are from cumulative data using mice obtained from several breeding efforts. Unpaired 2‐tailed Student t tests were used for comparisons of 2 groups. One‐way ANOVA was used to assess differences among multiple groups, as appropriate. A Tukey test was applied for the post hoc analysis to determine which pairwise differences are contributing to the significant ANOVA result. For the Kaplan‐Meier survival curves, the log‐rank test was used to determine the difference in animal rate of survival between groups. All quantitative data are presented as the mean±SD. P<0.05 was considered to be significant. All statistical analyses were performed with GraphPad Prism 7 and/or R Statistical Software Version 3.6.1.
RESULTS
Sorbs2 Is an ICD Protein in Cardiomyocytes
In mice, the Sorbs2 gene encodes a protein that is highly conserved with human SORBS2, especially in its N‐terminal sorbin homolog domain and C‐terminal SH3 domain, where they share >95% homology (Figure S1). The expression of Sorbs2 protein is highly enriched in the mouse heart (Figure 1A). The mouse cardiac Sorbs2 is ≈120 kDa in size, whereas its brain isoform is ≈150 kDa (Figure S2A), 33 suggesting there exists tissue‐specific alternative splicing events, posttranslational modification, or both. At the subcellular level, Sorbs2 is localized strongly in the ICDs of cardiomyocytes, with weaker localization in the perinuclear areas and the Z discs (Figure 1B). To resolve the Sorbs2 expression within ICD, we conducted double immunostaining using an Sorbs2 antibody and antibodies against β‐catenin, plakoglobin, or Cx43, markers of the adherens junction, adherens junction/desmosome, or gap junction structures, respectively. We found that Sorbs2 mostly colocalizes with β‐catenin, partially with plakoglobin, and slightly colocalizes with Cx43 (Figure 1C). The weighted colocalization efficiency of Sorbs2 is ≈60% with β‐catenin, 20% with plakoglobin, and 6% with Cx43, suggesting that Sorbs2 is mainly localized in the adherens junction and partially in the desmosome.
Figure 1. Sorbs2 (sorbin and SH3 domain‐containing 2) is a cardiac‐enriched intercalated disc and desmosome protein in mice.
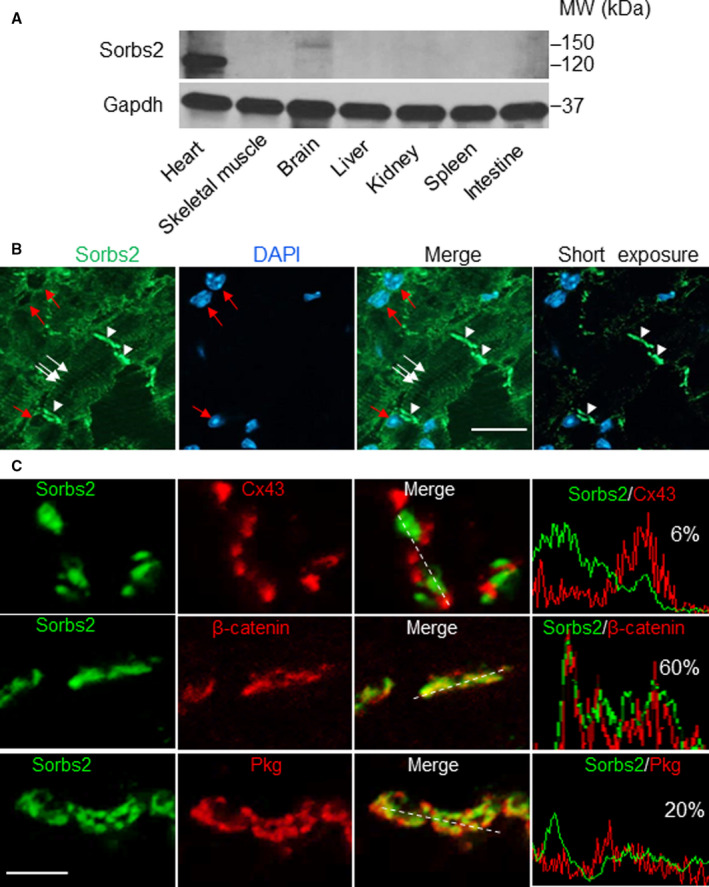
A, Western blot analysis of Sorbs2 protein expression in different mouse tissues. B, Images of immunostaining using an anti‐Sorbs2 antibody and counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) in sectioned wild‐type mouse hearts. Arrowheads indicate the intercalated disc. The arrows indicate Z‐disc localization. Red arrows indicate the perinucleus. Bar=20 μm. C, Immunostaining and pseudoline analysis of sectioned mouse hearts are shown, indicating that Sorbs2 protein mostly colocalizes with β‐catenin and plakoglobin (Pkg), but less with connexin 43 (Cx43). Bar=2 μm. MW indicates molecular weight.
Deleterious Modifying Effects of GBT002/Sorbs2 on DIC Are Cardiomyocyte Primary and Conserved in Mice
To elucidate the cardiac functions of Sorbs2, we studied the DIC modifying effects in the zebrafish GBT002 mutant. Because the 2 Loxp sites in the GBT vector enable cardiomyocyte‐specific genetic manipulation, 45 we crossed Tg(cmlc2:cre‐ER), a myocardium‐specific expressed and hydroxytamoxifen inducible Cre transgenic fish line, 46 into the GBT002/sorbs2b mutant for a cardiomyocyte‐specific rescue experiment. We found that the mutagenic insertion was effectively removed, and the sorbs2b transcript was fully restored in the myocardium of the GBT002−/−;Tg(cmlc2:cre‐ER) double‐mutant/transgenic fish on hydroxytamoxifen treatment (Figure S3). Although the GBT002−/− homozygous mutant manifests no visually noticeable phenotypes, after doxorubicin stress, the GBT002−/− mutant exhibited a significantly deleterious DIC modifying effect, as indicated by the increased fish mortality (Figure S3D). This deleterious DIC‐modifying effect of GBT002/sorbs2b was largely rescued in the GBT002−/−;Tg(cmlc2:cre‐ER) double‐mutant/transgenic fish, suggesting a critical contribution of myocardial expression of sorbs2b to its DIC‐modifying effects.
To show the DIC‐modifying effects in mammals, we obtained Sorbs2e8 knockout mice (B6N[Cg]‐Sorbs2/J), in which a targeted deletion of 1903‐bp nucleotides removes part of intron 7, the whole exon 8, and part of intron 8 from the Sorbs2 genomic locus (Figure 2A). Exon 8 encodes part of the N‐terminal Sorbin homolog domain (Figure S1), the removal of which results in aberrant splicing of exon 7 to an ectopic exon encoding LacZ (Figure 2A and 2B and Figure S2D and S2E), leading to a dramatic reduction in the level of the whole Sorbs2 transcript (Figure 2C). At the protein level, the Sorbs2 protein is ≈50% reduced in the Sorbs2e8/+ heterozygous and dramatically reduced in Sorbs2e8/e8 homozygous mice in all 4 cardiac chambers, as indicated by Western blotting using an antibody that recognizes a C‐terminal antigenic epitope of the Sorbs2 protein (Figure 2D and Figure S1). In the Sorbs2e8/e8 mice, Sorbs2 protein in the muscle and its larger isoform in the brain are also mostly ablated (Figure S2A). Consistent with the zebrafish data, we noted deleterious modifying effects of Sorbs2e8/+ heterozygous mice on DIC (Figure 3A). A mild but significant reduction in RV function was noted because of an enlarged end‐systolic diameter on doxorubicin stress. Other cardiac pump indexes remained unchanged (Table S1). We did not conduct detailed studies of the DIC‐modifying effects of Sorbs2e8/e8 homozygous mice because of the interference of severe cardiac phenotypes, as described below.
Figure 2. Targeted deletion of exon 8 in the Sorbs2 (sorbin and SH3 domain‐containing 2) gene led to near null depletion of the whole Sorbs2 transcript and protein in mice.
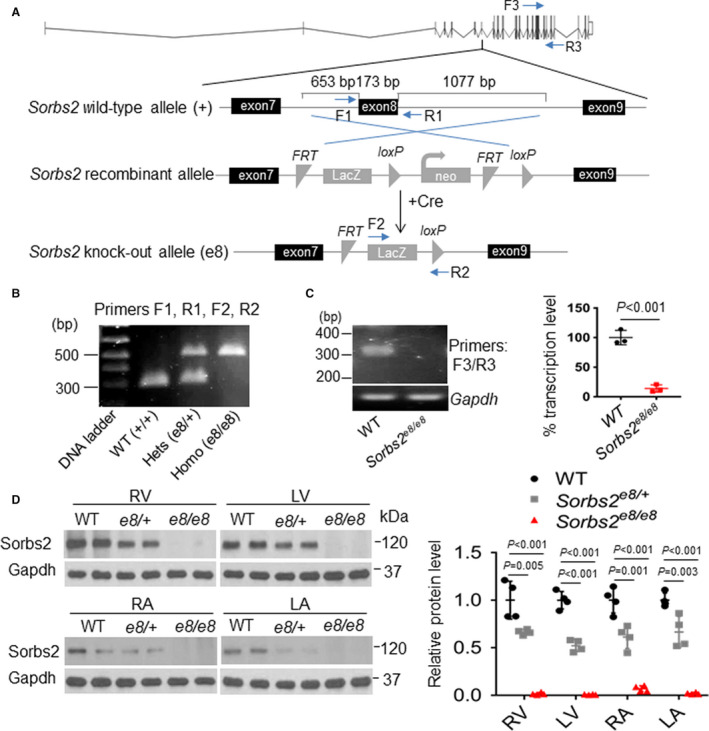
A, Schematic illustration of targeted deletion of Sorbs2 exon 8 in the Sorbs2e8/e8 mice and locations of primers used for genotyping polymerase chain reaction (PCR) and transcription analysis. B, Representative DNA gel images of PCR genotyping for identifying wild‐type (WT) (292 bp), Sorbs2e8/+ heterozygous (hets), and Sorbs2e8/e8 homozygous (homo) mutant alleles (500 bp). C, Representative DNA gel images and quantitative reverse transcription–PCR analysis of the Sorbs2 transcripts in WT and Sorbs2e8/e8 mutants. N=3, unpaired 2‐tailed Student t test. D, Western blotting and quantification of Sorbs2 protein expression in each cardiac chamber of WT and Sorbs2e8/e8 mutants. N=4. One‐way ANOVA. LA indicates left atrium; LV, left ventricle; RA, right atrium; and RV, right ventricle.
Figure 3. Sorbs2 (sorbin and SH3 domain‐containing 2) deficiency leads to right ventricle (RV) dilation, cardiac dysfunction, and premature death.
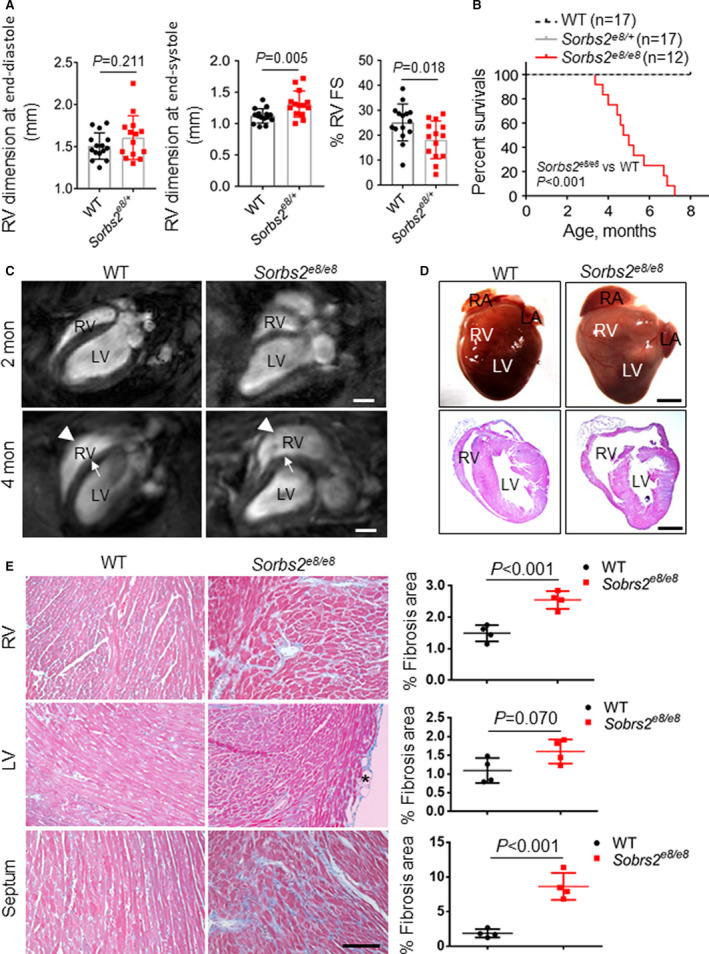
A, Echocardiography indexes in the RV of sorbs2e8/− and wild‐type (WT) control mice at 4 weeks postdoxorubicin injection (12 mg/kg body weight). N=14 to 15. Unpaired 2‐tailed Student t test. B, Kaplan‐Meier survival curves of Sorbs2e8/e8 and WT control mice. N=12 to 17. Log‐rank test. C, long‐axis views of magnetic resonance imaging images of 2‐ and 4‐month‐old mice. RV dilation (arrowheads) is prominent in the Sorbs2e8/e8 mouse hearts at 4 months old. Note that a septum bending defect (arrows) was also detected. D, Images of dissected whole hearts (top) and cross‐sectional view of hematoxylin and eosin–stained hearts (bottom). Dramatically enlarged RV and protuberant were noted in the Sorbs2e8/e8 mice. Note that both right atrium (RA) and left atrium (LA) enlargement was also frequently observed. E, Representative images of Masson trichrome staining and quantification analysis demonstrated significant myocardial fibrosis in the RV and septum but not in the LV of Sorbs2e8/e8 mice. Bar=50 μm. N=4, unpaired 2‐tailed Student t test. FS indicates fractional shortening; and LV, left ventricle.
Depletion of Sorbs2 Leads to RV Dilation, Cardiac Dysfunction, Premature Death, and Ventricular Arrhythmia in Mice
Sorbs2e8/e8 homozygous mice are fertile but started to die prematurely at 3.5 months old (Figure 3B). Cardiac MRI showed mild RV dilation in the Sorbs2e8/e8 mice as early as 2 months with continued progression at 4 months (Figure 3C). The RV bulges out along the short axis, and the overall shape of the ventricle changes from crescent to triangle‐like in the lateral view (Figure 3D). Cardiac function in both RV and LV decreased significantly (Table 1). Interestingly, in contrast to the straight ventricular septum of WT mice, the ventricular septum in Sorbs2e8/e8 mice becomes progressively bent. Both heart weight and LV posterior wall thickness at end diastole increased significantly, indicating the presence of LV hypertrophy (Table 1). In association with RV dilation and LV hypertrophy at 4 months, significant myocardium fibrosis was detected in the RV and septum but not in the LV (Figure 3E). Occasionally, adipose tissue deposition was noted in the LV but not in other chambers. The transcription levels of molecular markers for heart failure, such as Nppa, Nppb, and β‐Mhc, were markedly elevated in both LV and RV (Figure S4).
Table 1.
Quantification of Cardiac Function Indexes in Sorbs2e8/e8 and WT Control Mice via MRI at 4 Months Old
| Variable | WT Siblings | Sorbs2e8/e8 |
|---|---|---|
| Mice number, n | 5 | 9 |
| Body weight, g | 24.7±1.1 | 21.4±1.3* |
| Heart weight/body weight, mg/g | 6.4±0.6 | 9.2±0.8* |
| RVEDV, μL | 34.1±10.5 | 37.2±11.3 |
| RVESV, μL | 13.4±4.2 | 21.9±7.3* |
| RVEF, % | 58.7±12.7 | 41.2±9.9* |
| RVFACl, % | 46.8±6.1 | 32.4±5.6* |
| RVFACs, % | 57.1±15.7 | 29.9±14.9* |
| LVEDV, μL | 46.2±9.1 | 31.4±7.7* |
| LVESV, μL | 15.8±6.4 | 16.1±7.2 |
| LVEF, % | 66.2±9.1 | 50.3±16.1* |
| IVSs, mm | 1.4±0.1 | 1.1±0.1* |
| IVSd, mm | 1.0±0.1 | 0.8±0.1 |
| LVPWs, mm | 2.2±0.1 | 2.1±0.1 |
| LVPWd, mm | 1.1±0.1 | 1.6±0.1* |
IVSd indicates interventricular septum thickness at end diastole; IVSs, interventricular septum thickness at end systole; LVEDV, left ventricle end‐diastolic volume; LVEF, left ventricle ejection fraction; LVESV, left ventricle end‐systolic volume; LVPWd, left ventricular internal dimension at end diastole; LVPWs, left ventricular posterior wall thickness at end diastole; MRI, magnetic resonance imaging; RVEDV, right ventricle end‐diastolic volume; RVEF, right ventricle ejection fraction; RVESV, right ventricle end‐systolic volume; RVFACI, right ventricle fractional area change along long axis; RVFACs, right ventricle fractional area change along short axis; Sorbs2, sorbin and SH3 domain‐containing 2; and WT, wild type.
P<0.05, unpaired Student t test.
In addition to the profound cardiac structural remodeling, abnormal electrical remodeling was also striking in the Sorbs2e8/e8 mice. Significant increases in QRS and S‐wave durations with the development of right bundle branch block were commonly detected in Sorbs2e8/e8 mice at 4 months of age (Figure 4A). Interestingly, notched P waves, indicating left atrium enlargement, were also frequently observed. Random 2‐minute ECG recordings in anesthetized animals showed spontaneous arrhythmias, including premature ventricular contractions, nonsustained VTs, and polymorphic VTs, in Sorbs2e8/e8 mice (7/31) but not in WT controls (0/37) (Figure 4B) (χ 2, P<0.05). Electrophysiological studies in Langendorff‐perfused hearts showed that the VERP is markedly prolonged in Sorbs2e8/e8 mice compared with WT (Figure 4C). Sustained VTs with durations >30 seconds were induced by programmed electrical stimulation or burst pacing in 9 of 10 Sorbs2e8/e8 hearts (90%), but only in 3 of 12 WT hearts (25%) (χ 2, P<0.05). These results suggest that the Sorbs2e8/e8 mice have a greater propensity to develop spontaneous and induced ventricular tachyarrhythmias. Intracellular recordings of action potentials from the endocardial side of RV showed marked electrophysiological abnormalities in Sorbs2e8/e8 mice (Figure 4D). The resting potentials are significantly depolarized in Sorbs2e8/e8 mice compared with WT mice. Action potential amplitudes and upstroke velocities (dV/dt) are significantly reduced in Sorbs2e8/e8 mice (Figure 4E). There is remarkable prolongation of action potential durations at 50% and 90% repolarization, as well as lengthening the VERP in the Sorbs2e8/e8 mice.
Figure 4. Sorbs2 (sorbin and SH3 domain‐containing 2) deficiency leads to cardiac arrhythmia.
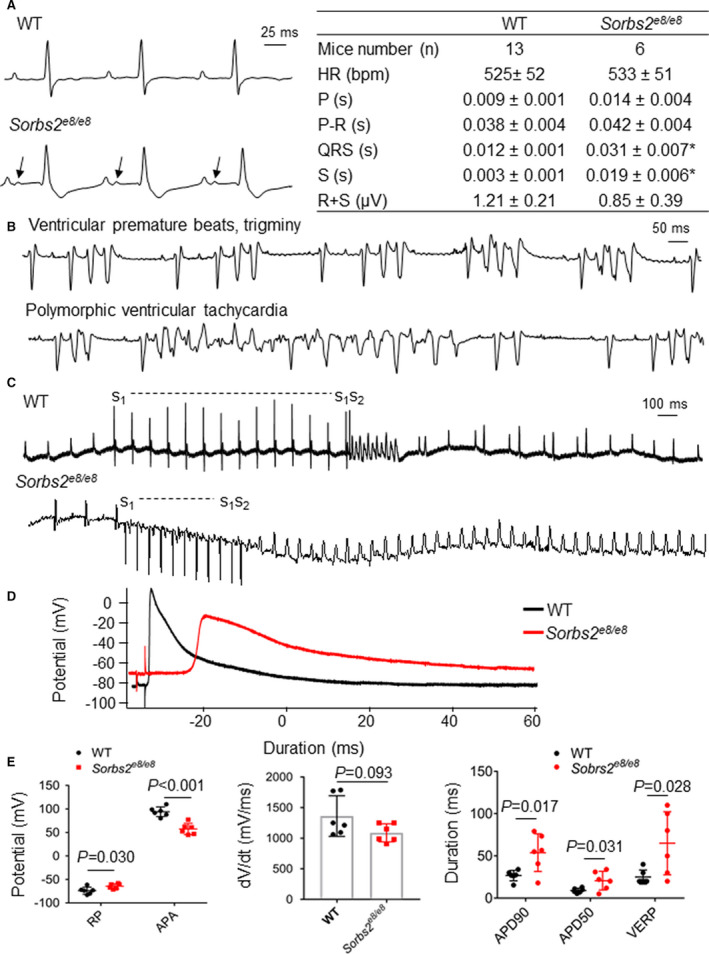
A, Shown are surface ECGs and quantification analysis in 4‐month‐old wild‐type (WT) and Sorbs2e8/e8 mice. N=6 to 13. Unpaired 2‐tailed Student t test. *P<0.05. B, Ventricular premature beats and trigeminy (top) and polymorphic ventricular tachycardia (VT) (bottom) were recorded in Sorbs2e8/e8 mice. C, Induction of nonsustained VT in Langendorff‐perfused WT mouse heart by programmed electrical stimulation (s1‐s1=100 ms, s1‐s2=30 ms) (top); induction of sustained monomorphic VTs in Langendorff‐perfused Sorbs2e8/e8 mouse heart by programmed stimulation (s1‐s1=100 ms, s1‐s2=80 ms) (bottom). D, Representative action potentials were recorded from the endocardial surface of isolated right ventricles (RVs) of Sorbs2e8/e8 and WT mice at a pacing cycle length of 200 ms. E, Compared with WT mice, Sorbs2e8/e8 mice RV action potentials have depolarized resting potentials (RPs), decreased action potential amplitudes (APAs), reduced upstroke velocity at phase 0 (Vmax, maximal velocity), and prolonged action potential durations at 50% (APD50) and 90% (APD90) repolarization, as well as a lengthened ventricular effective refractory period (VERP). N=6. Unpaired 2‐tailed Student t test. Bpm indicates beats per minute; HR, heart rate; P, duration of P waves; P‐R, duration of PR interval; QRS, duration of QRS complex; R+S, QRS amplitude; and S, duration of S wave.
Sorbs2e8/e8 Manifests Disrupted ICD Structure
Prompted by the subcellular expression of Sorbs2 in ICD, we examined whether Sorbs2 depletion affects the expression of other ICD proteins. Immunostaining of sectioned hearts confirmed the depletion of Sorbs2 expression in both ICD and Z discs in Sorbs2e8/e8 hearts at 4 months of age (Figure S5). Loss of Sorbs2 does not seem to affect the protein expression levels of desmosomal proteins, such as desmoglein 1/2 and plakoglobin (Figure 5A and Figure S5); however, the expression level of α‐catenin is significantly increased, whereas the gap function protein Cx34 is markedly reduced. Although the expression level of N‐cadherin remains largely unchanged, its dotted expression pattern, mostly observed in WT hearts, was changed to a continuous line in Sorbs2e8/e8 mutant hearts (Figure 5B) and from an alternating pattern of plakoglobin to a largely overlapping pattern (Figure S6). The downregulation of Cx43 impairs electrical impulse conduction and creates substrates for cardiac arrhythmias. 47 , 48 A recent study reported Sorbs2 as a potent RNA‐binding protein to stabilize tumor‐suppressor gene expression in ovarian cancer. 49 Because the protein level of Cx43 was markedly reduced in the Sorbs2e8/e8 mice hearts, we thus evaluated whether Sorbs2 protein could bind mRNA of Cx43/Gja. Indeed, using a targeted RNA immunoprecipitation assay, we detected drastic mRNA enrichment for Gja1 gene in the anti‐Sorbs2 antibody compared with the IgG control immunoprecipitated tissue lysates from a mouse heart (Figure 5C). In addition, the Gja1 transcript level was also significantly decreased in the Sorbs2e8/e8 mice hearts (Figure 5D), indicating that Sorbs2 protein might stabilize the Gja1 mRNA at normal condition. Together, these results implicated that Sorbs2 protein might regulate expression of other ICD proteins, such as Cx43, through functioning as an RNA binding protein.
Figure 5. Dual function of Sorbs2 (sorbin and SH3 domain‐containing 2) in maintaining intercalated disc (ICD) integrity and regulating the expression of α‐catenin and gap junction protein connexin 43 (Cx43).
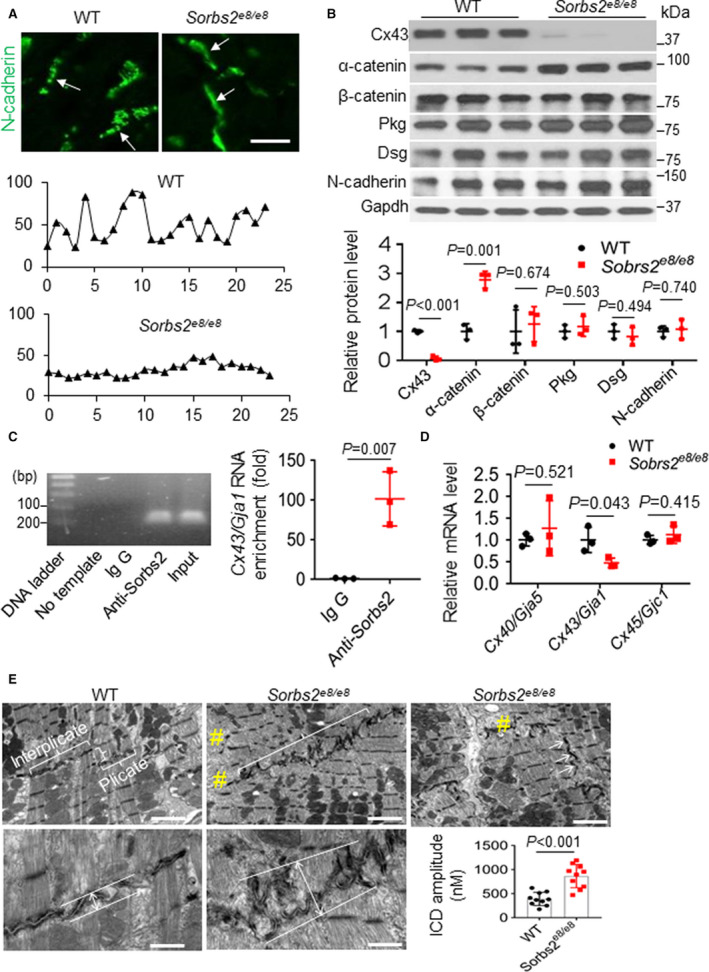
A, Shown are ICD images and represented pseudoline analysis of mouse hearts after immunostaining using an anti–N‐cadherin antibody. The expression of N‐cadherin in ICD manifests as discontinuous dots in wild‐type (WT) mice, which switched to continuous lines in Sorbs2e8/e8 mice (arrows). Bar=20 μm. The intensity distribution of the N‐cadherin signal along the ICD appears as wavy lines in WT mice, which largely changed to flat lines in the Sorbs2e8/e8 mice. B, Western blotting and quantification of ICD proteins in WT and Sorbs2e8/e8 knockout (KO) mice hearts at 4 months old. N=3. Unpaired 2‐tailed Student t test. C, A representative DNA gel and quantitative reverse transcription–polymerase chain reaction (RT‐PCR) analysis of the Cx43/Gja1 mRNA level in the anti‐Sorbs2 antibody and IgG control immunoprecipitated tissue lysates from right ventricle (RV) of WT mice. N=3. Unpaired 2‐tailed Student t test. D, Quantitative RT‐PCR analysis of the mRNA levels of the indicated genes in the RV tissue of Sorbs2e8/e8 mice compared with WT controls. N=3. Unpaired 2‐tailed Student t test. E, Transmission electron microscopy images and schematic illustration on measuring and quantifying the ICD amplitude in WT and Sorbs2e8/e8 KO mice hearts at 4 months old. #Disrupted myofibril. Bars for top panels=2 μm; bars for bottom panels=500 nm. N=10. Unpaired 2‐tailed Student t test. Dsg indicates desmoglein 1/2; and Pkg, plakoglobin.
At the ultrastructural level, ICD in a WT mouse heart is a unique structure at the longitudinal junction between 2 neighboring cardiomyocytes consisting of 2 regions: the interplicate region that is typically in parallel with Z discs along the myofibrils and the plicate region that connects the interplicate regions and is typically perpendicular to the interplicate and Z discs (Figure 5E). This highly organized ICD structure connects the myofibrils from 2 neighboring cardiomyocytes across the membrane, ensuring coordinated contraction. 50 In the Sorbs2e8/e8 mice hearts at 4 months old, however, there is no clear separation between the interplicate region and the plicate region (Figure 5E). The ICDs are no longer in parallel with the Z discs but are slanted and intersect with Z discs. The ICDs also appear less flat, and the amplitude of folding is significantly increased. As a consequence, the loss of sarcomere structures in the connecting region between myofibrils and ICDs is frequently detected (Figure 5E).
Identification and Functional Validation of Pathogenic Variants in the SORBS2 Gene Found in Human Patients With ACM
Given the cardiac‐enriched expression of Sorbs2 protein, its subcellular expression pattern in ICDs, and ACM‐like phenotype in Sorbs2e8/e8 mice, we postulated that SORBS2 may represent a previously unrecognized ACM‐susceptibility gene. To test this hypothesis, we performed Sanger sequencing in 59 unrelated Han Chinese patients with ACM and whole exon sequencing in 402 healthy controls to identify potential pathogenic variants in the coding region of the SORBS2 gene. Patients with ACM were significantly younger and featured larger RV interior diameter and right atrial interior diameter than ostensibly healthy controls (Table S2). Two rare variants (c.679+1G>T, c.869+1C>G) classified as likely pathogenic according to the 2015 American College of Medical Genetics and Genomics guidelines were identified in the SORBS2 gene in 2 unrelated patients with ACM who met the 2019 HRS expert consensus statement (Figure 6A, Table 2, Figure S7A, and Table S3). 42 , 43 Both likely pathogenic SORBS2 variants are splicing variants predicted to affect splicing donors and/or acceptors, potentially leading to the functional disruption of the SORBS2 protein. Neither of these variants was reported in the Human Gene Mutation Database nor in ClinVar database.
Figure 6. Identification and functional validation of an SORBS2 (sorbin and SH3 domain‐containing 2) splicing variant found in a human patient with arrhythmogenic cardiomyopathy (ACM).
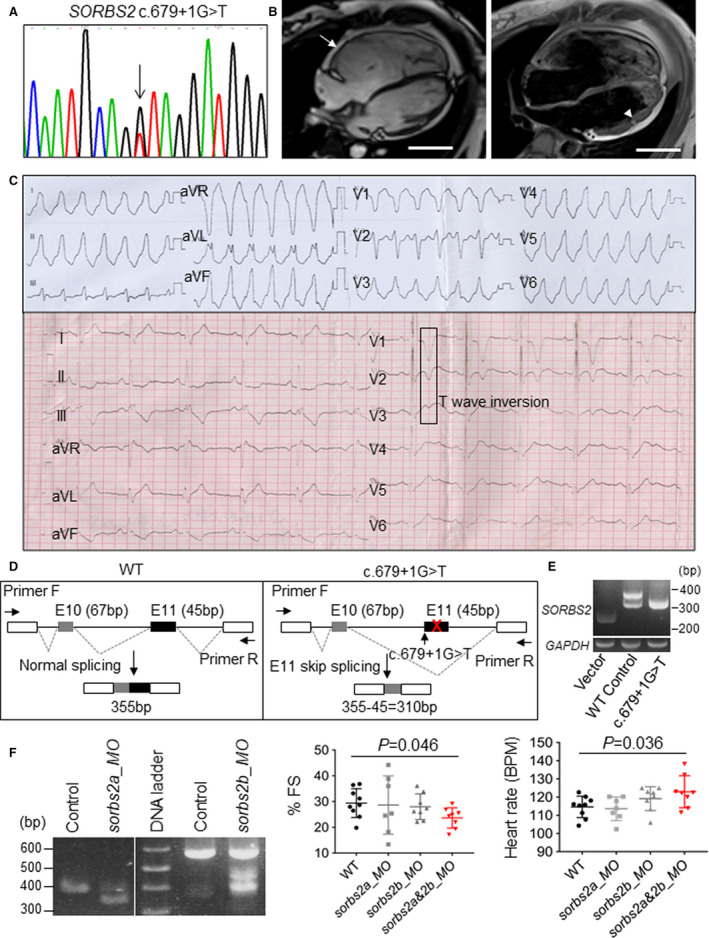
A, Chromagraph illustrates c.679+1G>T mutation identified in the SORBS2 gene from a patient with ACM. B, Magnetic resonance images of the patient harboring the c.679+1G>T mutation indicates right ventricle dilation (arrow) and fatty infiltration (arrowhead). Bar=5 cm. C, ECGs from the patient with ACM harboring the c.679+1G>T mutation. Ventricular tachycardia and T‐wave inversion were noted. D, Schematics of the minigene and reverse transcription–polymerase chain reaction (RT‐PCR) analysis to test that the splicing mutant c.679+1G>T leads to exon 11 skip in the HEK 293 cell culture system. E, RT‐PCR assessment of molecular lesions induced by sorbs2a or sorbs2b_ morpholino (MO) in zebrafish embryos. A predicted 352‐bp PCR band was detected in embryos injected with sorbs2a_MO because of an exon skipping effect. A predicted 447‐bp PCR band was detected in embryos injected with sorbs2b MO because of an intron retention effect. F, Fractional shortening (FS) and heart rate analysis in sorbs2a_MO‐, sorbs2b_MO‐, and sorbs2a&2b_MO‐injected morphants and uninjected controls. N=7 to 9. One‐way ANOVA. BPM indicates beats per minute; and WT, wild type.
Table 2.
Identification of Potentially Pathogenic SORBS2 Variants in 59 ACM Cases
| No. | Coding Change | Protein Change | Variant Type | gnomAD MAF | ACMG Classification | ACMG Criteria |
|---|---|---|---|---|---|---|
| 1 | c.679+1G>T | … | Splice site | Absent | Likely pathogenic | PVS1+PM2 |
| 1 | c.869+1C>G | … | Splice site | Absent | Likely pathogenic | PVS1+PM2 |
ACM indicates arrhythmogenic cardiomyopathy; ACMG, American College of Medical Genetics and Genomics; gnomAD, Genome Aggregation Database; MAF, minor allele frequency; No., number of related patients; PM, pathogenic moderate; PVS, pathogenic very strong; and SORBS2, sorbin and SH3 domain‐containing 2.
As a control, we performed both targeted sequencing, including 13 genes listed in Figure S7B, and Sanger sequencing for the CTNNA3 gene to scan known ACM genes for all the 59 patients. We identified variants for these 14 known ACM genes in 37 patients (data not shown). Among the 2 ACM patients carrying likely pathogenic SORBS2 splicing variants, the patient carrying the c.679+1G>T variant did not have any other pathogenic variants for known ACM causative genes (Table S4). This patient was a 42‐year‐old man who presented with palpitations during exercise. VT and right precordial lead (V1–V3) T‐wave inversions were detected by ECG, and enlarged RV with dyskinesia and RV fibrofatty infiltration were observed on MRI (Figure 6B and 6C). The other patient carrying the c.869+1C>G variant harbors a nonsense variant in the PKP2 gene (c.2421C>A, p. Tyr807Ter), a known ACM causative gene (Table S4). This latter patient was a 27‐year‐old man who presented with palpitations, manifesting significant RV dilation with dyskinesia and VT. In light of the marked incomplete penetrance and variable expressivity of PKP2 truncating variants and fully manifest ACM phenotype of this patient, we cannot rule out the possibility this patient's phenotype is secondary to digenic heterozygosity involving both PKP2 and SORBS2.
To functionally prove pathogenicity of the SORBS2 variants, we decided to focus on the c.679+1G>T splicing variant that was identified from the patient with ACM without other known ACM gene mutations. We first conducted a minigene hybridization assay and confirmed that the c.679+1G>T mutation is able to induce exon skipping events in HEK293 cells (Figure 6D). The loss of exon 11 putatively disrupts the Sorbin homolog domain and results in SORBS2 protein loss of function. To further assess the pathogenicity of the c.679+1G>T point mutation at the organ level, we mimicked the genetic lesion in a zebrafish embryo by using morpholino technology. The targeted sites are exon 6 in sorbs2a (ENSDART00000138475.2) and exon 10 in sorbs2b (ENSDART00000142605.3), 2 exons corresponding to exon 11 in human SORBS2, as evidenced by remarkable sequence conservation in the surrounding exons (Figure S8). The injection of the sorbs2a morpholino caused the skipping of exon 6 in the sorbs2a RNA transcript, whereas the injection of the sorbs2b morpholino caused intron retention, resulting in a premature stop codon that presumably leads to the truncation of the Sorbs2b protein (Figure 6D and 6E). Although the injection of sorbs2a and sorbs2b morpholino alone did not cause obvious abnormalities, the coinjection of both sorbs2a and sorbs2b morpholinos led to significantly decreased fractional shortening and increased heart rate (Figure 6F), confirming the pathogenicity of this splicing variant.
DISCUSSION
Sorbs2e8/e8 Manifests Several Key Features Reminiscent of Human ACM
Using both zebrafish and mouse models, we provided first in vivo evidence to reveal cardiac dominant expression and functions of Sorbs2. Prompted by our recent studies that identified GBT002/sorbs2b as a deleterious modifier for DIC in zebrafish, we demonstrated that this effect is mostly ascribed to disrupted expression of sorbs2b transcripts in the myocardium. We went on to show that this DIC‐modifying effect is conserved in the Sorbs2e8/+ knockout mouse, and uncovered additional phenotypes that cannot be revealed in the simple zebrafish model. Sorbs2e8/e8 knockout mice manifest RV remodeling and dysfunction, spontaneous sustained and nonsustained VT, and premature death at ≈3.5 months of age. These phenotypes resemble characteristic features of other well‐established mouse model of ACM, such as those based on Dsg2, Jup, and Dsp genes. 18 , 20 , 51 , 52 In patients with ACM, SCD and ventricular arrhythmia are often seen at the early phase when there is no obvious heart structural remodeling. 53 Similar progressive pathogenesis was observed in Sorbs2e8/e8 mice. Spontaneous and induced ventricular arrhythmias are noted as early as 2 months of age in Sorbs2e8/e8 mice (data not shown). Obvious RV remodeling occurs later, followed by LV dilation and dysfunction at ≈4 months. Electrophysiology experiments identified multiple levels of abnormalities. First, the ECG abnormalities with prolonged and bifid P waves and right bundle branch block were consistent with atrial and ventricular conduction abnormalities. Second, spontaneous atrial and ventricular arrhythmias were recorded in anesthetized Sorbs2e8/e8 mice. Third, microelectrode studies showed ventricular depolarized resting potentials, reduced action potential upstroke dV/dts, reduced action potential amplitudes, and prolonged ADP50s, ADP90s, and VERPs, indicating potential derangements in a multitude of ion channels, including those encoding IK1, INa, Ito, and IK. Fourth, Sorbs2‐deficient hearts are susceptible to the induction of VT, which may involve reentry and trigger activity in mechanisms. Thus, this study indicated that Sorbs2e8/e8 mouse might serve as a novel murine model for ACM with a rapid pathological progression, which can be used to further decipher underlying mechanism of a wide range of electrophysiological abnormalities that promote the development of arrhythmias, prominent RV remodeling, and premature death.
Although Sorbs2e8/e8 mice manifest several key features reminiscent of human ACM, fibrofatty infiltration, a pathogenic hallmark for human ACM, was not prominent in the Sorbs2e8/e8 mouse model. Notably, fibrofatty infiltration was not present in most of the mouse ACM models for other well‐established ACM genes, such as JUP, DSG2, and PKP. 20 , 51 The only mouse ACM model with a fibrofatty infiltration phenotype is the Dsp‐deficient mouse. 20 , 21 The species difference has been postulated to explain this discrepancy because a mouse heart does not show evidence of epicardial fat, as typically observed in a human heart. Different from other classic murine ACM models, our Sorbs2e8/e8 mouse also manifests unique phenotypes, such as septum bending and fibrosis and left atrium enlargement, whereas robust fibrosis was not detected in the RV. It would be interesting to investigate mechanisms underlying these unique phenotypes in the future. Currently, there are 2 small molecules: SB216763, a potent and selective inhibitor of glycogen synthase kinase‐3 β; and BAY117082, an inhibitor of nuclear factor‐κB signaling, known to correct phenotypes in both zebrafish and mouse ACM models with mutated plakoglobin or Dsg2 gene. 14 , 23 , 54 It would also be interesting to treat the Sorbs2e8/e8 mice with these 2 small molecules to see whether they can reverse and/or prevent the ACM‐like phenotypes resulted from Sorbs2 gene knockout as well.
ICD Expression and Function of Sorbs2 Could be Primary to the ACM‐Like Phenotypes
That the Sorbs2e8/e8 knockout mouse manifests several key features of ACM provides compelling evidence to support the concept that ACM is a disease of ICD. In contrast to the sarcomeric Z disc localization of Sorbs2 protein that has been reported before, herein, we detected its cardiac enriched expression pattern and a strong subcellular localization to the ICD/adherens junction within cardiomyocytes. Compared with other well‐established ACM‐susceptibility genes that encode desmosome proteins, Sorbs2 encodes an ICD protein that is predominantly localized to the adherens junction but imparts its effects on gap junctional proteins. Notably, the idea that these different ICD components are individual structures has been recently challenged, and they actually work together and share many accessory proteins. 55 New terms, such as area composita, that consist of both adherens junction and desmosome are favored. Consistent with the new concept, we noted similar ICD structural disruption in Sorbs2e8/e8 mice, as often observed in ACM mouse models and human patients, including the breakdown of the ICD structure, a widened desmosome gap, and a mislocated desmosome. 18 , 56 We also noted the loss of myofibrils in the proximity of the ICD region, as has been reported in Boxer dogs with ACM. 57 Thus, it is plausible that disruption of structural integrity of ICD is the primary damage that incurs sequential pathological events and leads to ACM phenotypes in the Sorbs2e8/e8 mice.
At the cellular and molecular levels, altered expression patterns and/or abundancy of other ICD proteins, such as a change of dotted subcellular localization pattern of N‐cadherin with plakoglobin to overlapping pattern, significantly elevated expression of α‐catenin, whereas downregulation of Cx43 protein was detected in the Sorbs2e8/e8 mouse heart. Given the well‐established role of α‐catenin as a core component of E‐cadherin–catenin complex that is required for maintaining the integrity of adherens junction, 58 and that a marked reduction in Cx43 has been found in many patients with ACM, 47 , 59 , 60 it is highly likely that the disturbed expression patterns and/or abundancy of N‐cadherin, α‐catenin, and Cx43 would contribute to the disrupted ICD structural integrity and subsequent arrhythmic phenotype in Sorbs2e8/e8 mouse. Mechanistically, our results from RNA immunoprecipitation assay provided evidence to support the hypothesis that Sorbs2, in addition to being an ICD structural protein, might also function as an RNA binding protein to regulate the transcripts of other ICD genes. Further studies along this direction are warranted. It would also be worth investigating whether the Sorbs2 regulates other known ACM pathways, such as Wnt/β‐catenin, Yes‐associated protein, and/or mechanical force transmission, in the future. 20 , 61
SORBS2 Is a Potential ACM‐Susceptibility Candidate Gene
Encouraged by the ACM‐like phenotypes of the Sorbs2e8/e8 mouse, we conducted preliminary human genetic studies to test whether SORBS2 is a candidate ACM‐susceptibility gene. Two rare SORBS2 variants, classified as likely pathogenic according to the 2015 American College of Medical Genetics and Genomics guidelines, were identified in a cohort of 59 Han Chinese patients with ACM. Functional validation studies in the zebrafish model support the potential pathogenicity of c.679+1G>T splice variant and suggest that SORBS2 loss‐of function variants may contribute to ACM pathogenesis.
However, we would like to emphasize that the human genetic studies in this work were conducted in sporadic ACM probands and should be considered preliminary in nature. Unfortunately, samples for family members of these patients are not available, which precludes cosegregation analysis. Future studies in much larger ACM cohorts are needed to definitively establish SORBS2 as an ACM‐susceptibility gene. If confirmed, SORBS2 would represent the first ACM gene that was initially identified from phenotypic studies in animal models, in contrast to other ACM genes that were mostly discovered through human genetic studies. Because mutant lines for most genes in either zebrafish or mouse genome have been already generated, it is anticipated that phenotyping efforts in these animal models could result in the identification of additional ACM‐susceptibility genes.
Sources of Funding
This work was supported by grants from National Institutes of Health (R01‐HL107304, R01‐HL081753, and R01‐GM63904) and the Mayo Foundation to Dr Xu; The Innovative Basic Science Award (1‐16‐IBS‐195) from the American Diabetes Association and the Prospective Research Award from the Mayo Clinic Department of Cardiovascular Diseases to Dr Lu; Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program to Drs Ackerman, Guidicessi, and Tester; and National Institutes of Health (R01‐HL074180) and the Mayo Foundation to Dr Lee.
Disclosures
None.
Supporting information
Tables S1–S5
Figures S1–S8
Acknowledgments
We thank Dr Prasanna Mishra of the Mayo Nuclear Magnetic Resonance Lab for guidance with magnetic resonance imaging in mice, Ronald May for performing echocardiography in mice, the Mayo Clinic Electron Microscopy Core Facility for transmission electron microscopy in mice, and the Mayo Histology Core Facility for performing Masson trichrome staining.
(J Am Heart Assoc. 2020;9:e017055 DOI: 10.1161/JAHA.119.017055.)
Preprint posted on BioRxiv August 5, 2019. doi: https://doi.org/10.1101/725077.
Jingchun Yang is currently located at the Gastrointestinal Department, Mayo Clinic.
For Sources of Funding and Disclosures, see page 17.
Contributor Information
Dao Wen Wang, Email: dwwang@tjh.tjmu.edu.cn.
Xiaolei Xu, Email: xu.xiaolei@mayo.edu.
References
- 1. Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72:784–804. [DOI] [PubMed] [Google Scholar]
- 2. Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593–1601. [DOI] [PubMed] [Google Scholar]
- 3. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. [DOI] [PubMed] [Google Scholar]
- 4. Bennett RG, Haqqani HM, Berruezo A, Della Bella P, Marchlinski FE, Hsu CJ, Kumar S. Arrhythmogenic cardiomyopathy in 2018–2019: ARVC/ALVC or both? Heart Lung Circ. 2019;28:164–177. [DOI] [PubMed] [Google Scholar]
- 5. McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119–2124. [DOI] [PubMed] [Google Scholar]
- 6. Roberts JD, Murphy NP, Hamilton RM, Lubbers ER, James CA, Kline CF, Gollob MH, Krahn AD, Sturm AC, Musa H, et al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J Clin Invest. 2019;129:3171–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vimalanathan AK, Ehler E, Gehmlich K. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophys Rev. 2018;10:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. [DOI] [PubMed] [Google Scholar]
- 9. Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. [DOI] [PubMed] [Google Scholar]
- 10. Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, SeN‐chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, Kotta MC, Chin A, Laing N, Ntusi NB, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10:e001605. [DOI] [PubMed] [Google Scholar]
- 13. van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, et al. Mutations in the area composita protein alphat‐catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–210. [DOI] [PubMed] [Google Scholar]
- 14. Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation. 2019;140:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 2014;114:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kant S, Holthofer B, Magin TM, Krusche CA, Leube RE. Desmoglein 2-dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ Cardiovasc Genet. 2015;8:553–563. [DOI] [PubMed] [Google Scholar]
- 17. Lodder EM, Rizzo S. Mouse models in arrhythmogenic right ventricular cardiomyopathy. Front Physiol. 2012;3:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet. 2014;23:1134–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Padron-Barthe L, Dominguez F, Garcia-Pavia P, Lara-Pezzi E. Animal models of arrhythmogenic right ventricular cardiomyopathy: what have we learned and where do we go? Insight for therapeutics. Basic Res Cardiol. 2017;112:50. [DOI] [PubMed] [Google Scholar]
- 20. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta‐catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–655. [DOI] [PubMed] [Google Scholar]
- 22. SeN‐chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927–935. [DOI] [PubMed] [Google Scholar]
- 23. Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding Y, Long PA, Bos JM, Shih YH, Ma X, Sundsbak RS, Chen J, Jiang Y, Zhao L, Hu X, et al. A modifier screen identifies dnajb6 as a cardiomyopathy susceptibility gene. JCI Insight. 2016;1:e88797 DOI: 10.1172/jci.insight.88797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang B, Golemis EA, Kruh GD. ArgBP2, a multiple Src homology 3 domain‐containing, Arg/Abl-interacting protein, is phosphorylated in v-Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J Biol Chem. 1997;272:17542–17550. [DOI] [PubMed] [Google Scholar]
- 27. Anekal PV, Yong J, Manser E. Arg kinase-binding protein 2 (ArgBP2) interaction with alpha-actinin and actin stress fibers inhibits cell migration. J Biol Chem. 2015;290:2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cestra G, Toomre D, Chang S, De Camilli P. The Abl/Arg substrate ArgBP2/nArgBP2 coordinates the function of multiple regulatory mechanisms converging on the actin cytoskeleton. Proc Natl Acad Sci USA. 2005;102:1731–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ronty M, Taivainen A, Moza M, Kruh GD, Ehler E, Carpen O. Involvement of palladin and alpha-actinin in targeting of the Abl/Arg kinase adaptor ArgBP2 to the actin cytoskeleton. Exp Cell Res. 2005;310:88–98. [DOI] [PubMed] [Google Scholar]
- 30. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, et al. Cardiac fibroblast-derived microrna passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Molck MC, Simioni M, Paiva Vieira T, Sgardioli IC, Paoli Monteiro F, Souza J, Fett-Conte AC, Felix TM, Lopes Monlleo I, Gil-da-Silva-Lopes VL. Genomic imbalances in syndromic congenital heart disease. J Pediatr (Rio J). 2017;93:497–507. [DOI] [PubMed] [Google Scholar]
- 32. Kakimoto Y, Ito S, Abiru H, Kotani H, Ozeki M, Tamaki K, Tsuruyama T. Sorbin and SH3 domain‐containing protein 2 is released from infarcted heart in the very early phase: proteomic analysis of cardiac tissues from patients. J Am Heart Assoc. 2013;2:e000565 DOI: 10.1161/JAHA.113.000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Q, Gao X, Li C, Feliciano C, Wang D, Zhou D, Mei Y, Monteiro P, Anand M, Itohara S, et al. Impaired dendritic development and memory in Sorbs2 knock-out mice. J Neurosci. 2016;36:2247–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Hoage T, Bai P, Ding Y, Chen Z, Zhang R, Huang W, Jahangir A, Paw B, Li YG, et al. Cardiac hypertrophy involves both myocyte hypertrophy and hyperplasia in anemic zebrafish. PLoS One. 2009;4:e6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu W, Chen J, Ji S, Allen JS, Bayly PV, Wickline SA, Yu X. Harmonic phase MR tagging for direct quantification of lagrangian strain in rat hearts after myocardial infarction. Magn Reson Med. 2004;52:1282–1290. [DOI] [PubMed] [Google Scholar]
- 36. Jiang K, Jiao S, Vitko M, Darrah R, Flask CA, Hodges CA, Yu X. The impact of cystic fibrosis transmembrane regulator disruption on cardiac function and stress response. J Cyst Fibros. 2016;15:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kovalova S, Necas J, Cerbak R, Malik P, Vespalec J. Echocardiographic volumetry of the right ventricle. Eur J Echocardiogr. 2005;6:15–23. [DOI] [PubMed] [Google Scholar]
- 38. Lu T, Jiang B, Wang XL, Lee HC. Coronary arterial bk channel dysfunction exacerbates ischemia/reperfusion-induced myocardial injury in diabetic mice. Appl Physiol Nutr Metab. 2016;41:992–1001. [DOI] [PubMed] [Google Scholar]
- 39. Yi F, Ling TY, Lu T, Wang XL, Li J, Claycomb WC, Shen WK, Lee HC. Down-regulation of the small conductance calcium-activated potassium channels in diabetic mouse atria. J Biol Chem. 2015;290:7016–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart: a challenge to the critical mass hypothesis. Circ Res. 1999;85:174–181. [DOI] [PubMed] [Google Scholar]
- 41. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Panchatcharam M, Bhuiyan MAN, Peretik JM, Orr AW, James J, Osinska H, et al. Cardiac dysfunction in the sigma 1 receptor knockout mouse associated with impaired mitochondrial dynamics and bioenergetics. J Am Heart Assoc. 2018;7:e009775 DOI: 10.1161/JAHA.118.009775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: executive summary. Heart Rhythm. 2019;16:e373–e407. [DOI] [PubMed] [Google Scholar]
- 43. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoage T, Ding Y, Xu X. Quantifying cardiac functions in embryonic and adult zebrafish. Methods Mol Biol. 2012;843:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clark KJ, Balciunas D, Pogoda HM, Ding Y, Westcot SE, Bedell VM, Greenwood TM, Urban MD, Skuster KJ, Petzold AM, et al. In vivo protein trapping produces a functional expression codex of the vertebrate proteome. Nat Methods. 2011;8:506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fontes MS, van Veen TA, de Bakker JM, van Rijen HV. Functional consequences of abnormal Cx43 expression in the heart. Biochim Biophys Acta. 2012;1818:2020–2029. [DOI] [PubMed] [Google Scholar]
- 48. van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. [DOI] [PubMed] [Google Scholar]
- 49. Zhao L, Wang W, Huang S, Yang Z, Xu L, Yang Q, Zhou X, Wang J, Shen Q, Wang C, et al. The RNA binding protein SORBS2 suppresses metastatic colonization of ovarian cancer by stabilizing tumor-suppressive immunomodulatory transcripts. Genome Biol. 2018;19:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wilson AJ, Schoenauer R, Ehler E, Agarkova I, Bennett PM. Cardiomyocyte growth and sarcomerogenesis at the intercalated disc. Cell Mol Life Sci. 2014;71:165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kirchhof P, Fabritz L, Zwiener M, Witt H, Schafers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–1806. [DOI] [PubMed] [Google Scholar]
- 53. Asimaki A, Kleber AG, Saffitz JE. Pathogenesis of arrhythmogenic cardiomyopathy. Can J Cardiol. 2015;31:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D, Jasti R, MacRae CA, Leber R, Kleber AG, et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. 2016;1:e85923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vermij SH, Abriel H, van Veen TA. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc Res. 2017;113:259–275. [DOI] [PubMed] [Google Scholar]
- 56. Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon A, Wozniek O, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. [DOI] [PubMed] [Google Scholar]
- 57. Oxford EM, Danko CG, Kornreich BG, Maass K, Hemsley SA, Raskolnikov D, Fox PR, Delmar M, Moise NS. Ultrastructural changes in cardiac myocytes from boxer dogs with arrhythmogenic right ventricular cardiomyopathy. J Vet Cardiol. 2011;13:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–514. [DOI] [PubMed] [Google Scholar]
- 59. Asimaki A, Protonotarios A, James CA, Chelko SP, Tichnell C, Murray B, Tsatsopoulou A, Anastasakis A, te Riele A, Kleber AG, et al. Characterizing the molecular pathology of arrhythmogenic cardiomyopathy in patient buccal mucosa cells. Circ Arrhythm Electrophysiol. 2016;9:e003688 DOI: 10.1161/CIRCEP.115.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Saffitz JE, Asimaki A, Huang H. Arrhythmogenic right ventricular cardiomyopathy: new insights into mechanisms of disease. Cardiovasc Pathol. 2010;19:166–170. [DOI] [PubMed] [Google Scholar]
- 61. Hu Y, Pu WT. Hippo activation in arrhythmogenic cardiomyopathy. Circ Res. 2014;114:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figures S1–S8