

Abstract
Background
The development of pathological cardiac hypertrophy involves the coordination of a series of transcription activators and repressors, while their interplay to trigger pathological gene reprogramming remains unclear. NULP1 (nuclear localized protein 1) is a member of the basic helix‐loop‐helix family of transcription factors and its biological functions in pathological cardiac hypertrophy are barely understood.
Methods and Results
Immunoblot and immunostaining analyses showed that NULP1 expression was consistently reduced in the failing hearts of patients and hypertrophic mouse hearts and rat cardiomyocytes. Nulp1 knockout exacerbates aortic banding‐induced cardiac hypertrophy pathology, which was significantly blunted by transgenic overexpression of Nulp1. Signal pathway screening revealed the nuclear factor of activated T cells (NFAT) pathway to be dramatically suppressed by NULP1. Coimmunoprecipitation showed that NULP1 directly interacted with the topologically associating domain of NFAT3 via its C‐terminal region, which was sufficient to suppress NFAT3 transcriptional activity. Inactivation of the NFAT pathway by VIVIT peptides in vivo rescued the aggravated pathogenesis of cardiac hypertrophy resulting from Nulp1 deficiency.
Conclusions
NULP1 is an endogenous suppressor of NFAT3 signaling under hypertrophic stress and thus negatively regulates the pathogenesis of cardiac hypertrophy. Targeting overactivated NFAT by NULP1 may be a novel therapeutic strategy for the treatment of pathological cardiac hypertrophy and heart failure.
Keywords: aortic banding, heart failure, NFAT3 signaling, nuclear localized protein 1, pathological cardiac hypertrophy
Subject Categories: Heart Failure, Hypertrophy, Remodeling
Nonstandard Abbreviations and Acronyms
- AB
aortic banding
- Ad
adenoviral
- Ang II
angiotensin II
- CAT
chloramphenicol acetyltransferase
- Col1
Collagen Type I
- Col3
Collagen Type III
- Ctgf
connective tissue growth factor
- Gapdh
glyceraldehyde‐3‐phosphate dehydrogenase
- LVEDd
left ventricular end‐diastolic diameter
- MCM
MerCreMer
- Myh7
β‐myosin heavy chain
- NFAT
nuclear factor of activated T cells
- NRVM
neonatal rat ventricular myocyte
- NULP1
nuclear localized protein 1
- PE
phenylephrine
- WGA
wheat germ agglutinin
- α/β‐MHC
α/β‐myosin heavy chain
Clinical Perspective
What Is New?
NULP1 (nuclear localized protein 1) expression is consistently decreased in heart samples from patients with heart failure and in heart tissues from mice with aortic banding‐induced cardiac hypertrophy.
NULP1 negatively regulates nuclear factor of activated T cells (NFAT) signaling independent of calcineurin via specifically interacts with NFAT3 and suppresses its overactivated transcriptional activity under hypertrophic stress.
Inactivation of NFAT by inhibitor VIVIT reversed the exacerbated hypertrophic pathology after Nulp1 deficiency.
What Are the Clinical Implications?
The development of cardiac hypertrophy involves the coordination of a series of transcription activators and repressors, whereas their interplay to trigger pathological gene reprogramming remains unclear.
We identify a transcription repressor, NULP1, acts as a negative regulator of pathological cardiac hypertrophy by suppressing NFAT3 transcriptional activity.
Targeting overactivated NFAT by NULP1 may be a novel therapeutic strategy for the treatment of pathological cardiac hypertrophy and heart failure.
Cardiac hypertrophy occurs in response to a wide array of stimuli, including neurohumoral factors, hemodynamic stress, myocardial infarction, and viral infection. However, sustained cardiac hypertrophy leads to maladaptive gene expression reprogramming, myocardial remodeling, and finally to heart failure, 1 , 2 a disease currently lacking efficient therapy. Although considerable progress has been made in elucidating the molecular mechanisms underlying the pathophysiology of cardiac hypertrophy, much remains unknown.
The development of cardiac hypertrophy is characterized by pathological cardiomyocyte growth driven by reprogrammed gene expression. This process involves the activation and modulation of numerous signaling pathways and transcription factors, 3 , 4 among which the calcineurin/nuclear factor of activated T cells (NFAT) pathway plays a central role in coordinating complex signaling events, 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 In human hypertrophic myocardium, the calcineurin/NFAT signaling pathway is activated as a result of abnormal Ca2+ homeostasis. 13 Transgenic induction of calcineurin or NFAT expression in hearts causes concentric hypertrophy, 5 , 14 which can be reversed by pharmaceutically inhibiting calcineurin or NFAT activity. 15 , 16 , 17 , 18 However, global deletion of the regulatory subunit (B1) of calcineurin or the NFAT3/NFAT4 isoforms leads to embryonic lethality, 19 and cardiac‐specific knockout of calcineurin B1 impairs heart development and function, 20 suggesting an indispensable physiological function of the calcineurin/NFAT pathway. Thus, complete inhibition of calcineurin/NFAT would not be a promising drug‐targeting strategy to treat cardiac hypertrophy. How calcineurin/NFAT activity is activated and how it can be fine‐tuned in the context of hypertrophic cardiomyopathy remain to be elucidated.
NULP1 (nuclear localized protein 1, also called transcription factor 25, TCF25) is a member of the basic helix‐loop‐helix family of transcription factors that are important in embryonic development. 21 NULP1 expression is highly enriched in adult hearts but is substantially decreased in postischemic heart failure. 22 , 23 As a transcription repressor, NULP1 has been shown to modulate the transcriptional activity of serum response factor, 23 a transcription factor involved in heart development and heart diseases. Here, we identify NULP1 as a negative regulator of cardiac hypertrophy by gain‐and‐loss‐of‐function analyses. This effect is independent of calcineurin but depends on a direct interaction with NFAT3 and subsequent repression of its transcriptional activity. The findings of this study reveal an endogenous suppressor of NFAT signaling that is dynamically regulated under stress and provide a novel therapeutic strategy to target overactivated NFAT in cardiac hypertrophy.
Methods
In adherence to the Transparency and Openness Promotion Guidelines, the data that support the findings of this study are available from the corresponding author upon reasonable request.
Human Heart Samples
Failing human heart specimens were procured from patients with end‐stage heart failure secondary to dilated cardiomyopathy during heart transplantation. Normal heart samples were obtained from donors who died in accidents, and their hearts were unsuitable for transplantation for noncardiac reasons (Table S1). Written informed consents were obtained from all participants before heart sample collection. All procedures involving human tissue use were approved by the ethical review board of Renmin Hospital of Wuhan University and were in accordance with the principles outlined in the Declaration of Helsinki.
Genetically Engineered Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee of Renmin Hospital of Wuhan University and were in accordance with the criteria of the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
To generate Nulp1 knockout (Nulp1‐KO) mice, we predicted and constructed the guiding sequences that target exon 1 of the Nulp1 gene in the mouse genome by using an online CRISPR design tool (http://crispr.mit.edu). A pair of oligomers (oligo1: TAGGGCTCAAACCGGTTGTTGACA and oligo2: AAACTGTCAACAACCGGTTTGAGC) were annealed and cloned into the BsaI restriction site of the pUC57‐sgRNA expression vector (Addgene 51132). After purification, the sgRNA was transcribed using the MEGAshortscript™ T7 Kit (Ambion, AM1354) and subsequently purified with the miRNeasy Micro Kit (Qiagen, 217084). The Cas9 expression plasmid (Addgene 44758) was linearized by PmeI and served as the template for in vitro transcription by using the T7 Ultra Kit (Ambion, AM1345). Cas9 mRNA was purified with the RNeasy Mini Kit (Qiagen, 74104). Subsequently, the mixture of Cas9 and sgRNA mRNA was injected into 1‐cell embryos (C57BL/6J) with the FemtoJet 5247 microinjection system. Founder identification was performed via polymerase chain reaction (PCR) of mouse tail genomic DNA. A 318‐bp DNA fragment including the sgRNA target site was amplified by PCR using the following primers: Nulp1‐forward (5′‐TGTCCTCCTTGATGACGATG‐3′) and Nulp1‐reverse (5′‐TTTCTTCCACCTTCCACTCG‐3′). The purified PCR product was denatured and reannealed in NEB Buffer 2 to constitute heteroduplex DNA, followed by digestion with T7EN (NEB, M0302L) and analysis through agarose gel electrophoresis. Sanger sequencing was applied to identify precise mutations of the indels in the engineered gene. Then, the appropriate founder was used to produce offspring. Finally, the F1 and F2 offspring were further analyzed via the PCR by using following primers: forward(GAGGAAGAGGGACCAAAACC)and reverse(GTCATGCCCCCACCATTT)and Sanger sequencing method.
To generate transgenic mice with cardiac‐restricted overexpression of Nulp1 (Nulp1‐transgenic [TG]), the full‐length mouse Nulp1 cDNA was inserted downstream of the CAG‐CAT cassette expressing the chloramphenicol acetyltransferase (CAT) gene flanked by 2 loxP sites driven by the CAG promoter. Subsequently, the vector was linearized and microinjected into fertilized mouse embryos (C57BL/6J background) to produce the CAG‐CAT‐Nulp1 transgenic mice, which were identified via PCR analysis of their tail genomic DNA. Transgenic mice expressing a tamoxifen‐inducible Cre recombinase flanked on each end by a mutated murine estrogen receptor (Mer) ligand binding domain (MerCreMer; MCM) under the control of the cardiomyocyte‐specific α‐MHC (α‐myosin heavy chain) promoter were purchased from the Jackson Laboratory (α‐MHC‐MCM, 005650).The obtained CAG‐CAT‐Nulp1 mice were then crossed with α‐MHC‐MCM mice to generate α‐MHC‐MCM/CAG‐CAT‐Nulp1 double transgenic mice. To achieve cardiac‐specific Nulp1 overexpression, 6‐week‐old offspring were injected with tamoxifen (Sigma‐Aldrich, T5648, 25 mg/kg per day) for 5 consecutive days to induce Cre‐mediated excision of the loxP‐flanked CAT gene located between the CAG promoter and the Nulp1 gene.
Male mice aged 8 to 10 weeks and weighing 24 to 27 g were used for all subsequent experiments. All mice were maintained in a controlled environment with food and water available ad libitum.
Aortic Banding Surgery in Mouse
A mouse model of cardiac hypertrophy induced by pressure overload was established by aortic banding (AB) operation as previously described. First, mice anesthetization was achieved by intraperitoneal injection with pentobarbital (Sigma‐Aldrich, P3761). The left chest of each mouse was subsequently opened, and the thoracic aorta was accessed through the second intercostal space by blunt dissection. AB was achieved by banding the thoracic aorta against a 27 G (for body weights of 24–25 g) or 26 G (for body weights of 26–27 g) needle using a 7‐0 silk suture. Then, the needle was removed, and the chest cavity, muscle, and skin were closed layer by layer. A similar surgical procedure without aortic constriction were performed in the sham‐operated mice. The mice were continuously monitored until recovery.
VIVIT Administration
A synthetic cell‐permeable peptide VIVIT (Millipore, 480401) was administered to suppress the activity of NFAT as previously reported. 16 , 24 The protein powder was dissolved and then filtered with a 0.22‐μm membrane. VIVIT (10 mg/kg) was continuously administered every 2 days via subcutaneous injection immediately after AB surgery for 4 weeks. Mice given an equal volume of saline served as controls.
Echocardiographic Measurements
Echocardiographic analyses were performed using a Mylab 30CV ultrasound system (Biosound Esaote Inc.) equipped with a 15‐MHz transducer as previously described. First, the surviving mice were anesthetized by 2% isoflurane inhalation. Then, 2‐dimensional guided M‐mode tracings of cross sections of the minor axis of the left ventricle at the level of the papillary muscles were obtained to measure the parameters indicated below. The left ventricular end‐diastolic diameter (LVEDd) and left ventricular end‐systolic diameter (LVESd) were measured at the times of the largest and smallest left ventricular areas, respectively. These echocardiographic measurements were performed in triplicate and then averaged. Left ventricular fractional shortening (LVFS) was calculated using the following equation: LVFS (%)=(LVEDd−LVESd)/LVEDd×100%.
Histological Analyses
At the indicated times after sham or AB surgery, the mice were sacrificed and their hearts were removed. The removed mouse hearts were fixed in 10% formalin, embedded in paraffin, and subsequently transversely sectioned at a thickness of 5 µm. The heart sections were then subjected to hematoxylin and eosin staining for morphological analyses and picrosirius red staining for collagen deposition assessments. Fluorescein isothiocyanate‐conjugated WGA (wheat germ agglutinin; Invitrogen) staining was performed to demarcate cardiomyocyte boundaries, and DAPI was used to label the nuclei. The myocyte cross‐sectional area and LV collagen volume were measured using a quantitative digital image analysis system (Image‐Pro Plus 6.0). At least 100 circular‐to‐oval‐shaped LV cardiomyocytes were traced to determine the cardiomyocyte cross‐sectional area. The LV collagen volume was measured as the positive area of the picrosirius red staining and was expressed as a percentage of the total area. More than 40 fields in each group were analyzed.
Immunohistochemical Analysis
For immunohistochemistry staining, paraffin‐embedded human hearts were cut transversely into 5‐µm sections. After a 5‐minute high‐pressure antigen retrieval process in citrate buffer with a pH of 6.0, the heart sections were blocked with 10% bovine serum albumin for 60 minutes and then incubated overnight at 4°C with a NULP1‐specific antibody. Binding was visualized with the appropriate peroxidase‐conjugated secondary antibody for 60 minutes at 37°C.
Primary Neonatal Rat Ventricular Myocyte Culture, Adenovirus Transfection, and Immunofluorescence Staining
Primary neonatal rat ventricular myocytes (NRVMs) were isolated from 1‐ to 2‐day‐old Sprague Dawley rat hearts and seeded in 6‐well culture plates coated with gelatin. To overexpress wild‐type Nulp1, we cloned the full‐length rat Nulp1 under the control of a cytomegalovirus promoter into a replication‐defective adenoviral vector (AdNulp1). An empty adenoviral vector (AdVector) was used as a control. We used a rat short hairpin RNA targeting Nulp1 and Nfat3 adenoviral constructs (AdshNulp1 and AdshNfat3) to silence Nulp1 and Nfat3 gene expression. AdshRNA was used as a non‐targeting control. NRVMs were infected with the corresponding adenoviruses at a multiplicity of infection of 100 for 24 hours. After 48 hours of cultivation, the culture medium cells were deprived of serum‐free DMEM/F12 for another 12 hours and then subjected to angiotensin II (Ang II, 1 μmol/L) or phenylephrine (PE, 100 μmol/L) stimulation.
Immunofluorescence staining was performed to evaluate the morphology of NRVMs. We fixed the cardiomyocytes with 3.7% formaldehyde for 15 minutes at room temperature. After being washed 3 times, the cells were permeabilized with 0.1% Triton X‐100 in PBS for 5 minutes and stained with α‐actinin (A7811, Sigma‐Aldrich, 1:100 dilution). Binding was visualized with the indicated fluorescent secondary antibody (donkey anti‐mouse IgG [H+L] secondary antibody, A21202, Invitrogen, 1:200). DAPI stain marked the position of nuclei. The cell surface areas were measured using a quantitative digital image analysis system (Image‐Pro Plus 6.0).
Cignal 45‐Pathway Reporter Array
A Cignal 45‐Pathway Reporter Array (SABiosciences, CCA‐901L) was used to rapidly identify relevant pathways as previously described. 24 In brief, 200 ng GFP (green fluorescent protein) or NULP1 plasmid with 50 μL Opti‐MEM® was added to each well of the Cignal Finder Array plate. Subsequently, 0.6 μL of Attractene Transfection Reagent (Qiagen) in 50 μL of Opti‐MEM® per well was used for each individual transfection. After incubation for 5 minutes at room temperature, we added 50 μL of diluted Attractene into each well containing 50 μL of the diluted nucleic acids (1:1 ratio). The plate was set at room temperature for 20 minutes to allow complex formation. Subsequently, 50 μL of prepared H9C2 cell suspension was added to each well that contained constructs‐Attractene complexes. The cells were incubated for 16 hours, and the medium was then changed to complete growth medium. After another 24 hours cotransfection, the luciferase assay was performed using the Dual‐Luciferase Reporter Assay System from Promega.
Luciferase Reporter Assays
The luciferase reporter assay in NRVM was performed as previously described. 24 Adenovirus encoding an NFAT‐dependent luciferase reporter construct (Ad‐NFAT‐luc), which encodes 3 copies of an NFAT binding site (from the interleukin‐2 promoter) followed by the firefly luciferase reporter gene, was constructed. The Ad‐NFAT‐luc was used to infect NRVMs in combination with AdNulp1 or AdshNulp1 and their corresponding control virus for 24 hours. Cells were subsequently treated with Ang II for 24 hours, harvested, washed 3 times, and lysed in passive lysis buffer (Promega). The luciferase activity was determined using the Dual‐Luciferase Reporter Assay System (Promega).
The luciferase reporter assay in H9C2 cells was performed using established protocols. Briefly, plasmids encoding FLAG‐caNFAT1/NFAT3, NFAT‐responsive firefly luciferase reporter plasmids, and internal control Renilla luciferase reporter plasmids were cotransfected with plasmids expressing full‐length or truncated NULP1 using lipofectamine 2000 (11668500, Thermo Fisher Scientific). In the same experiment, empty control plasmids were added to ensure that each transfection received the same amount of total DNA. After 24 hours of transfection, cells were lysed in passive lysis buffer (Promega) and assayed for both firefly and Renilla luciferase activities using the Dual‐Luciferase Assay Kit (Promega). The fluorescent light emission was determined using GloMax® 20/20 Luminometer (Promega). Firefly luciferase units were normalized to Renilla luciferase controls.
Calcineurin Activity Assay
Calcineurin phosphatase activity was measured using the Calcineurin Phosphatase Activity Assay Kit (Abcam, ab139464), according to manufacturer's protocol. 25 Briefly, left ventricular tissues were lysed and desalted to prepare the test sample. Then, the total phosphatase activity was measured at OD620 nm and calculated against a phosphate standard curve. Calcineurin activity was determined by subtracting the “ethylene glycol tetraacetic acid buffer”‐sensitive phosphatase activity from the total phosphatase activity.
Coimmunoprecipitation Assay
A coimmunoprecipitation assay to determine protein‐protein interaction was performed as previously described. 26 Cultured HEK293T cells were cotransfected with plasmids encoding the indicated proteins using Turbo transfection reagent (Thermo Fisher Scientific, R0531). After 24 hours of transfection, the cells were lysed in an ice‐cold immunoprecipitation buffer (20 mmol/L Tris‐HCl [pH 7.4], 150 mmol/L NaCl, 1 mmol/L EDTA, and 1% Triton X‐100) supplemented with a protease inhibitor cocktail (Roche, 04693132001) and were subsequently centrifuged at 13 000g for 15 minutes. The obtained cell lysates were incubated with the indicated antibody and Protein G‐conjugated agarose beads (Roche, 11719416001) overnight at 4°C. Then, the immunocomplex was collected and washed with cold immunoprecipitation buffer and eluted from agarose beads. Finally, eluted proteins were subjected to immunoblotting using the indicated primary antibodies and the corresponding secondary antibodies.
Quantitative Real‐Time PCR
Total RNA was extracted from left ventricular tissues or cultured cells using TRIzol reagent (Invitrogen, 15596‐026). The obtained RNA samples were reverse‐transcribed to cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche, 04896866001). SYBR Green (Roche, 04887352001) was applied to quantify the mRNA levels of the indicated genes by quantitative real‐time PCR amplification. The mRNA levels of the target genes were normalized to the gene expression of glyceraldehyde‐3‐phosphate dehydrogenase (Gapdh) or β‐actin. The real‐time PCR primer pairs used in this study are listed in Table S2.
Western Blot Assay
Total protein from the heart tissue specimens or the cultured cells was extracted by RIPA lysis buffer. Highly enriched fractions of cytoplasmic and nuclear proteins were extracted using NE‐PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, 78833). The protein concentrations were determined using a BCA Protein Assay Kit (Pierce, 23225). The protein extracts were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore). After blocking with 5% fat‐free milk, the membranes were incubated with the indicated primary antibodies on a rocking platform overnight at 4°C. The membranes were then incubated with the corresponding peroxidase‐conjugated secondary antibodies for 1 hour at room temperature. The immunoreactive bands were visualized using an enhanced chemiluminescence detection kit (Bio‐Rad, 170‐5061), and the signals were detected and quantified using a Bio‐Rad ChemiDoc™ XRS+System (Bio‐Rad). Specific protein expression levels were normalized to GAPDH for total cell and cytoplasmic lysates or to Lamin B for the nuclear proteins.
Antibodies
Antibodies against the following proteins were purchased from Santa Cruz Biotechnology: atrial natriuretic peptide (sc‐20158), β‐MHC (myosin heavy chain beta; sc‐53090), and Lamin B (sc‐6217). Antibody against GAPDH was obtained from Bioworld Technology (MB001). Antibodies against FLAG were obtained from Sigma‐Aldrich (F3165). Antibodies against HA were purchased from Cell Signaling Technology (#3724). Antibodies against NULP1 were obtained from Santa Cruz (sc‐514203) and BETHYL (A303‐087A).
Statistical Analysis
All values are presented as the mean±SD. Statistical differences among the groups were determined using Student t tests for comparisons between 2 groups and ANOVA for multiple groups. Briefly, for data that showed a normal distribution and homogeneity of variance, differences between 2 groups were compared with a 2‐tailed Student t test. One‐way ANOVA was applied for multiple comparisons, followed by Bonferroni analysis (for data meeting homogeneity of variance) or Tamhane's T2 analysis (for data demonstrating heteroscedasticity). For data sets with a skewed distribution, a nonparametric statistical analysis was performed using the Kruskal–Wallis test followed by Dunn's test for multiple comparisons. P<0.05 was considered statistically significant.
Results
NULP1 Expression Is Decreased in Cardiac Hypertrophy
As NULP1 is highly enriched in the adult heart and regulates the transcriptional activity of serum response factor, 23 a transcription factor involved in heart development and heart diseases, we examined NULP1 expression in hearts from normal donors and patients with heart failure (Table S1). Protein levels of NULP1 were significantly lower in failing hearts than in nonfailing hearts, whereas the expression levels of hypertrophic markers, atrial natriuretic peptide and β‐MHC, were remarkably elevated (Figure 1A). To validate whether the deregulation of NULP1 is consistent in animal disease models, we induced cardiac hypertrophy with AB surgery in mice and treated isolated NRVMs with Angiotensin II (Ang II, 1 μmol/L) or PE (100 μmol/L). We found that the expression of NULP1 levels was significantly reduced in the hearts at 4 and 8 weeks after AB surgery (Figure 1B) as well as in the NRVMs after Ang II or PE treatment (Figure 1C). Moreover, the decrease of NULP1 protein levels in human hearts from patients with heart failure was further validated by immunohistochemistry staining (Figure 1D). In addition, quantitative PCR analyses were performed to evaluate the mRNA alteration of NULP1 subjected to hypertrophic stimuli. Intriguingly, there were no significant differences in the mRNA levels of NULP1, neither in human failing hearts nor mouse hypertrophy models, indicating that the decrease of NULP1 protein mediated by hypertrophic stimuli was not due to changes in transcription (Figure S1A and S1B). Taken together, the dramatic downregulation of NULP1 protein level in response to hypertrophic stress suggested a potential role of NULP1 in pathological cardiac hypertrophy.
Figure 1. NULP1 (nuclear localized protein 1) expression is decreased upon hypertrophic stimuli.
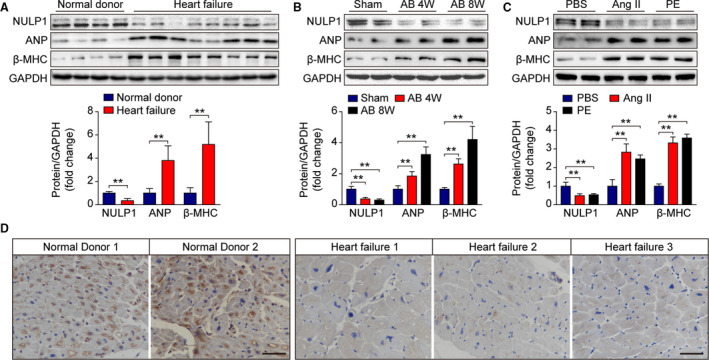
A, Representative western blots and quantification of NULP1, atrial natriuretic peptide (ANP), and β‐myosin heavy chain (β‐MHC) protein levels in human heart tissues from normal control donors (n=4) and from patients with heart failure (n=8). B, Immunoblot analysis and quantification showing NULP1, ANP, and β‐MHC expression in heart samples from mice subjected to a sham operation or AB (aortic banding) surgery for the indicated times. n=4 mice per group. C, Representative immunoblots and quantitative results of NULP1, ANP, and β‐MHC expression in 48 hours PBS‐, Ang II (angiotensin II ,1 μmol/L)‐, or PE (phenylephrine, 100 μmol/L)‐treated NRVMs. D, Representative images of immunohistochemical staining for NULP1 in normal control donors and in patients with heart failure. Scale bar, 50 μm. For all statistical plots, the data are presented as the mean±SD; in (A) **P<0.01; the statistical analysis was performed using a Student t test; in (B and C) **P<0.01; the statistical analysis was performed using a 1‐way ANOVA or 2‐tailed Student t test. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase.
NULP1 Protects Against Cardiomyocyte Hypertrophy In Vitro
To evaluate the impact of NULP1 on the development of cardiac hypertrophy, we manipulated NULP1 expression in NRVMs using reconstituted adenoviruses expressing either the wild‐type Nulp1 or an shRNA targeting endogenous Nulp1 (Figure 2A). Nulp1 knockdown in NRVMs had no effect at the basal level but significantly potentiated the enlarged cell size (Figure 2B) and enhanced the induction of Anp and β‐MHC (Myh7) expressions after Ang II or PE treatment (Figure 2C). By contrast, Nulp1 overexpression significantly suppressed cardiomyocyte hypertrophy and the mRNA levels of Anp and Myh7 induced by Ang II or PE treatment without any effect at the basal level (Figure 2D and 2E), suggesting an antihypertrophic role of NULP1.
Figure 2. NULP1 (nuclear localized protein 1) protects against Angiotensin II‐ (Ang II‐) and phenylephrine‐ (PE‐)induced cardiomyocyte hypertrophy in vitro.
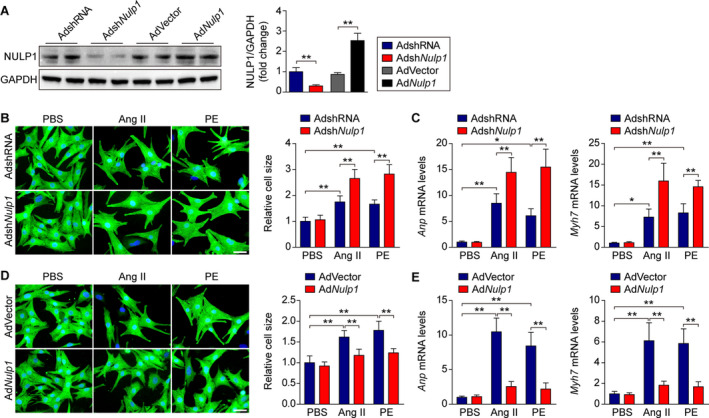
A, Immunoblots and quantification of NULP1 protein levels in isolated neonatal rat ventricular myocytes (NRVMs) infected with the indicated adenoviruses (AdshRNA and AdshNulp1, AdVector and AdNulp1). n=4 samples per group. B, Left, representative images of immunofluorescence staining for α‐actinin (green) and DAPI (blue) in cultured NRVMs infected with adenoviruses expressing shRNA (AdshRNA) or shNulp1 (AdshNulp1) followed by 48 hours of PBS, Ang II (1 μmol/L) or PE (100 μmol/L) treatment. Scale bar, 20 μm. Right, relative cell size of cultured NRVMs infected with AdshRNA or AdshNulp1 in response to PBS, Ang II, or PE stimuli. n>50 cells per group. C, quantitative polymerase chain reaction (qPCR) analyses of the mRNA levels of atrial natriuretic peptide (Anp) and Myh7 in PBS‐, Ang II‐, or PE‐treated NRVMs infected with AdshRNA or AdshNulp1. n=4 samples per group. D, Left, representative images of immunofluorescence staining for α‐actinin (green) and DAPI (blue) in cultured NRVMs infected with empty adenoviral vector (AdVector) or adenovirus expressing Nulp1 (AdNulp1) followed by 48 hours of PBS, Ang II (1 μmol/L), or PE (100 μmol/L) treatment. Scale bar, 20 μm. Right, relative cell size of cultured NRVMs infected with AdVector or AdNulp1 in response to PBS, Ang II, or PE stimuli. n>50 cells per group. E, qPCR analyses of the mRNA levels of Anp and Myh7 in PBS‐, Ang II‐, or PE‐treated NRVMs infected with AdVector or AdNulp1. n=4 samples per group. For all statistical plots, the data are presented as the mean±SD; in (A through E) *P<0.05, **P<0.01; the statistical analysis was performed using a 1‐way ANOVA. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase.
Nulp1 Deletion Exacerbates AB‐Induced Cardiac Hypertrophy In Vivo
Nulp1 KO mice were generated using the CRISPR‐cas9 system to elucidate the physiological function of NULP1 in vivo (Figure 3A). Among the 6 offspring founders established, 4 were found to be specifically mutated at exon 1 of Nulp1 (Figure 3B and 3C), and founder #5‐6 with a frame‐shift 11‐bp deletion was used to generate homozygous Nulp1 KO mice, which was validated by PCR and western blot assays (Figure 3D and 3E). Nulp1 deficiency did not cause any morphological difference at the basal level; however, it significantly increased the heart weight gain 4 weeks after AB surgery (Figure 3F). Echocardiography revealed a larger LVEDd and worse left ventricular fractional shortening after AB surgery in Nulp1‐KO mice compared with the wild‐type controls (Figure 3G). The hypertrophic heart morphology and enlarged cardiomyocyte cross‐section area were markedly potentiated in Nulp1‐KO mice (Figure 3H and 3I). Concurrently, the AB‐induced expression of hypertrophic markers Anp, brain natriuretic peptide (Bnp), and Myh7 was further enhanced in Nulp1‐KO mice (Figure 3J). Moreover, Nulp1 deficiency promoted post‐AB myocardial fibrosis, as evidenced by picrosirius red staining and qPCR measurements of fibrosis markers Col1 (Collagen Type 1), Col3 (Collagen Type 3), and connective tissue growth factor (Ctgf) (Figure 3K and 3L). These results indicated a protective role of Nulp1 during cardiac hypertrophy.
Figure 3. Nulp1 deficiency in mice exacerbates aortic banding (AB)‐induced cardiac hypertrophy.
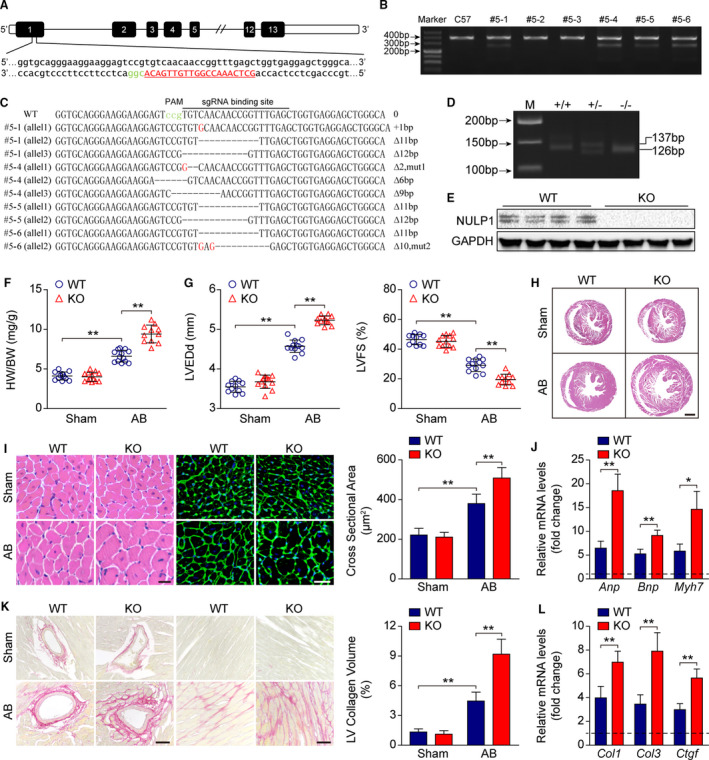
A, Single guide RNA was designed and constructed to target exon 1 of the Nulp1 gene. B, T7 endonuclease 1 assays were used to screen live births of F0 mice. Cleavage products were identified for founders #5‐1, #5‐4, #5‐5, and #5‐6. C, The polymerase chain reaction (PCR) products of the 4 mutant founders were TA cloned and sequenced so that the precise mutations of the indels could be verified. D, Genotyping of Nulp1‐knockout (KO) mice via PCR and 3.0% agarose gel electrophoresis. A 137‐bp band indicated the wild‐type (WT) allele, and a 126‐bp band indicated the mutated Nulp1 allele. E, Nulp1 deletion was validated by western blot analysis. F, Comparison of heart weight (HW)/body weight (BW) ratios in wild‐type (WT) and KO mice subjected to sham or AB surgery for 4 weeks. n=11 to 13 mice per group. G, Echocardiography analyses showing left ventricular end‐diastolic diameter (LVEDd) and left ventricular fractional shortening (LVFS) of WT and KO mice subjected to sham or AB (aortic banding) operation for 4 weeks. n=11 to 12 mice per group. H, hematoxylin and eosin (H&E) staining of whole‐heart sections along the short axes showing the gross morphology of the hearts from WT and KO mice 4 weeks after sham or AB surgery. n=6 to 8 mice per group. Scale bar, 1000 μm. I, Left, heart sections stained with H&E and WGA (wheat germ agglutinin) showing the individual myofibril morphology in the indicated groups. n=6 to 8 mice per group. Scale bars, 20 μm. Right, quantification of the average cross‐sectional areas of cardiomyocytes from the indicated groups. n>100 cells per group. J, mRNA levels of hypertrophic marker genes atrial natriuretic peptide (Anp), brain natriuretic peptide (Bnp), and Myh7 as determined by quantitative PCR (qPCR) in the indicated groups. n=4 mice per group. K, Left, picrosirius red (PSR) staining of heart sections from WT and KO mice subjected to sham or AB operation for 4 weeks to visualize collagen deposition. n=6 to 8 mice per group. Scale bars, 50 μm. Right, quantitative results of LV collagen volume in the indicated groups. n>40 fields per group. L, qPCR analyses of the transcript levels of fibrotic marker genes collagen I (Col1), collagen III (Col3), and connective tissue growth factor (Ctgf) in the indicated groups. n=4 mice per group. For all statistical plots, the data are presented as the mean±SD; in (F, G, I through L) *P<0.05, **P<0.01; the statistical analysis was performed using1‐way ANOVA or two‐tailed student's t test. In (J and L), the dotted line indicates the mRNA levels in the WT sham group, which were normalized to 1. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase.
Nulp1 Overexpression Reverses AB‐Induced Cardiac Hypertrophy In Vivo
We then generated Nulp1 TGmice with cardiac‐specific overexpression of NULP1 (Figure 4A). No morphological difference was observed in Nulp1 transgenic mice at the basal level; however, Nulp1 overexpression significantly blunted the pathogenesis of cardiac hypertrophy induced by AB surgery, as evidenced by less heart weight gain (Figure 4B), improved cardiac performance (Figure 4C), subtler hypertrophic morphology (Figure 4D and 4E), repressed pathological gene expression (Figure 4F), and alleviated myocardial fibrosis (Figure 4G and 4H) after AB surgery compared with wild‐type controls. These data indicated that NULP1 protects the heart against pressure‐overload‐induced cardiac hypertrophy.
Figure 4. Cardiac‐specific Nulp1 overexpression in mice reverses aortic banding (AB)‐induced cardiac hypertrophy.
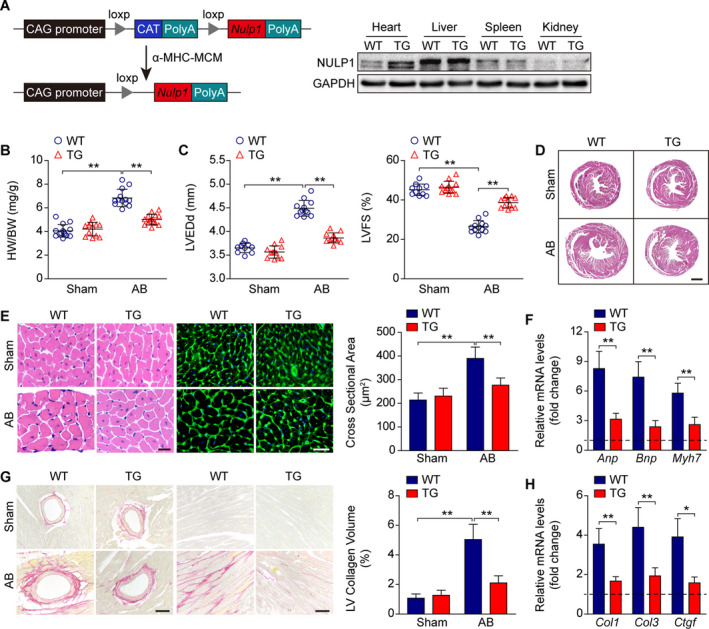
A, Left, schematic diagram depicting the construction of cardiac‐restricted Nulp1 transgenic (TG) mice. Right, representative western blots showing the NULP1 (nuclear localized protein 1) protein levels in different organ tissues from wild‐type (WT) and TG mice. B, Comparison of heart weight (HW)/body weight (BW) ratios in WT and TG mice subjected to sham or AB surgery for 4 weeks. n=12 to 14 mice per group. C, Echocardiography analyses showing left ventricular end‐diastolic diameter (LVEDd) and left ventricular fractional shortening (LVFS) of WT and TG mice subjected to sham or AB operation for 4 weeks. n=10 to 12 mice per group. D, Hematoxylin and eosin (H&E) staining of whole‐heart sections along the short axes showing the gross morphology of the hearts from WT and TG mice 4 weeks after sham or AB surgery. n=7 to 8 mice per group. Scale bar, 1000 μm. E, Left, heart sections stained with H&E and WGA (wheat germ agglutinin) showing the individual myofibril morphology in the indicated groups. n=7 to 8 mice per group. Scale bars, 20 μm. Right, quantification of the average cross‐sectional areas of cardiomyocytes from the indicated groups. n>100 cells per group. F, mRNA levels of hypertrophic marker genes atrial natriuretic peptide (Anp), brain natriuretic peptide (Bnp), and Myh7 as determined by quantitative polymerase chain reaction (qPCR) in the indicated groups. n=4 mice per group. G, Left, picrosirius red (PSR) staining of heart sections from WT and TG mice subjected to sham or AB operation for 4 weeks to visualize collagen deposition. n=7 to 8 mice per group. Scale bars, 50 μm. Right, quantitative results of LV collagen volume in the indicated groups. n>40 fields per group. H, qPCR analyses of the transcript levels of fibrotic marker genes collagen I (Col1), collagen III (Col3), and connective tissue growth factor (Ctgf) in the indicated groups. n=4 mice per group. For all statistical plots, the data are presented as the mean±SD; in (B, C, E through H, *P<0.05, **P<0.01; the statistical analysis was performed using 1‐way ANOVA or two‐tailed student's t test. In (F and H) the dotted line indicates the mRNA levels in the WT sham group, which were normalized to 1. GAPDH indicates glyceraldehyde‐3‐phosphate dehydrogenase.
NULP1 Regulates NFAT Signaling Independent of Calcineurin
To elucidate the molecular mechanism underlying the antihypertrophic effect of NULP1, we performed a dual‐luciferase screening analysis using a Cignal 45‐Pathway Reporter array (SABiosciences). Strikingly, the NFAT signaling pathway stood out among 45 pathways as negatively regulated by overexpressed NULP1 (Figure 5A). Previous study reported that NULP1 was able to suppress the transcriptional activity of serum response factor in COS‐7 cells. 23 However, the luciferase screening analysis demonstrated that NULP1 seemed to have no inhibitory effect on the SRE‐Luc activity in cardiomyocytes. This discrepancy may be attributed mainly to the diverse cell types and specific hypertrophic stimuli. When introduced into NRVMs, the NFAT‐responsive luciferase reporter assay demonstrated that the increased NFAT transcriptional activity after Ang II treatment was enhanced by Nulp1‐silence but was suppressed by Nulp1‐overexpression (Figure 5B).
Figure 5. NULP1 (nuclear localized protein 1) regulates nuclear factor of activated T cell (NFAT) signaling during cardiac hypertrophy independent of calcineurin.
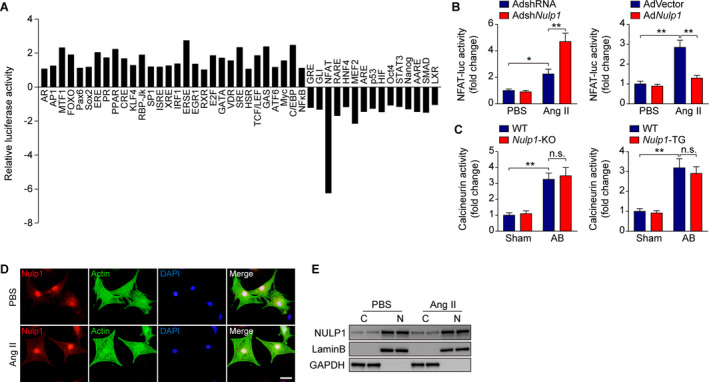
A, The results of the Cignal 45‐Pathway Reporter Array in H9C2 cells transfected with plasmids overexpressing green fluorescent protein (GFP) or NULP1. B, NFAT‐luciferase activity in neonatal rat ventricular myocytes (NRVMs) infected with adenoviruses AdshRNA and AdshNulp1 (left) or AdVector and AdNulp1 (right) in response to PBS), Ang II (angiotensin II, 1 μmol/L) stimulation. n=3 per group. C, Calcineurin phosphatase activity in the lysates of the left ventricle from the indicated experimental groups. n=6 mice per group. D, Representative images of immunofluorescence staining for NULP1 (red) and α‐actinin (green) in NRVMs followed by 6 hours of PBS or Ang II (1 μmol/L) treatment. DAPI stain (blue) marked the position of nuclei. E, Representative western blots showing the NULP1 protein levels in the cytoplasm (C) and nucleus (N) of NRVMs in response to PBS or Ang II (1 μmol/L) stimuli for 6 hours. For all statistical plots, the data are presented as the mean±SD; in (B and C) *P<0.05, **P<0.01 and n.s. indicates no significance; the statistical analysis was performed using a 1‐way ANOVA. AB indicates aortic banding; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; KO, knockout; and WT, wild type.
NFAT proteins are phosphorylated and reside in the cytoplasm in resting cells; upon hypertrophic stimulation, they are dephosphorylated by the Ca2+/calmodulin‐dependent serine phosphatase calcineurin, translocate to the nucleus, and become transcriptionally active. 5 Interestingly, the activity of the upstream regulator calcineurin was not affected by Nulp1‐KO or Nulp1 transgenic in either sham or AB groups (Figure 5C). Immunofluorescence imaging showed that NULP1 predominantly localized inside the nucleus in NRVMs, and the localization was not affected under hypertrophic stimulation (Figure 5D and 5E), excluding the possible involvement of a shuttle step of the dephosphorylated and activated NFAT in mediating NULP1 function.
NULP1 Interacts With and Inactivates NFAT3 During Cardiac Hypertrophy
Four NFAT isoforms, NFAT1‐4, have been shown to be involved in cardiac remodeling. 24 , 27 , 28 , 29 To test the hypothesis that NULP1 directly interacts with NFAT, we performed a coimmunoprecipitation assay with tagged NULP1 paired with different NFAT isoforms. As shown in Figure 6A, NULP1 specifically bound to NFAT1 and NFAT3 but not to NFAT2 or NFAT4. However, the interaction did not affect the transcriptional activity of NFAT1 but substantially suppressed that of NFAT3 (Figure 6B and 6C). A truncation mapping assay showed that NFAT3 directly bound to the C‐terminal region (241‐676) of NULP1 with its 2 independent topologically associating domains (1‐260 and 613‐901; Figure 6D and 6E). In line with the interaction basis, the NFAT transcriptional activity was significantly suppressed by the C‐terminal region (195‐676 and 241‐676) but not by the N‐terminal region of NULP1 (1‐194, Figure 6F). Further, we overexpressed Flag‐tagged NULP1 in NRVMs by adenovirus infection, followed by PBS or Ang II challenge for 6h. Immunoprecipitation was then performed using Flag or IgG antibodies. As demonstrated in Figure S2, binding of NULP1 and endogenous NFAT3 was identified in NRVMs under basal condition, which was reduced in response to hypertrophic stimuli. Collectively, these data suggested NFAT3 to be a direct target of NULP1 during cardiac hypertrophy.
Figure 6. NULP1 (nuclear localized protein 1) interacts with and inactivates nuclear factor of activated T cells 3 (NFAT3) during cardiac hypertrophy.
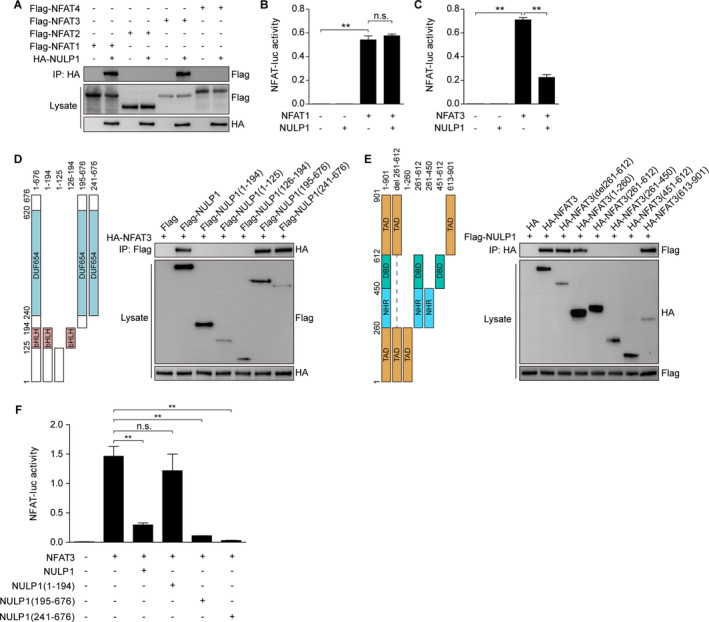
A, Representative western blots for coimmunoprecipitation (co‐IP) assays screening the binding of NULP1 with different NFATs by incubating HA‐NULP1 and the indicated NFAT plasmids in vitro. B, NFAT‐luciferase (NFAT‐luc) activity in H9C2 cells transfected with NFAT1 alone or in combination with NULP1. C, NFAT‐luciferase activity in H9C2 cells transfected with NFAT3 alone or in combination with NULP1. D and E, Schematic of full‐length and truncated NULP1 (D, left) and NFAT3 (E, left) and representative western blots for mapping analyses showing the binding domains of NULP1 to NFAT3 (D and E, right). F, NFAT‐luciferase activity in H9C2 cells transfected with NFAT3 alone or in combination with full‐length and truncated NULP1. For all statistical plots, the data are presented as the mean±SD; in (B, C, and F) **P<0.01 and n.s. indicate no significance; the statistical analysis was performed using a 1‐way ANOVA.
The Regulatory Effect of NULP1 on Cardiac Hypertrophy is Dependent on NFAT3
To validate that the NFAT pathway mediates the function of NULP1 in cardiac hypertrophy, we applied an NFAT inhibitor, VIVIT, which competitively blocks the activation of NFAT and its translocation into nucleus. 16 The results showed that, compared with the saline controls, pharmacological inactivation of NFAT by VIVIT treatment alleviated the AB‐induced cardiac hypertrophy and neutralized the exacerbated pathology in Nulp1‐KO mice, as evidenced by lower heart weight gain (Figure 7A), smaller LVEDd (Figure 7B), improved left ventricular fractional shortening (Figure 7B), and attenuated myocardial remodeling (Figure 7C through 7E).
Figure 7. Nuclear factor of activated T cells (NFAT) inactivation rescues the aggravated pathology by Nulp1 deletion in vivo.
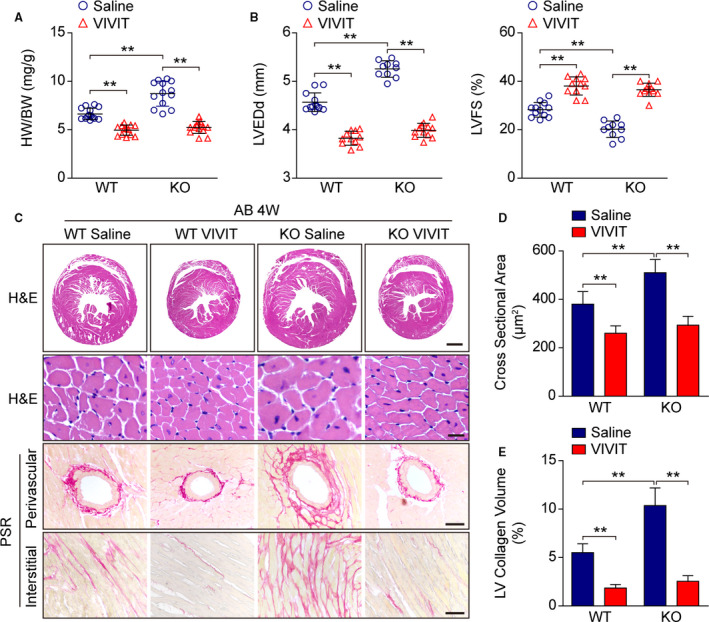
A, Comparison of heart weight (HW)/body weight (BW) ratios in wild‐type (WT) and Nulp1 knockout (KO) mice subjected to 4 weeks of aortic banding (AB) surgery with or without NFAT inhibitor (VIVIT) treatment. n=13 to 14 mice per group. B, Echocardiographic parameters left ventricular end‐diastolic diameter (LVEDd) and left ventricular fractional shortening (LVFS) in the indicated groups. n=10 to 12 mice per group. C, Histological analyses by hematoxylin and eosin (H&E) staining of whole‐heart short‐axis cross sections (the first row; scale bar, 1000 μm) and individual myofibril cross sections (the second row; scale bar, 50 μm) and representative images of cardiac fibrosis, as visualized by picrosirius red (PSR) staining of the perivascular area (the third row; scale bar, 50 μm) and interstitial area (the fourth row; scale bar, 50 μm) in the indicated groups. n=6 to 8 mice per group. D, Results pertaining to the cross‐sectional areas of the cardiomyocytes in the indicated groups (n≥100 cells per group). E, Quantification of LV collagen volumes in the indicated groups (n≥40 fields per group). For all statistical plots, the data are presented as the mean±SD; in (A, B, D, and E) **P<0.01; the statistical analysis was performed using a 1‐way ANOVA.
To further confirm the regulatory effect of NULP1 on pathological cardiac hypertrophy is NFAT3 dependent, NRVMs were infected with AdshNulp1 alone or in combination with AdshNfat3, followed by Ang II treatment. The results showed that Nfat3 knockdown rescued the exacerbated cardiomyocyte hypertrophic response induced by Nulp1 deficiency (Figure 8A and 8B), which is in agreement with the results in vivo. We then infected NRVMs with AdNulp1 alone or in combination with AdNfat3, followed by Ang II stimuli. Conversely, the reduced cardiomyocyte size and mRNA levels of the fetal genes in AdNulp1‐infected cells were fully abolished by overexpression of Nfat3 (revised figure and shown as Figure 8C and 8D).
Figure 8. The regulatory effect of NULP1 (nuclear localized protein 1) on cardiac hypertrophy is dependent on nuclear factor of activated T cells 3 (NFAT3).
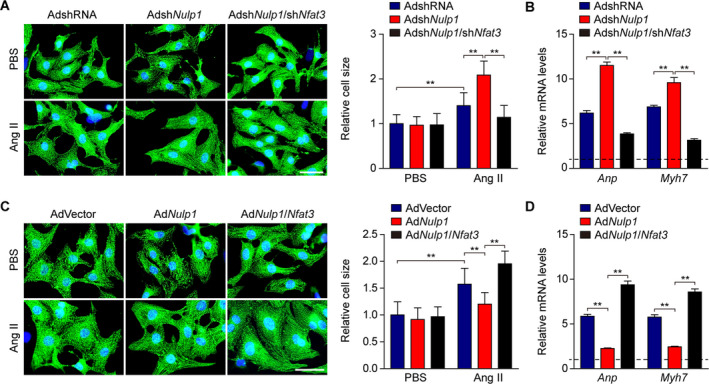
A, Left, representative images of immunofluorescence staining for α‐actinin (green) and DAPI (blue) in cultured neonatal rat ventricular myocytes (NRVMs) infected with adenovirus AdshNulp1 alone or in combination with AdshNfat3 in response to PBS (phosphate buffer saline) Ang II (angiotensin II, 1 μmol/L) stimuli for 48 hours. Scale bar, 20 μm. Right, relative cell size of cultured NRVMs in the indicated groups. n>50 cells per group. B, quantitative polymerase chain reaction (qPCR) analyses of the mRNA levels of atrial natriuretic peptide (Anp) and Myh7 in the indicated groups. n=3 samples per group. C, Left, representative images of immunofluorescence staining for α‐actinin (green) and DAPI (blue) in cultured NRVMs infected with AdNulp1 alone or in combination with AdNfat3 followed by 48 hours of PBS or Ang II treatment. Scale bar, 20 μm. Right, relative cell size of cultured NRVMs in the indicated groups. n>50 cells per group. D, qPCR analyses of the mRNA levels of Anp and Myh7 in the indicated groups. n=3 samples per group. For all statistical plots, the data are presented as the mean±SD; in (A through D) **P<0.01; the statistical analysis was performed using a 1‐way ANOVA. In (B and D) the dotted line indicates the mRNA levels in AdshRNA PBS or AdVector PBS group, which were normalized to 1.
Discussion
Pathological gene reprogramming contributes to the development of cardiac hypertrophy. This process is governed by coordinated transcription remodeling involving multiple factors for or against pathological cardiac remodeling. 30 Here, we identified a transcription repressor, NULP1, which negatively regulates cardiac hypertrophy by directly interacting with and inactivating NFAT3 (Figure S3). Neither knockout nor overexpression of Nulp1 affects normal heart development and function, suggesting that Nulp1 is a feasible drug target to potentially block the progression of cardiac hypertrophy and heart failure in the clinic.
The calcineurin/NFAT pathway is a pivot for the coordinated signaling cascades in cardiac hypertrophy. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 Although inhibition of calcineurin/NFAT may be beneficial, global deletion or complete inactivation of these genes impairs heart development and its function. 19 , 20 Thus, it is of pivotal importance to elucidate the molecular mechanism underlying how calcineurin/NFAT is precisely regulated under stress. Four NFAT isoforms differentially contribute to cardiac remodeling in this progression. 24 , 27 , 28 , 29 Our previous findings revealed that IRF8, an effector of the innate immune network, protected the heart from pathological hypertrophy by inhibiting the translocation of NFAT2 (also named NFATc1) into the nucleus. 24 In this study, we find that NULP1 directly interacts with NFAT1 and NFAT3 but not NFAT2 or NFAT4. Strikingly, the transcriptional activity of NFAT3, but not NFAT1, is dramatically suppressed by NULP1, suggesting differential regulation of different NFAT homologs during cardiac hypertrophy. Interestingly, estrogen receptors have also been reported to repress NFAT3 transcriptional activity. 31 , 32 These findings imply a scavenging mechanism that exists to control NFAT signaling spatially and temporally under pathophysiological conditions.
Transgenic mice expressing activated form of NFAT3 in the heart developed cardiac hypertrophy that progressed to dilated cardiomyopathy and heart failure. 5 The blockade of cardiac hypertrophy by inactivating NFAT3 as unveiled in the current study is supported by previous reports 33 , 34 ; however, Wilkins et al 29 found that deletion of NFAT3 (also named NFATc4) did not compromise the ability of myocardium to undergo hypertrophy under calcineurin overexpression. This may be attributed to a compensatory remodeling of this pathway by other NFAT isoforms under deficiency of NFAT3. On the other hand, individually blocking NFAT1 or NFAT4 is sufficient to reverse the progression of cardiac hypertrophy induced by calcineurin. 24 , 27 , 29 The distinct functions of and the interplay among different NFAT isoforms particularly under disease conditions remain unclear. The specificity of NULP1 to modify NFAT3 function but not the others suggests different interactomes that probably underlying the differential functions of NFAT isoforms in cardiac hypertrophy.
NULP1 is highly expressed in the brain in embryos but becomes enriched in adult hearts. 23 In particular, NULP1 has been shown to regulate the transcriptional activity of serum response factor, a key transcription factor in heart development, 23 suggesting that downregulation of NULP1 during cardiac hypertrophy may be another instance of fetal gene reprograming. 35 Intriguingly, there were no significant changes in the mRNA levels of NULP1 in response to hypertrophic stress, neither in human failing hearts nor mouse hypertrophy models, although dramatic protein downregulation was observed. These results indicated that the decrease of NULP1 protein may be dependent on protein degradation. In fact, previous study reported that NULP1 could be ubiquitinated in N2A cells, which implied protein stability regulation of NULP1. 21 Further studies are needed to investigate whether hypertrophic stimuli promote NULP1 ubiquitination and resultant degradation in cardiomyocytes and to identify the E3 ligase of NULP1. Expression of NULP1 negatively correlates with innate immune signaling underlying the antiviral effects of ribavirin, 36 and the innate immune system has been recognized as a crucial regulatory circuit in cardiovascular diseases. 37 The function of NULP1 in the innate immune network needs to be further examined. High‐throughput profiling implicates NULP1 as a promising cancer marker and vaccine target in prostate tumors. 38 It would be intriguing to further investigate the role of NULP1 in other pathological settings such as cancer.
Taken together, our findings revealed NULP1 as a negative regulator of pathological cardiac hypertrophy that functions through directly interacting with and inactivating NFAT3. These findings shed light on the mechanism of transcription remodeling during cardiac hypertrophy and provide novel molecular targets to treat hypertrophic cardiomyopathy and heart failure.
Sources of Funding
This work was supported by grants from the Youth Program of National Natural Science Foundation of China (No. 81600387) the National Science Fund for Distinguished Young Scholars (No. 81425005), the Key Project of the National Natural Science Foundation (No. 81330005 and No. 81630011), the National Key Research and Development Program (No. 2013YQ030923‐05 and 2016YFF0101500), and the Key Collaborative Project of the National Natural Science Foundation (No. 91639304).
Disclosures
None.
Supporting information
Tables S1–S2
Figures S1–S3
(J Am Heart Assoc. 2020;9:e016419 DOI: 10.1161/JAHA.120.016419.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016419
For Sources of Funding and Disclosures, see page 15.
Contributor Information
Xiao‐Dong Zhang, Email: zhangxd@whu.edu.cn.
Peng Zhang, Email: zhp@whu.edu.cn.
References
- 1. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. [DOI] [PubMed] [Google Scholar]
- 2. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. [DOI] [PubMed] [Google Scholar]
- 3. Sugden PH. Signalling pathways in cardiac myocyte hypertrophy. Ann Med. 2001;33:611–622. [PubMed] [Google Scholar]
- 4. MacLellan WR. Advances in the molecular mechanisms of heart failure. Curr Opin Cardiol. 2000;15:128–135. [DOI] [PubMed] [Google Scholar]
- 5. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin‐dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Molkentin JD. Calcineurin‐NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. [DOI] [PubMed] [Google Scholar]
- 7. Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. J Biol Chem. 2001;276:3524–3530. [DOI] [PubMed] [Google Scholar]
- 8. Tokudome T, Horio T, Kishimoto I, Soeki T, Mori K, Kawano Y, Kohno M, Garbers DL, Nakao K, Kangawa K. Calcineurin‐nuclear factor of activated T cells pathway‐dependent cardiac remodeling in mice deficient in guanylyl cyclase A, a receptor for atrial and brain natriuretic peptides. Circulation. 2005;111:3095–3104. [DOI] [PubMed] [Google Scholar]
- 9. Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, Cheng J, Lu G, Morris DJ, Castrillon DH, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114:1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, Kuro OM, Rothermel BA, Hill JA. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci USA. 2007;104:20517–20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA. Physical and functional interaction between calcineurin and the cardiac L‐type Ca2+ channel. Circ Res. 2009;105:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hojayev B, Rothermel BA, Gillette TG, Hill JA. FHL2 binds calcineurin and represses pathological cardiac growth. Mol Cell Biol. 2012;32:4025–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diedrichs H, Hagemeister J, Chi M, Boelck B, Muller‐Ehmsen J, Schneider CA. Activation of the calcineurin/NFAT signalling cascade starts early in human hypertrophic myocardium. J Int Med Res. 2007;35:803–818. [DOI] [PubMed] [Google Scholar]
- 14. Berry JM, Le V, Rotter D, Battiprolu PK, Grinsfelder B, Tannous P, Burchfield JS, Czubryt M, Backs J, Olson EN, et al. Reversibility of adverse, calcineurin‐dependent cardiac remodeling. Circ Res. 2011;109:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aramburu J, Yaffe MB, Lopez‐Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity‐driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. [DOI] [PubMed] [Google Scholar]
- 16. Kuriyama M, Matsushita M, Tateishi A, Moriwaki A, Tomizawa K, Ishino K, Sano S, Matsui H. A cell‐permeable NFAT inhibitor peptide prevents pressure‐overload cardiac hypertrophy. Chem Biol Drug Des. 2006;67:238–243. [DOI] [PubMed] [Google Scholar]
- 17. Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. [DOI] [PubMed] [Google Scholar]
- 18. Hill JA, Rothermel B, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, Kutschke W, Bassel‐Duby R, Williams RS. Targeted inhibition of calcineurin in pressure‐overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem. 2002;277:10251–10255. [DOI] [PubMed] [Google Scholar]
- 19. Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–875. [DOI] [PubMed] [Google Scholar]
- 20. Schaeffer PJ, Desantiago J, Yang J, Flagg TP, Kovacs A, Weinheimer CJ, Courtois M, Leone TC, Nichols CG, Bers DM, et al. Impaired contractile function and calcium handling in hearts of cardiac‐specific calcineurin b1‐deficient mice. Am J Physiol Heart Circ Physiol. 2009;297:H1263–H1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steen H, Lindholm D. Nuclear localized protein‐1 (Nulp1) increases cell death of human osteosarcoma cells and binds the X‐linked inhibitor of apoptosis protein. Biochem Biophys Res Commun. 2008;366:432–437. [DOI] [PubMed] [Google Scholar]
- 22. Lachtermacher S, Esporcatte BL, Montalvao F, Costa PC, Rodrigues DC, Belem L, Rabischoffisky A, Faria Neto HC, Vasconcellos R, Iacobas S, et al. Cardiac gene expression and systemic cytokine profile are complementary in a murine model of post‐ischemic heart failure. Braz J Med Biol Res. 2010;43:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai Z, Wang Y, Yu W, Xiao J, Li Y, Liu L, Zhu C, Tan K, Deng Y, Yuan W, et al. Hnulp1, a basic helix‐loop‐helix protein with a novel transcriptional repressive domain, inhibits transcriptional activity of serum response factor. Biochem Biophys Res Commun. 2006;343:973–981. [DOI] [PubMed] [Google Scholar]
- 24. Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K, Gao L, Zhou H, Zhu XH, Liu PP, et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun. 2014;5:3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X, Fang J, Gong J, Guo JH, Zhao GN, Ji YX, Liu HY, Wei X, Li H. Cardiac‐specific EPI64C blunts pressure overload‐induced cardiac hypertrophy. Hypertension. 2016;67:866–877. [DOI] [PubMed] [Google Scholar]
- 26. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, et al. Suppressor of IKKvarepsilon is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bourajjaj M, Armand AS, da Costa Martins PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH, De Windt LJ. NFATc2 is a necessary mediator of calcineurin‐dependent cardiac hypertrophy and heart failure. J Biol Chem. 2008;283:22295–22303. [DOI] [PubMed] [Google Scholar]
- 28. Guan XH, Hong X, Zhao N, Liu XH, Xiao YF, Chen TT, Deng LB, Wang XL, Wang JB, Ji GJ, et al. CD38 promotes angiotensin II‐induced cardiac hypertrophy. J Cell Mol Med. 2017;21:1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin‐mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li L, Chen Y, Li J, Yin H, Guo X, Doan J, Molkentin JD, Liu Q. TAK1 regulates myocardial response to pathological stress via NFAT, NFkappaB, and Bnip3 pathways. Sci Rep. 2015;5:16626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang H, Wang X, Li J, Zhu J, Xie X, Yuan B, Yang Z, Zeng M, Jiang Z, Li J, et al. Tissue type‐specific modulation of ER transcriptional activity by NFAT3. Biochem Biophys Res Commun. 2007;353:576–581. [DOI] [PubMed] [Google Scholar]
- 32. Qin X, Wang XH, Yang ZH, Ding LH, Xu XJ, Cheng L, Niu C, Sun HW, Zhang H, Ye QN. Repression of nfat3 transcriptional activity by estrogen receptors. Cell Mol Life Sci. 2008;65:2752–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le K, Li R, Xu S, Wu X, Huang H, Bao Y, Cai Y, Lan T, Moss J, Li C, et al. PPARalpha activation inhibits endothelin‐1‐induced cardiomyocyte hypertrophy by prevention of NFATc4 binding to GATA‐4. Arch Biochem Biophys. 2012;518:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bai S, Kerppola TK. Opposing roles of FoxP1 and Nfat3 in transcriptional control of cardiomyocyte hypertrophy. Mol Cell Biol. 2011;31:3068–3080. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. [DOI] [PubMed] [Google Scholar]
- 36. Thomas E, Feld JJ, Li Q, Hu Z, Fried MW, Liang TJ. Ribavirin potentiates interferon action by augmenting interferon‐stimulated gene induction in hepatitis C virus cell culture models. Hepatology. 2011;53:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang XJ, Zhang P, Li H. Interferon regulatory factor signalings in cardiometabolic diseases. Hypertension. 2015;66:222–247. [DOI] [PubMed] [Google Scholar]
- 38. Alsøe L, Stacy JE, Fosså A, Funderud S, Brekke OH, Gaudernack G. Identification of prostate cancer antigens by automated high‐throughput filter immunoscreening. J Immunol Methods. 2008;330:12–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figures S1–S3