

Abstract
BACKGROUND
Vascular healing response associated with adjunctive n‐3 polyunsaturated fatty acid therapy therapy in patients receiving strong statin therapy remains unclear. The aim of this study was to evaluate the effect of polyunsaturated fatty acid therapy with eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA) in addition to strong statin therapy on coronary atherosclerotic plaques using optical coherence tomography.
METHODS AND RESULTS
This prospective multicenter randomized controlled trial included 130 patients with acute coronary syndrome treated with strong statins. They were assigned to either statin only (control group, n=42), statin+high‐dose EPA (1800 mg/day) (EPA group, n=40), statin+EPA (930 mg/day)+DHA (750 mg/day) (EPA+DHA group, n=48). Optical coherence tomography was performed at baseline and at the 8‐month follow‐up. The target for optical coherence tomography analysis was a nonculprit lesion with a lipid plaque. Between baseline and the 8‐month follow‐up, fibrous cap thickness (FCT) significantly increased in all 3 groups. There were no significant differences in the percent change for minimum FCT between the EPA or EPA+DHA group and the control group. In patients with FCT <120 µm (median value), the percent change for minimum FCT was significantly higher in the EPA or EPA+DHA group compared with the control group.
CONCLUSIONS
EPA or EPA+DHA therapy in addition to strong statin therapy did not significantly increase FCT in nonculprit plaques compared with strong statin therapy alone, but significantly increased FCT in patients with thinner FCT.
Registration
URL: https://www.umin.ac.jp/ctr/; Unique identifier: UMIN 000012825.
Keywords: fatty acid, fibrous cap, optical coherence tomography
Subject Categories: Acute Coronary Syndromes, Atherosclerosis, Coronary Artery Disease, Optical Coherence Tomography (OCT)
Nonstandard Abbreviations and Acronyms
- ACS
acute coronary syndrome
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- FCT
fibrous cap thickness
- LDL‐C
low‐density lipoprotein cholesterol
- OCT
optical coherence tomography
- PCI
percutaneous coronary intervention
- PUFA
polyunsaturated fatty acid
- TCFA
thin‐cap fibroatheroma
CLINICAL PERSPECTIVE
What Is New?
The present study is a prospective, multicenter, randomized controlled trial to examine the effect of eicosapentaenoic acid (EPA) or EPA+docosahexaenoic acid as an adjunct to strong statin therapy on coronary plaque using optical coherence tomography.
EPA or EPA+docosahexaenoic acid therapy in addition to strong statin therapy did not significantly increase fibrous cap thickness in nonculprit plaques compared with strong statin therapy alone, but significantly increased fibrous cap thickness in patients with thinner fibrous cap thickness.
What Are the Clinical Implications?
In the present study, EPA or EPA+docosahexaenoic acid therapy in addition to strong statin therapy did not significantly affect coronary plaque regression in nonculprit plaques compared with strong statin therapy alone, but might provide a greater stabilizing effect on coronary plaques with thinner fibrous cap thickness or thin‐cap fibroatheroma.
Many large‐scale clinical trials have shown that lipid‐lowering statin therapy can be used as primary or secondary prevention for ischemic cardiovascular events. 1 However, despite successful lowering of low‐density lipoprotein cholesterol (LDL‐C) levels with statins, patients with atherosclerotic cardiovascular disease remain at elevated risk for cardiovascular events. 2
Epidemiological and clinical studies have shown that long‐term intake of long‐chain n‐3 polyunsaturated fatty acids (PUFAs), especially eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), can reduce the incidence of cardiovascular events. 3 , 4 A large‐scale intervention clinical trial (JELIS [Japan EPA Lipid Intervention Study]) that included patients with hypercholesterolemia showed that long‐term use of EPA (1800 mg/day) in addition to statin therapy significantly reduced the incidence of secondary coronary events. 5
The presence of vulnerable plaques, which contain a thin fibrous cap, large lipid or necrotic core, and increased macrophage density, has been demonstrated to predispose patients to plaque rupture and subsequent acute coronary syndrome (ACS). 6 , 7 Intravascular imaging studies have demonstrated that aggressive lipid‐lowering therapy with strong statins leads to vulnerable plaque stabilization. 8 , 9 However, the vascular healing response associated with adjunctive PUFA therapy in patients receiving strong statin therapy remains unclear.
Intravascular optical coherence tomography (OCT) is a high‐resolution imaging technique for plaque characterization. OCT allows for the measurement of fibrous cap thickness (FCT) and lipid volume, which are considered major factors in plaque vulnerability. 10 The aim of the present study was to evaluate the effect of adding PUFA therapy (EPA or EPA+DHA) to strong statin therapy on coronary atherosclerotic plaques in patients with ACS using OCT.
METHODS
The data, analytic methods, and study materials will not be made available to any researchers for purposes of reproducing the results or replicating the procedure.
Study Design and Protocol
This study was a prospective, randomized, open‐label, multicenter study involving 5 hospitals. The objective was to evaluate the effect of adding PUFAs to strong statin therapy on coronary atherosclerotic plaques using OCT. Patients were randomly assigned to either rosuvastatin alone (control group, n=42), rosuvastatin+high‐dose EPA (1800 mg/day) (EPA group, n=40), or rosuvastatin+EPA (930 mg/day)+DHA (750 mg/day) (EPA+DHA group, n=48) after successful percutaneous coronary intervention (PCI) for ACS. PUFA therapy was initiated after randomization. The dose of rosuvastatin was increased to the maximum prescribed dose with a target LDL‐C value of <100 mg/dL. 11 Treatments lasted for 12 months after randomization. All patients underwent OCT‐guided PCI for ACS caused by the culprit lesion. All patients also had OCT examination in the treated coronary vessel and untreated coronary vessels immediately after the index PCI. OCT examination at the 8‐month follow‐up was performed in the same vessels as during baseline OCT. All patients received aspirin (100 mg/day) and clopidogrel (75 mg/day) for 12 months after PCI. Clinical follow‐up visits to assess adverse events were scheduled every 6 months for up to 12 months. Blood samples for fasting lipid profile and fatty acid fractions were obtained in the early morning at baseline and at the 8‐month follow‐up. The present study was approved by the institutional ethics committee of Nara Medical University. Written informed consent was given by all participants before enrollment. This study was registered with the University Hospital Medical Information Network (UMIN000012825).
Patient Populations
Eligibility criteria were as follows: (1) successful PCI for ACS (ST‐segment–elevation acute myocardial infarction) or non–ST‐segment–en elevation acute coronary syndrome, (2) nonculprit vessels suitable for OCT examination, (3) scheduled coronary catheterization at 8 months after the index procedure, and (4) statin treatment before the onset of ACS or initiation of statin treatment immediately after PCI because of serum LDL‐C levels >100 mg/dL. Patients with any of the following were excluded from the study: (1) hypersensitivity to PUFAs, (2) current use of any PUFAs, (3) bleeding tendency, or (4) cardiogenic shock or congestive heart failure.
Randomization
Randomization was centralized at the independent center from investigators. After the investigator obtained the consent from the participant, the treatment assignment was informed to the investigator by the randomization center. All recruited patients were randomly allocated in a 1:1:1 ratio to the control group, EPA group, or EPA+DHA group. Randomization was performed by simple randomization using a random number table during a stable period of hospitalization when statin administration had been confirmed.
OCT Image Acquisition
OCT imaging was obtained using a frequency domain OCT system (C8 system, Dragonfly imaging catheter and ILUMIEN OPTIS; St. Jude Medical, St. Paul, MN). Briefly, the OCT imaging catheter was advanced distal to the target lesion over a conventional 0.014‐inch angioplasty guidewire through a 6 or 7 Fr guiding catheter. After intracoronary administration of 1 mg of isosorbide dinitrate, pullbacks were performed during continuous injection of x‐ray contrast media or lactated low‐molecular‐weight dextran at 2.5 to 3.5 mL/s through the guiding catheter using an injection pump to remove blood from the field of view and allow for clear visualization of the vessel wall. Images were acquired at 5 frames/mm and an automated pullback speed of 20 mm/s. 12
OCT Image Analysis
For the OCT image acquisition of target lesions in nonculprit lesions, 1 pullback was performed from distal site as far as possible to the tip of guiding catheter in each of the treated coronary vessel and untreated coronary vessels immediately after the index PCI. OCT examination at the 8‐month follow‐up was performed within the same observation range as the baseline. OCT images at baseline and at the 8‐month follow‐up were centralized and analyzed in a blinded fashion using a dedicated offline review system (St. Jude Medical) at the independent core laboratory (Department of Cardiovascular Medicine, Nara Medical University) after the end of clinical follow‐up.
Plaque tissue characterization was performed using previously validated criteria. 10 , 13 Lipid core was defined as a signal‐poor region with diffuse borders. Fibrous cap was identified as a signal‐rich band overlying the lipid core. Plaque rupture was defined as an intimal interruption and cavity formation in the plaque. Macrophage infiltration was defined as bright spots with high OCT backscattering signal variances. A microchannel was defined as a no‐signal tubular structure without a connection to the vessel lumen recognized on ≥3 consecutive cross‐sectional images. Cholesterol crystals were defined as thin, linear regions of high signal intensity within the lipid plaque without backscattering.
For the selection of target lesions at baseline, de novo, a nonculprit lesion with lipid core detected by OCT examination was selected as the target lesion. When there were 2 or more lesions with lipid core in a treated or untreated coronary vessel, the lesion that presented the thinnest FCT in lesions with lipid core was selected as the target lesion. The target lesion could be in a coronary artery that was treated or not treated with PCI. In a PCI‐treated coronary artery, the target lesion had to be located >10 mm from the lesion treated with PCI.
Three candidate frames were selected to determine the minimum FCT by visually screening for all contiguous frames. 14 FCT was defined as the distance from the arterial lumen to inner border of the underlying lipid core. 15 FCT was measured 3 times at the thinnest part of the fibrous cap on each frame, and then the average value was calculated. 10 Minimum FCT was defined as the smallest FCT in the candidate frames. Maximum lipid arc was determined as the largest lipid arc in 3 candidate frames selected by visual screening within the target lesion. 14 When lipid was present in ≥90° of any cross‐sectional image within the plaque, it was considered a lipid‐rich plaque. In lipid‐rich plaques, the lipid arc was measured at 1‐mm intervals throughout the entire length of the lesion, and measurements were averaged. Lipid length was calculated based on the number of frames with lipid‐rich plaque. Lipid volume index was defined as average lipid arc multiplied by lipid length. 16
To ensure reliable comparisons between baseline and 8‐month follow‐up, the target lesion or target frame at the 8‐month follow‐up was selected at the same location as the baseline based on the distance from landmarks such as branches and calcifications. Serial OCT images at baseline and at the 8‐month follow‐up were reviewed side by side.
Two observers who were blinded to each patient's clinical characteristics and treatment allocation made OCT measurements independently. The intraobserver and interobserver reproducibility measured by Pearson coefficient were r=0.98 and r=0.88 for minimum FCT and r=0.96 and r=0.86 for maximal lipid arc, respectively.
Absolute change and percent change for OCT measurements (minimum FCT, maximal lipid arc, average lipid arc, lipid length, and lipid volume index) during the observation period were calculated as follows:
In subgroup analysis, patients were divided into 2 groups according to the median minimum FCT (120 µm) (thinner FCT; patients with minimum FCT <120 µm, thicker FCT; patients with minimum FCT ≥120 µm). Thin‐cap fibroatheroma (TCFA) was defined as a thickness of ≤65 μm in the thinnest fibrous cap in a lipid‐rich plaque on a cross‐sectional image.
Blood Sample Analysis
Serum levels of total cholesterol, high‐density lipoprotein cholesterol, triglycerides, glucose, hemoglobin A1c, EPA, DHA, and arachidonic acid were measured in the early morning after an overnight fast. Serum levels of LDL‐C were calculated using the following Friedewald formula: LDL‐C (mg)=total cholesterol−high‐density lipoprotein cholesterol−triglycerides/5.
End Points
The primary end point of the study was the change and the percent change for minimum FCT between baseline and 8‐month follow‐up. Secondary end points were the change and the percent change for maximum lipid arc, lipid length, and lipid volume index between baseline and 8‐month follow‐up as well as major cardiovascular events, which included all‐cause death, cardiac death, nonfatal myocardial infarction, stroke, and coronary revascularization. During study protocol development, the change in minimum FCT, maximal lipid arc, average lipid arc, lipid length, and lipid volume index were prespecified as primary or secondary end point. During manuscript preparation, post hoc analyses about plaque rupture, macrophage infiltration, microchannel, and cholesterol crystals were added to further evaluate the effect of adding PUFA therapy to strong statin therapy.
Statistical Analysis
During study protocol development, no data about the effects of PUFAs on FCT changes evaluated by OCT were available to guide sample size calculations. Therefore, it was impossible to calculate correctly the sample size for testing the assumption that PUFA therapy on top of the strong statin therapy significantly increase FCT compared with strong statin therapy alone. At development of the protocol of the present study, to determine the sample size, we referred to the previous study that evaluated the change in FCT by OCT. 17 That study reported that FCT after statin therapy for 9 months increased by 130 µm because of LDL‐C reduction of ≈36%. Therefore, referring these findings, we calculated the number of enrolled patients with the assumption that the change in FCT after administration of statin was 100 µm, and the SD of the distribution was 200 µm. With a 2‐sided α level of 0.05 and a power of 90%, 44 patients were required in each group. Considering a 25% rate for possible missing investigations or withdrawals, the sample size was set at 60 patients per group.
All analyses were performed using JMP statistics, version 11 (SAS Institute Inc., Cary, NC). All values are expressed as mean±SD or median with interquartile interval (25th–75th percentile) for continuous variables and counts and percentages for categorical variables. Testing for significant differences in each parameter between baseline and 8‐month follow‐up was performed using the paired Student t test. Continuous variables were compared using the parametric statistical 1‐way analysis of variance or the nonparametric Kruskal–Wallis test on the basis of the variable's distribution. When one‐way analysis of variance or the Kruskal–Wallis test showed significant differences, continuous variables were compared between the control group and other groups using post hoc Dunnett or Steel tests. Categorical data were compared using the Pearson chi‐squared test. A P<0.05 was considered statistically significant.
RESULTS
Patient Populations
Figure 1 shows the study profile. Between May 2014 and March 2017, a total of 130 patients who were eligible based on the inclusion criteria were enrolled in this study. During the 8‐month follow‐up period, 16 patients were excluded and 114 patients underwent follow‐up coronary angiography. A total of 17 patients were excluded from OCT analysis because of (1) absence of lipid plaques at baseline (control group, 5 patients; EPA group, 3 patients; EPA+DHA group, 1 patient), (2) no follow‐up OCT examination (control group, 1 patient; EPA+DHA group, 1 patient), and (3) inadequate OCT imaging (control group, 1 patient; EPA group, 4 patient; EPA+DHA group, 1 patient). Ultimately, 97 patients were included in the analysis (control group, 31 patients; EPA group, 31 patients; EPA+DHA group, 35 patients).
Figure 1. Study profile.
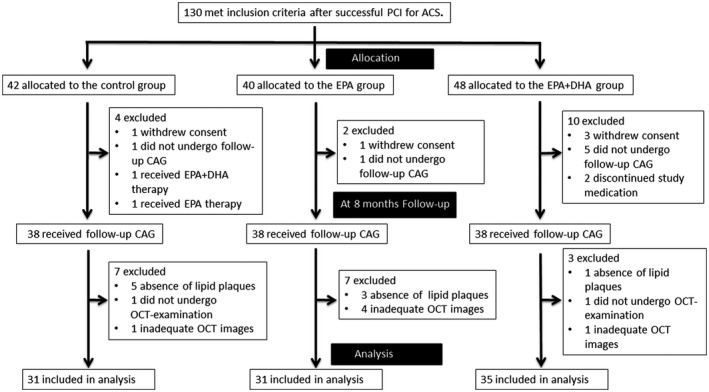
ACS indicates acute coronary syndrome; CAG, coronary angiography; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; OCT, optical coherence tomography; and PCI, percutaneous coronary intervention.
Baseline Clinical Characteristics and Laboratory Data
Clinical characteristics, target lesions, and concomitant medications at baseline are shown in Tables 1 and 2. Despite the randomized controlled trial study design, significant differences were observed in the prevalence of diabetes mellitus between the control and EPA groups (P=0.015). There were no significant differences in laboratory data between the groups. Oral hypoglycemic agents were more frequently used in the EPA group compared with the control group (P=0.003).
Table 1.
Baseline Clinical Characteristics and Laboratory Data
| Control Group (n=31) | EPA Group (n=31) | EPA+DHA Group (n=35) | P Value | |
|---|---|---|---|---|
| Age, y | 63 (55–73) | 66 (57–70) | 67 (59–70) | 0.862 |
| Male sex | 27 (87) | 24 (77) | 28 (81) | 0.596 |
| Hypertension | 22 (71) | 18 (58) | 16 (46) | 0.117 |
| Diabetes mellitus | 11 (35) | 3 (10)* | 11 (31) | 0.043 |
| Current smoking | 16 (52) | 13 (42) | 13 (37) | 0.488 |
| Body mass index, kg/m2 | 24.7 (22.5–25.7) | 24.3 (23.3–26.7) | 25.0 (23.1–26.9) | 0.533 |
| Clinical presentation | 0.168 | |||
| STEMI | 22 (71) | 25 (81) | 29 (83) | |
| NSTACS | 9 (29) | 6 (20) | 6 (17) | |
| Target vessel | 0.860 | |||
| LAD | 15 (48) | 11 (36) | 2 (34) | |
| LCX | 7 (23) | 7 (23) | 10 (29) | |
| RCA | 9 (29) | 13 (42) | 13 (37) | |
| Location of target plaque | 0.243 | |||
| Culprit vessel | 10 (32) | 14 (45) | 9 (26) | |
| Nonculprit vessel | 21 (68) | 17 (55) | 26 (74) | |
| Laboratory data | ||||
| FBS, mg/dL | 113 (92–167) | 122 (107–143) | 124 (103–168) | 0.526 |
| HbA1c, % | 5.9 (5.6–7.1) | 5.9 (5.6–6.0) | 6.0 (5.7–6.5) | 0.585 |
| SCr, mg/dL | 0.9 (0.7–1.0) | 0.8 (0.7–0.9) | 0.8 (0.78–1.0) | 0.536 |
| Triglycerides, mg/dL | 106 (73–141) | 117 (91–135) | 96 (67–131) | 0.316 |
| Total cholesterol, mg/dL | 183 (166–215) | 195 (162–217) | 182 (163–207) | 0.851 |
| LDL‐C, mg/dL | 125 (105–140) | 120 (99–142) | 118 (99–145) | 0.953 |
| HDL‐C, mg/dL | 43 (36–48) | 45 (38–51) | 46 (38–54) | 0.600 |
| EPA, mg/dL | 43.0 (29.2–62.5) | 37.0 (23.5–66.1) | 45.2 (32.9–66.3) | 0.848 |
| AA, mg/dL | 165.1±41.7 | 163.4±36.3 | 163.1±46.5 | 0.980 |
| DHA, mg/dL | 112.8±32.8 | 112.5±36.7 | 113.1±29.9 | 0.997 |
| EPA/AA ratio | 0.31 (0.20–0.43) | 0.26 (0.15–0.43) | 0.33 (0.18–0.40) | 0.739 |
Values are presented as mean±SD, median (interquartile interval), or n (percentage). AA indicates arachidonic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FBS, fasting blood sugar; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; LAD, left anterior descending artery; LCx, left circumflex artery; LDL‐C, low‐density lipoprotein cholesterol; NSTACS, non–ST‐elevation acute coronary syndrome; RCA, right coronary artery; RCA, right coronary artery; SCr, serum creatinine; and STEMI, ST‐segment–elevation myocardial infarction.
P<0.05 vs control group.
Table 2.
Medications at Baseline and Follow‐Up
| Control Group (n=31) | EPA Group (n=31) | EPA+DHA Group (n=35) | P Value | |
|---|---|---|---|---|
| Baseline | ||||
| Strong statin | 4 (13) | 2 (6) | 9 (26) | 0.087 |
| Standard statin | 1 (3) | 1 (3) | 0 (0) | 0.562 |
| Aspirin | 4 (13) | 3 (10) | 4 (11) | 0.923 |
| Calcium channel blocker | 13 (42) | 7 (23) | 9 (26) | 0.199 |
| ACE inhibitor or ARB | 10 (32) | 8 (26) | 8 (23) | 0.683 |
| Beta blocker | 5 (16) | 2 (6) | 1 (3) | 0.134 |
| Insulin | 0 (0) | 0 (0) | 1 (3) | 0.409 |
| Oral hypoglycemic agent | 10 (32) | 1 (3)* | 7 (20) | 0.013 |
| Follow‐up | ||||
| Calcium channel blocker | 6 (19) | 8 (26) | 4 (11) | 0.322 |
| ACE inhibitor or ARB | 26 (84) | 28 (90) | 32 (91) | 0.589 |
| Beta blocker | 23 (74) | 25 (81) | 23 (66) | 0.389 |
| Insulin | 0 (0) | 0 (0) | 1 (2) | 0.409 |
| Oral hypoglycemic agent | 10 (32) | 2 (6)* | 7 (20) | 0.038 |
Values are n (percentage). ACE indicates angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; DHA, docosahexaenoic acid; and EPA, eicosapentaenoic acid.
P<0.05 vs control group.
Table 3 shows changes in the lipid profile and fatty acid fractions. Serum total cholesterol and LDL‐C levels significantly decreased between baseline and 8‐month follow‐up in all 3 groups. Serum EPA levels increased significantly between baseline and 8‐month follow‐up in all 3 groups. Serum DHA levels increased significantly in the EPA+DHA group only. Serum EPA levels and the EPA/arachidonic acid ratio at the 8‐month follow‐up were significantly higher in the EPA and EPA+DHA groups compared with the control group. Serum DHA levels at follow‐up was significantly higher in the EPA+DHA group compared with the control group.
Table 3.
Change in Lipid Profile and Fatty Acid Fractions
| Control Group (n=31) | EPA Group (n=31) | EPA+DHA Group (n=35) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow‐Up | P Value | Baseline | Follow‐Up | P Value | Baseline | Follow‐Up | P Value | |
| Triglycerides, mg/dL | 106 (73–141) | 152 (107–172) | 0.003 | 117 (91–135) | 119 (85–162) | 0.425 | 96 (67–131) | 133 (99–164) | 0.123 |
| Total cholesterol, mg/dL | 183 (166–215) | 145 (138–172) | <0.0001 | 195 (162–217) | 152 (140–165) | <0.0001 | 182 (163–207) | 164 (135–177) | <0.0001 |
| LDL‐C, mg/dL | 125 (105–140) | 78 (61–96) | <0.0001 | 120 (99–142) | 78 (65–95) | <0.0001 | 118 (99–145) | 82 (60–103) | <0.0001 |
| HDL‐C, mg/dL | 43 (36–48) | 43 (36–51) | 0.985 | 45 (38–51) | 49 (42–56) | 0.053 | 46 (38–54) | 44 (37–52) | 0.880 |
| EPA, mg/dL | 43.0 (29.2–62.5) | 55.7 (43.0–76.5) | 0.014 | 37.0 (29.3–66.1) | 170.1 (145.4–212.0)* | <0.0001 | 45.2 (32.9–66.3) | 122.3 (100.5–147.4)* | 0.0001 |
| AA, mg/dL | 165.1±41.7 | 184.8±52.5 | 0.013 | 163.4±36.3 | 159.7±39.5* | 0.082 | 163.1±46.5 | 164.5±37.3 | 0.503 |
| DHA, mg/dL | 112.8±32.8 | 114.9±31.3 | 0.848 | 112.5±36.7 | 106.7±31.6 | 0.568 | 113.1±29.9 | 140.7±44.6* | <0.0001 |
| EPA/AA ratio | 0.31 (0.20–0.43) | 0.35 (0.21–0.50) | 0.328 | 0.26 (0.15–0.43) | 1.15 (0.88–1.61)* | <0.0001 | 0.33 (0.18–0.40) | 0.75 (0.60–0.98)* | <0.0001 |
Values are presented as mean±SD or median (interquartile interval). AA indicates arachidonic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; HDL‐C, high‐density lipoprotein cholesterol; and LDL‐C, low‐density lipoprotein cholesterol.
P<0.05 vs control group.
OCT Analysis
Figure 2 shows representative OCT images in each of the 3 groups. Table 4 shows change in OCT findings between baseline and 8‐month follow‐up. Minimum FCT increased significantly and maximal lipid arc decreased significantly at the 8‐month follow‐up compared with baseline values in all 3 groups. Lipid length and lipid volume index decreased significantly at the 8‐month follow‐up compared with baseline values in the EPA and EPA+DHA groups. However, significant changes in those OCT measurements were not observed in the control group. Significant changes in the incidence of macrophage infiltration, cholesterol crystal, microchannel, and plaque rupture were not observed in each of the allocation groups. There was no significant difference in the incidence of macrophage infiltration, cholesterol crystal, microchannel, or plaque rupture at baseline among 3 groups, but significant differences were observed in the incidence of cholesterol crystal at the 8‐month follow‐up between the control and EPA groups (P=0.0116). There was no significant difference in OCT measurements (minimum FCT, maximum lipid arc, average lipid arc, lipid length, and lipid volume index) at baseline between culprit vessel and nonculprit vessel in each of the allocation groups (Table S1).
Figure 2. Representative cases of optical coherence tomography analysis.
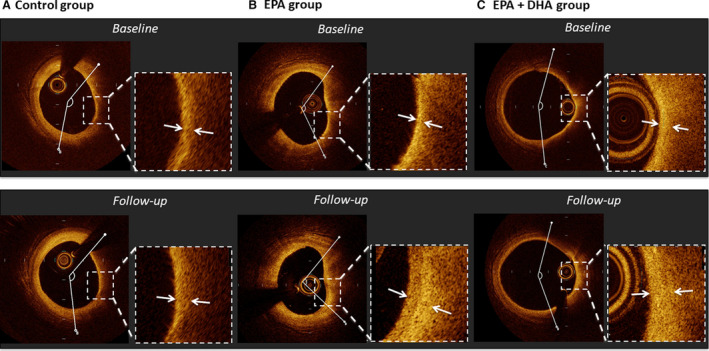
A, In a case from the control group, fibrous cap thickness slightly increased (130–150 μm) and lipid arc changed very slightly (134.9–145.0°) over 8 months. B, In a case from the EPA group, fibrous cap thickness increased (60–210 μm) and lipid arc decreased (118.3 to 95.1°) over 8 months. C, In a case from the EPA+DHA group, fibrous cap thickness increased (60–150 μm) and lipid arc decreased slightly (158.7 to 145.3°) over 8 months. DHA indicates docosahexaenoic acid; and EPA, eicosapentaenoic acid.
Table 4.
Change in Optical Coherence Tomography Findings
| Control Group (n=31) | EPA Group (n=31) | EPA+DHA Group (n=35) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow‐Up | P Value | Baseline | Follow‐Up | P Value | Baseline | Follow‐Up | P Value | |
| Minimal FCT, μm | 130 (70–190) | 160 (100–250) | 0.0121 | 120 (70–160) | 150 (120–210) | 0.0029 | 100 (60–150) | 200 (130–260) | <0.0001 |
| Maximal lipid arc, ° | 202.5 (162.2–234.4) | 165.9 (137.9–196.9) | 0.0003 | 176.9 (149.5–247.3) | 159.0 (121.1–241.4) | 0.0079 | 175.2 (138.4–218.2) | 157.9 (127.2–213.4) | 0.0141 |
| Average lipid arc, ° | 152.0 (125.5–164.3) | 144.4 (127.6–171.3) | 0.4790 | 150.9 (121.6–185.7) | 153.7 (113.3–207.6) | 0.9522 | 147.6 (122.2–195.1) | 134.0 (103.9–183.7) | 0.9522 |
| Lipid length, mm | 4.6 (2.8–7.4) | 4.4 (2.8–7.0) | 0.5440 | 4.2 (2.4–5.6) | 3.6 (2.4–5.0) | 0.0016 | 3.4 (2.2–5.6) | 2.8 (2.0–4.4) | 0.0107 |
| Lipid volume index | 766.0 (435.2–1148.8) | 723.8 (360.4–1017.0) | 0.5051 | 626.6 (462.4–932.6) | 558.9 (317.2–835.6) | 0.0484 | 462.1 (355.7–876.8) | 384.2 (225.1–735.4) | 0.0119 |
| Macrophage infiltration | 28 (90) | 27 (87) | 0.5722 | 27 (87) | 25 (81) | 0.1607 | 33 (94) | 30 (86) | 0.083 |
| Cholesterol crystal | 8 (26) | 8 (26) | 1.000 | 3 (10) | 1 (3)*, * | 0.1607 | 6 (17) | 6 (17) | 1.000 |
| Microchannel | 6 (19) | 6 (19) | 1.000 | 5 (16) | 4 (13) | 0.3253 | 6 (17) | 6 (17) | 1.000 |
| Plaque rupture | 3 (10) | 3 (10) | 1.000 | 1 (3) | 1 (3) | 1.000 | 0 (0) | 0 (0) | 1.000 |
Values are presented as median (interquartile interval) or n (percentage). DHA indicates docosahexaenoic acid; EPA, eicosapentaenoic acid; and FCT, fibrous cap thickness.
P<0.05 vs control group at follow‐up.
Table 5 shows the percent changes in OCT measurements. There were no significant differences in the percent change for minimum FCT, maximal lipid arc, average lipid arc, lipid length, and lipid volume index between the EPA or EPA+DHA group and the control group. Figure 3 shows correlations between minimum FCT at baseline and the percent change for minimum FCT. A significant negative correlation was found between minimum FCT at baseline and the percent change for minimum FCT in the EPA and EPA+DHA groups, but not in the control group. Next, we evaluated the effect of treatment with EPA or EPA+DHA in subgroups by FCT severity. As shown in Figure 4, in the subgroup with minimum FCT <120 µm, the percent change for minimum FCT was significantly greater in the EPA group (P=0.0230) and the EPA+DHA group (P=0.0140) than in the control group (P=0.0102, Kruskal–Wallis test). However, in 51 patients with minimum FCT ≥120 µm, there was no significant difference in the percent change for minimum FCT between the control group and the other groups (P=0.1083, Kruskal–Wallis test). In 22 patients with TCFA, the percent change for minimum FCT was significantly higher in the EPA group (P=0.0185) and the EPA+DHA group (P=0.0240) than in the control group (P=0.0118, Kruskal–Wallis test).
Table 5.
Absolute Change and Percent Change for optical coherence tomography Measurements
| Control Group (n=31) | EPA Group (n=31) | EPA+DHA Group (n=35) | P Value | |
|---|---|---|---|---|
| Minimum FCT | ||||
| Absolute change | 20 (0 to 70) | 20 (0 to 90) | 60 (10 to 120) | 0.1491 |
| Percent change | 20.0 (0 to 60.0) | 14.3 (0 to 128.6) | 58.3 (7.1 to 150.0) | 0.1075 |
| Maximal lipid arc | ||||
| Absolute change | −24.5 (−42.6 to 4.4) | −8.2 (−38.4 to 0) | −12.8 (−27.3 to −0.5) | 0.1928 |
| Percent change | −12.3 (−28.5 to 1.6) | −5.4 (−13.9 to 0) | −7.6 (−17.8 to −0.4) | 0.3122 |
| Average lipid arc | ||||
| Absolute change | −2.0 (−20.3 to 17.8) | −1.6 (−8.9 to 16.7) | −7.9 (−32.6 to 11.8) | 0.3121 |
| Percent change | −1.5 (−14.1 to 11.4) | −1.3 (−7.3 to 10.8) | −5.1 (−18.5 to 8.4) | 0.4044 |
| Lipid length | ||||
| Absolute change | 0 (−0.6 to −0.8) | −0.6 (−1.6 to −0.2) | −0.4 (−1.6 to 0.2) | 0.0719 |
| Percent change | 0 (−21.1 to 16.7) | −15.8 (−33.3 to −4.8) | −12.5 (−30.6 to 7.1) | 0.1233 |
| Lipid volume index | ||||
| Absolute change | −28.1 (−198.4 to 188.4) | −77 (−236.4 to 35.0) | −82.2 (−276.3 to 286.9) | 0.3570 |
| Percent change | −3.8 (−25.5 to 28.1) | −16.6 (−40.8 to 5.3) | −16.6 (−43.9 to 16.6) | 0.1810 |
Values are presented as median (interquartile interval). DHA indicates docosahexaeonic acid; EPA, eicosapentaenoic acid; and FCT, fibrous cap thickness.
Figure 3. Correlations between the percent change for minimum FCT and minimum FCT at baseline.
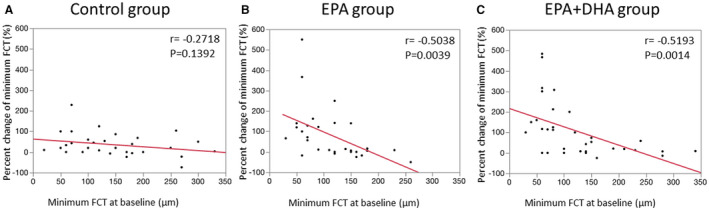
.
A, No correlation was found in the control group (r=−0.2718, P=0.1392). B, A significant negative correlation was found in the EPA group (r=−0.5368, P=0.0018). C, A significant negative correlation was found in the EPA+DHA group (r=−0.5193, P=0.0014). DHA indicates docosahexaenoic acid; EPA, eicosapentaenoic acid; and FCT, fibrous cap thickenss.
Figure 4. Comparison of the percent change for minimum FCT at the 8‐month follow‐up relative to baseline in the control, EPA, and EPA+DHA groups in subgroup analysis.
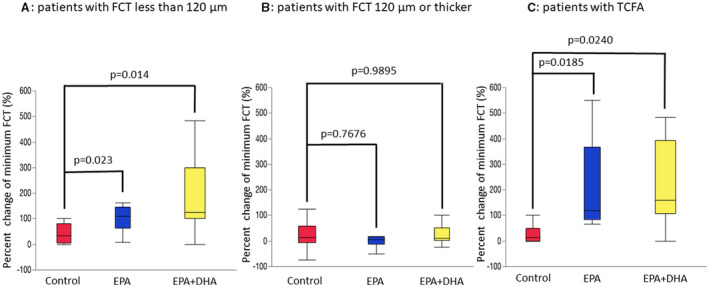
A, In patients with minimum FCT <120 µm, percent change for FCT was significantly lower in the control group (33.3; interquartile interval, 5.0–80.0) compared with the EPA group (110; interquartile interval, 64.3–145.6; P=0.0230 vs control) or the EPA+DHA group (125; interquartile interval, 100.0–300.0; P=0.0140 vs control). B, In patients with minimum FCT ≥120 µm, there were no significant differences between the control group (14.2; interquartile interval, −6.0 to 57.5) and the EPA group (5.6; interquartile interval, −13.0 to 15.5; P=0.768 vs control) or the EPA+DHA group (10.9; interquartile interval, 1.8–10.9, P=0.990 vs control). C, In patients with TCFA, percent change for minimum FCT was significantly higher in the EPA group (120.0; interquartile interval, 83.3–366.7; P=0.0185 vs control) or the EPA+DHA group (160; interquartile interval, 108.3–391.7; P=0.0240 vs control) compared with the control group (15.0; interquartile interval, 0–50.0). DHA indicates docosahexaenoic acid; EPA; eicosapentaenoic acid; FCT, fibrous cap thickenss; interquartile interval, interquartile range; and TCFA, thin‐cap fibroatheroma.
Clinical Outcomes
During the follow‐up period of 12 months, there were no significant differences in major cardiovascular events, all‐cause death, cardiac death, nonfatal myocardial infarction, stroke, coronary revascularization, or target lesion revascularization between the control group and the other 2 groups (Table S2).
DISCUSSION
This prospective randomized study evaluated the vascular healing response in coronary atherosclerotic plaques in patients with ACS treated with a strong statin alone, EPA therapy in addition to strong statin therapy, or EPA and DHA therapy in addition to strong statin therapy using OCT. There were 3 principal findings of the present study. First, between baseline and the 8‐month follow‐up, minimum FCT significantly increased in all 3 groups. Second, a significant negative correlation was observed between baseline minimum FCT and percent change for minimum FCT in the EPA and EPA+DHA groups, whereas no correlation was found with strong statin therapy alone. Third, there were no significant differences in the percent change for minimum FCT between the EPA or EPA+DHA group and the control group. However, in patients with thinner FCT, the percent change for minimum FCT was significantly higher in the EPA and EPA+DHA groups compared with strong statin therapy alone.
Current guidelines focus on LDL‐C as the primary target of therapy, tailoring the level of optimal LDL‐C reduction to the individual's level of risk. 18 , 19 , 20 However, cardiovascular risk in patients on statin therapy remains high; this risk has been called residual risk. Results from a meta‐analysis of statin trials involving 90 056 individuals found that the rate of major cardiovascular events occurring during 5 years of follow‐up among patients taking statins was 21.7% for those with prior cardiovascular disease and 9.5% for those without prior disease. 21 Residual risk was lower after low LDL‐C levels (70–100 mg/dL) were achieved, but remained high in the Treating to New Targets trial, with 8.7% of the group allocated to atorvastatin 80 mg experiencing a major event during a 5‐year period. 2 Therefore, treatments in addition to strong stains are necessary for reducing the residual risk for cardiovascular events.
Several epidemiological and clinical studies showed that long‐term intake of PUFAs such as EPA and DHA can reduce cardiovascular events. 1 , 2 In the JELIS trial, 3 18 645 patients with hypercholesterolemia were randomized to receive statin alone or statin plus highly purified EPA. At the end of 5 years, those randomized to statin plus EPA had a 19% lower occurrence of major cardiovascular events. Recently, the Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial, 22 which enrolled 8179 patients with hyperglyceridemia randomized to receive EPA (total daily dose, 4 g) or placebo, demonstrated that EPA treatment reduced the risk of cardiovascular events by 25% compared with placebo with a median follow‐up of 4.9 years and that EPA treatment even in patients receiving strong or moderate statin therapy at baseline was associated with a significant reduction in cardiovascular events. As shown in both large‐scale randomized controlled trials, EPA in addition to statin treatment is considered an effective approach for reducing the residual risk for cardiovascular events. Regarding the effect of EPA+DHA therapy on cardiovascular events, the Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico‐Prevenzione trial, 23 a large, randomized controlled trial, demonstrated that EPA+DHA supplements are associated with a significant reduction in cardiovascular events compared with placebo. However, the effect of EPA+DHA therapy in addition to statin therapy on cardiovascular events has not been reported to date. Therefore, larger randomized controlled studies are needed to elucidate the effect of adjunctive EPA+DHA treatment in addition to strong statin treatment on cardiovascular events.
Vulnerable plaques, which contain a thin fibrous cap, large lipid or necrotic core, and increased number of inflammatory cells such as macrophages, has been demonstrated to predispose patients to plaque rupture and subsequent ACS. 6 , 7 Inflammation is now recognized to play an important role in atherosclerotic plaque progression. 24 , 25 The anti‐inflammatory actions of marine PUFAs may stabilize atherosclerotic plaques by decreasing inflammatory and immune cell infiltration and activity in the plaque. 26 Matsumoto et al 27 demonstrated that EPA treatment resulted in increased collagen content, more smooth muscle cells, and less macrophage infiltration. Wu et al 28 showed that a diet rich in EPA or DHA reduces the levels of apoptosis induced by oxidized LDL and prevents atherosclerotic progression. Therefore, PUFAs including EPA and DHA stabilize vulnerable plaques through anti‐inflammatory effects and antioxidant effects in atherosclerotic lesions. In the present study, the association between anti‐inflammatory effects of PUFAs and plaque stabilization was not clear because macrophage accumulation was not evaluated with OCT. A recent optical frequency domain imaging study reported that minimum FCT is inversely correlated with macrophage grade. 29 Our study demonstrated a significant negative correlation between baseline minimum FCT and increased FCT in the EPA and EPA+DHA groups, whereas no correlation was found with strong statin therapy alone. These results suggest that PUFAs have a potential benefit on atherosclerotic plaque with substantial inflammatory cell infiltration.
Several intravascular imaging studies 30 , 31 , 32 have demonstrated the effect of EPA treatment in addition to strong statin therapy on coronary atherosclerotic plaques. In a study using integrated backscatter intravascular ultrasound, which can classify plaque components based on a color‐coded map, 33 Niki et al 30 reported that lipid volume is significantly lower in patients treated with EPA in addition to strong statin therapy compared with patients treated with strong stain therapy alone. The change in lipid volume was significantly correlated with the change in pentraxin 3 or monocyte chemoattractant protein‐1 levels in patients who received adjunctive EPA therapy. This study suggested that EPA plus strong statin therapy stabilizes plaque components better than strong statin therapy alone through the suppression of coronary inflammation. Rupture of a vulnerable plaque, which consists of thin FCT, large lipid pools, and macrophage infiltration around the fibrous cap, is considered a major cause of ACS. 6 OCT is a high‐resolution intravascular imaging modality that can provide detailed vulnerable plaque visualization. 10 In particular, OCT has the advantage of being able to accurately measure FCT and detect the accumulation of macrophages compared with intravascular ultrasound. An OCT study targeting lesions with TCFA reported that the change in FCT is significantly higher and the incidence of macrophage accumulation at follow‐up is significantly lower with combined EPA and strong statin treatment compared with strong statin treatment alone. 32 In the present study, the percent change for minimum FCT was significantly higher in the EPA and EPA+DHA groups than in the control group among patients with thinner FCT (<120 µm) or TCFA. However, there was no significant difference in the percent change for minimum FCT in patients with thicker FCT (≥120 µm). These results suggest that adjunctive EPA treatment might have a more favorable benefit on atherosclerotic plaques with vulnerable features than those without vulnerable features.
The difference in anti‐atherosclerotic effects between EPA and DHA has not been clearly identified. Erkkilä et al 34 reported that higher plasma levels of DHA, but not EPA, are associated with less progression of coronary atherosclerosis in postmenopausal women with coronary artery disease. In a virtual histology intravascular ultrasound study that evaluated the determinants of coronary plaque progression in patients with diabetes mellitus taking statins, Nozue et al 35 reported that low‐serum DHA, but not EPA, is an independent predictor of coronary plaque progression. Takashima et al 36 reported that EPA+DHA treatment more effectively reduces atherogenesis than EPA treatment alone in apolipoprotein E‐deficient mice fed a Western diet. These results suggested that DHA itself exerts a protective effect on coronary plaque, and the administration of DHA might reduce cardiovascular events. In this study, the EPA+DHA group, despite receiving less than half the amount of EPA received by the EPA group, had a similar FCT increase compared with the EPA group in a subgroup analysis targeting coronary plaques with thin FCT or TCFA. These results suggested that DHA itself might contribute to coronary plaque stabilization by increasing FCT. To the best of our knowledge, the present study is the first to examine the effect of EPA+DHA as an adjunct to strong statin therapy on atherosclerotic coronary plaque.
Study Limitations
There are several limitations in this study. First, because before our study there had been no data about the effects of PUFAs on FCT changes evaluated by OCT, we could not correctly calculate the sample size for testing the assumption that PUFA therapy in addition to strong statin therapy significantly increase FCT compared with strong statin therapy alone. Second, the present study was planned and conducted before the current guideline 18 , 19 , 20 was published. The current European Society of Cardiology guideline 20 recommended LDL‐C level of <55 mg/dL in patients with very high risk such as ACS. In the present study, the dose of rosuvastatin was not adjusted to achieve a target LDL‐C level <55 mg/day. Third, EPA content varied between the EPA group (1800 mg/day) and the EPA+DHA group (930 mg/day). Fourth, OCT is less suitable for accurate lipid core size measurement compared with intravascular ultrasound because of its relatively shallow penetration depth. Fifth, randomization did not lead to a similar prevalence of diabetes mellitus that might affect atherosclerotic plaque progression. Because diabetes mellitus has played an important role for the progressions of atherosclerotic plaque, a lower prevalence of diabetes mellitus in the EPA group might have affected coronary plaque regression. However, a recent OCT study reported that the response of coronary plaques to statin therapy is similar between patients with and without diabetes mellitus. 37 Similarly, in the present study, there was no significant difference in the change in OCT measurements between patients with and without diabetes mellitus (Table S3). Therefore, it is considered that the impact of diabetes mellitus on the change in OCT measurements have little in this study. Sixth, the present study was conducted at multiple centers within Japan, where people generally have a higher dietary intake of PUFAs than people in many other countries. Seventh, for testing the assumption that the change in FCT after statin administration was 100 µm, 44 patients were required in each group to provide a statistical power of 90% in the present study. However, the number of enrolled patients did not reach the target, and 97 patients were ultimately included in the analysis. Therefore, if the number of enrolled patients was set at 32 patients per group, a statistical power was calculated as 78.2% in the present study. Eighth, the analyses did not adhere to the intention‐to‐treat principle. Finally, given that there are several limitations, especially the small sample size analyzed and the insufficient target LDL‐C level, it should be better that the results of the present study are considered as hypothesis generating. The results of subgroup analysis (Figure 4) should be interpreted with caution because primary end point was not met.
CONCLUSIONS
In this study of patients with ACS, EPA or EPA+DHA therapy in addition to strong statin therapy did not significantly increase FCT in nonculprit plaques compared with strong statin therapy alone, but significantly increased FCT in patients with thinner FCT. EPA or EPA+DHA therapy in addition to strong statin therapy might provide a greater stabilizing effect on coronary plaques with vulnerable features.
Sources of Funding
None.
Disclosures
None.
Supporting information
Tables S1–S3
Acknowledgments
We thank the following physicians who participated as investigators in this study and who made this work possible: Yu Sugawara, Yutaka Goryo, and Takuya Isojima.
(J Am Heart Assoc. 2020;9:e05331 DOI: 10.1161/JAHA.119.015593.)
For Sources of Funding and Disclosures, see page 12.
References
- 1. O'Keefe JH Jr, Cordain L, Harris WH, Moe RM, Vogel R. Optimal low‐density lipoprotein is 50 to 70 mg/dl: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43:2142–2146. [DOI] [PubMed] [Google Scholar]
- 2. LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, Gotto AM, Greten H, Kastelein JJ, Shepherd J, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435. [DOI] [PubMed] [Google Scholar]
- 3. Daviglus ML, Stamler J, Orencia AJ, Dyer AR, Liu K, Greenland P, Walsh MK, Morris D, Shekelle RB. Fish consumption and the 30‐year risk of fatal myocardial infarction. N Engl J Med. 1997;336:1046–1053. [DOI] [PubMed] [Google Scholar]
- 4. Kromhout D, Bosschieter EB, de Lezenne CC. The inverse relation between fish consumption and 20‐year mortality from coronary heart disease. N Engl J Med. 1985;312:1205–1209. [DOI] [PubMed] [Google Scholar]
- 5. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open‐label, blinded endpoint analysis. Lancet. 2007;369:1090–1098. [DOI] [PubMed] [Google Scholar]
- 6. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. [DOI] [PubMed] [Google Scholar]
- 7. Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, et al. A prospective natural‐history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. [DOI] [PubMed] [Google Scholar]
- 8. Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, Crowe T, Howard G, Cooper CJ, Brodie B, et al. Effect of intensive compared with moderate lipid‐lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–1080. [DOI] [PubMed] [Google Scholar]
- 9. Komukai K, Kubo T, Kitabata H, Matsuo Y, Ozaki Y, Takarada S, Okumoto Y, Shiono Y, Orii M, Shimamura K, et al. Effect of atorvastatin therapy on fibrous‐cap thickness in coronary atherosclerotic plaque as assessed by optical coherence tomography: the EASY‐FIT study. J Am Coll Cardiol. 2014;64:2207–2217. [DOI] [PubMed] [Google Scholar]
- 10. Jang IK, Tearney GJ, MacNeill B, Takano M, Moselewski F, Iftima N, Shishkov M, Houser S, Aretz HT, Halpern EF, et al. In vivo characterization of coronary atherosclerotic plaque by use of optical coherence tomography. Circulation. 2005;111:1551–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teramoto T, Sasaki J, Ishibashi S, Birou S, Daida H, Dohi S, Egusa G, Hiro T, Hirobe K, Iida M, et al. Comprehensive risk management for the prevention of cardiovascular disease: executive summary of the Japan Atherosclerosis Society (JAS) guidelines for the diagnosis and prevention of atherosclerotic cardiovascular diseases in Japan—2012. J Atheroscler Thromb. 2013;20:603–615. [DOI] [PubMed] [Google Scholar]
- 12. Watanabe M, Uemura S, Sugawara Y, Ueda T, Soeda T, Takeda Y, Kawata H, Kawakami R, Saito Y. Side branch complication after a single‐stent crossover technique: prediction with frequency domain optical coherence tomography. Coron Artery Dis. 2014;25:321–329. [DOI] [PubMed] [Google Scholar]
- 13. Tearney GJ, Regar E, Akasaka T, Adriaenssens T, Barlis P, Bezerra HG, Bouma B, Bruining N, Cho JM, Chowdhary S, et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: a report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J Am Coll Cardiol. 2012;59:1058–1072. [DOI] [PubMed] [Google Scholar]
- 14. Yamano T, Kubo T, Shiono Y, Shimamura K, Orii M, Tanimoto T, Matsuo Y, Ino Y, Kitabata H, Yamaguchi T, et al. Impact of eicosapentaenoic acid treatment on the fibrous cap thickness in patients with coronary atherosclerotic plaque: an optical coherence tomography study. J Atheroscler Thromb. 2015;22:52–61. [DOI] [PubMed] [Google Scholar]
- 15. Kini AS, Vengrenyuk Y, Yoshimura T, Matsumura M, Pena J, Baber U, Moreno P, Mehran R, Maehara A, Sharma S, et al. Fibrous cap thickness by optical coherence tomography in vivo. J Am Coll Cardiol. 2017;69:644–657. [DOI] [PubMed] [Google Scholar]
- 16. Kato K, Yonetsu T, Kim SJ, Xing L, Lee H, McNulty I, Yeh RW, Sakhuja R, Zhang S, Uemura S, et al. Nonculprit plaques in patients with acute coronary syndromes have more vulnerable features compared with those with non‐acute coronary syndromes: a 3‐vessel optical coherence tomography study. Circ Cardiovasc Imaging. 2012;5:433–440. [DOI] [PubMed] [Google Scholar]
- 17. Takarada S, Imanishi T, Kubo T, Tanimoto T, Kitabata H, Nakamura N, Tanaka A, Mizukoshi M, Akasaka T. Effect of statin therapy on coronary fibrous‐cap thickness in patients with acute coronary syndrome: assessment by optical coherence tomography study. Atherosclerosis. 2009;202:491–497. [DOI] [PubMed] [Google Scholar]
- 18. Kinoshita M, Yokote K, Arai H, Iida M, Ishigaki Y, Ishibashi S, Umemoto S, Egusa G, Ohmura H, Okamura T, et al. Japan Atherosclerosis Society (JAS) Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases 2017. J Atheroscler Thromb. 2018;25:846–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, et al. 2018 AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2019;73:3168–3209. [DOI] [PubMed] [Google Scholar]
- 20. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. [DOI] [PubMed] [Google Scholar]
- 21. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, et al. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 22. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr, Juliano RA, Jiao L, Granowitz C, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380:11–22. [DOI] [PubMed] [Google Scholar]
- 23. Valagussa F, Franzosi MG, Geraci E. Dietary supplementation with n‐3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI‐Prevenzione trial. Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- 24. van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. [DOI] [PubMed] [Google Scholar]
- 25. Bogaty P, Poirier P, Simard S, Boyer L, Solymoss S, Dagenais GR. Biological profiles in subjects with recurrent acute coronary events compared with subjects with long‐standing stable angina. Circulation. 2001;103:3062–3068. [DOI] [PubMed] [Google Scholar]
- 26. Calder PC. The role of marine omega‐3 (n‐3) fatty acids in inflammatory processes, atherosclerosis and plaque stability. Mol Nutr Food Res. 2012;56:1073–1080. [DOI] [PubMed] [Google Scholar]
- 27. Matsumoto M, Sata M, Fukuda D, Tanaka K, Soma M, Hirata Y, Nagai R. Orally administered eicosapentaenoic acid reduces and stabilizes atherosclerotic lesions in ApoE‐deficient mice. Atherosclerosis. 2008;197:524–533. [DOI] [PubMed] [Google Scholar]
- 28. Wu T, Geigerman C, Lee YS, Wander RC. Enrichment of LDL with EPA and DHA decreased oxidized LDL‐induced apoptosis in U937 cells. Lipids. 2002;37:789–796. [DOI] [PubMed] [Google Scholar]
- 29. Konishi T, Sunaga D, Funayama N, Yamamoto T, Murakami H, Hotta D, Nojima M, Tanaka S. Eicosapentaenoic acid therapy is associated with decreased coronary plaque instability assessed using optical frequency domain imaging. Clin Cardiol. 2019;42:618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niki T, Wakatsuki T, Yamaguchi K, Taketani Y, Oeduka H, Kusunose K, Ise T, Iwase T, Yamada H, Soeki T, et al. Effects of the addition of eicosapentaenoic acid to strong statin therapy on inflammatory cytokines and coronary plaque components assessed by integrated backscatter intravascular ultrasound. Circ J. 2016;80:450–460. [DOI] [PubMed] [Google Scholar]
- 31. Watanabe T, Ando K, Daidoji H, Otaki Y, Sugawara S, Matsui M, Ikeno E, Hirono O, Miyawaki H, Yashiro Y, et al. A randomized controlled trial of eicosapentaenoic acid in patients with coronary heart disease on statins. J Cardiol. 2017;70:537–544. [DOI] [PubMed] [Google Scholar]
- 32. Nishio R, Shinke T, Otake H, Nakagawa M, Nagoshi R, Inoue T, Kozuki A, Hariki H, Osue T, Taniguchi Y, et al. Stabilizing effect of combined eicosapentaenoic acid and statin therapy on coronary thin‐cap fibroatheroma. Atherosclerosis. 2014;234:114–119. [DOI] [PubMed] [Google Scholar]
- 33. Okubo M, Kawasaki M, Ishihara Y, Takeyama U, Yasuda S, Kubota T, Tanaka S, Yamaki T, Ojio S, Nishigaki K, et al. Tissue characterization of coronary plaques: comparison of integrated backscatter intravascular ultrasound with virtual histology intravascular ultrasound. Circ J. 2008;72:1631–1639. [DOI] [PubMed] [Google Scholar]
- 34. Erkkilä AT, Matthan NR, Herrington DM, Lichtenstein AH. Higher plasma docosahexaenoic acid is associated with reduced progression of coronary atherosclerosis in women with CAD. J Lipid Res. 2006;47:2814–2819. [DOI] [PubMed] [Google Scholar]
- 35. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Nozato T, Miyake S, et al. Low serum docosahexaenoic acid is associated with progression of coronary atherosclerosis in statin‐treated patients with diabetes mellitus: results of the treatment with statin on atheroma regression evaluated by intravascular ultrasound with virtual histology (TRUTH) study. Cardiovasc Diabetol. 2014;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takashima A, Fukuda D, Tanaka K, Higashikuni Y, Hirata Y, Nishimoto S, Yagi S, Yamada H, Soeki T, Wakatsuki T, et al. Combination of n‐3 polyunsaturated fatty acids reduces atherogenesis in apolipoprotein E‐deficient mice by inhibiting macrophage activation. Atherosclerosis. 2016;254:142–150. [DOI] [PubMed] [Google Scholar]
- 37. Kurihara O, Thondapu V, Kim HO, Russo M, Sugiyama T, Yamamoto E, Fracassi F, Minami Y, Wang Z, Lee H, et al. Comparison of vascular response to statin therapy in patients with versus without diabetes mellitus. Am J Cardiol. 2019;123:1559–1564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3