

Abstract
Sodium‐glucose cotransporter 2 (SGLT2) inhibitors reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure. Initially, these drugs were believed to have a profile similar to diuretics or hemodynamically active drugs, but they do not rapidly reduce natriuretic peptides or cardiac filling pressures, and they exert little early benefit on symptoms, exercise tolerance, quality of life, or signs of congestion. Clinically, the profile of SGLT2 inhibitors resembles that of neurohormonal antagonists, whose benefits emerge gradually during sustained therapy. In experimental models, SGLT2 inhibitors produce a characteristic pattern of cellular effects, which includes amelioration of oxidative stress, mitigation of mitochondrial dysfunction, attenuation of proinflammatory pathways, and a reduction in myocardial fibrosis. These cellular effects are similar to those produced by angiotensin converting enzyme inhibitors, β‐blockers, mineralocorticoid receptor antagonists, and neprilysin inhibitors. At a molecular level, SGLT2 inhibitors induce transcriptional reprogramming of cardiomyocytes that closely mimics that seen during nutrient deprivation. This shift in signaling activates the housekeeping pathway of autophagy, which clears the cytosol of dangerous cytosolic constituents that are responsible for cellular stress, thereby ameliorating the development of cardiomyopathy. Interestingly, similar changes in cellular signaling and autophagic flux have been seen with inhibitors of the renin‐angiotensin system, β‐blockers, mineralocorticoid receptor antagonists, and neprilysin inhibitors. The striking parallelism of these molecular, cellular, and clinical profiles supports the premise that SGLT2 inhibitors should be regarded as neurohormonal antagonists when prescribed for the treatment of heart failure with a reduced ejection fraction.
Keywords: heart failure, neurohormonal antagonists, SGLT2 inhibitors
Subject Categories: Cardiomyopathy, Heart Failure, Oxidant Stress, Mechanisms, Growth Factors/Cytokines
Nonstandard Abbreviations and Acronyms
- ACE
angiotensin‐converting enzyme
- Akt
protein kinase B
- AMPK
adenosine monophosphate‐activated protein kinase
- AMPKα2
adenosine monophosphate‐activated protein kinase isoform alpha 2
- DAPA‐HF
Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure
- EMPEROR‐Reduced
Empagliflozin Outcome Trial in Chronic Heart Failure With Reduced Ejection Fraction
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- mTORC2
mammalian target of rapamycin complex 2
- SGLT2
sodium‐glucose cotransporter 2
- SIRT1
sirtuin‐1
First proposed in 1992, the neurohormonal hypothesis postulates that heart failure with a reduced ejection fraction should be regarded as a neurohormonal disorder and that these patients should benefit from the use of drugs that interfere with the deleterious effects of neurohormonal systems.1 At the time of its formulation, angiotensin‐converting enzyme (ACE) inhibitors were the only neurohormonal antagonist that had been approved for use in patients with chronic heart failure. However, since 1992, numerous large‐scale clinical trials have demonstrated the benefits of β‐blockers, mineralocorticoid receptor antagonists, and sacubitril/valsartan.2 These drugs interfere with the deleterious effects of excessive activation of the sympathetic nervous system, aldosterone, and neprilysin that characterizes patients with heart failure and impaired systolic function.
Combination therapy with multiple neurohormonal antagonists represents the cornerstone of class I recommendations in current heart failure guidelines based on compelling evidence that these drugs prolong survival in a broad spectrum of patients with heart failure and a reduced ejection fraction in trials that recorded a meaningful number of serious cardiovascular events.3 Other drugs that are recommended for use in chronic heart failure (eg, digoxin, ivabradine, and hydralazine/isosorbide dinitrate) act primarily to reduce the risk of heart failure hospitalizations or have been reported to reduce the risk of death based only on small numbers of events or in select groups.3, 4
In recent years, sodium‐glucose cotransporter 2 (SGLT2) inhibitors were shown to reduce the risk of heart failure hospitalizations (and often cardiovascular death) in high‐risk patients with type 2 diabetes mellitus who generally did not have heart failure at the time of enrollment in the trials.5 Furthermore, in the DAPA‐HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, dapagliflozin reduced the risk of cardiovascular death in patients with established heart failure and a reduced ejection fraction, including those without diabetes mellitus. A second large‐scale trial (EMPEROR‐Reduced [Empagliflozin Outcome Trial in Chronic Heart Failure With Reduced Ejection Fraction]) that is evaluating the effects of empagliflozin in patients with advanced disease is nearing completion.6 If the DAPA‐HF and EMPEROR‐Reduced trials yield concordant findings, then SGLT2 inhibitors will likely join the ranks of the current class I recommended drugs for heart failure; but should SGLT2 inhibitors be regarded as neurohormonal antagonists, akin to how we currently think about angiotensin receptor neprilysin inhibitors, β‐blockers, and mineralocorticoid receptor antagonists?
WHAT FEATURES OF A DRUG IDENTIFY IT AS A NEUROHORMONAL ANTAGONIST?
In the 1970s and 1980s, heart failure was regarded primarily as a hemodynamic disorder.7 Decreases in cardiac output and increases in left ventricular filling pressures were attributed to an impairment in cardiac contractility and constriction of arterial resistance and venous capacitance vessels. Treatment focused on the use of diuretics, systemic vasodilators, and positive inotropic agents; these drugs produced immediate changes in hemodynamic variables, often with rapid relief of dyspnea. However, the immediate actions of positive inotropic and systemic vasodilator drugs often failed to predict their long‐term effects. Sustained treatment with hemodynamically active drugs often stimulated deleterious neurohormonal systems or mimicked their adverse actions on the myocardium.8 As a result, the short‐term benefits of these agents were frequently not sustained,9 and prolonged therapy often acted to accelerate progression of the underlying disease and increase the risk of hospitalization and death.10, 11
Neurohormonal antagonists (which interfere with the actions of deleterious endogenous mechanisms) exhibit a strikingly different pattern of response. The benefits of these drugs emerge slowly and typically require prolonged treatment to become apparent. In many instances, these drugs produce no immediate benefit on cardiac output or left ventricular filling pressures or lead to unwanted hemodynamic responses, for example, hypotension with ACE inhibitors, carvedilol and neprilysin inhibitors, and worsening heart failure with β‐blockers. Any favorable effect on left ventricular remodeling or ejection fraction is often delayed for 6 to 12 months or longer.12 During the first weeks or months of treatment, patients often report little improvement in quality of life or exercise tolerance.13, 14 Nevertheless, long‐term treatment is accompanied by striking effects to reduce the risk of death and hospitalization. Interestingly, prior treatment with ACE inhibitors and β‐blockers has not precluded the benefits of mineralocorticoid receptor antagonists and neprilysin inhibitors,2 suggesting that simultaneous antagonism of different deleterious pathways is needed to produce optimal effects on survival.
The features and time course of the response to treatment distinguish neurohormonal antagonists from diuretics or hemodynamically active agents. The benefits of neurohormonal interventions emerge slowly, presumably because the heart needs time to recover once it is shielded from endogenous influences that are injurious to cardiomyocyte function and survival. Viewed from this perspective, neurohormonal antagonism in chronic heart failure represents a form of pharmacological cardioprotection.
SGLT2 INHIBITORS DO NOT EXERT THEIR BENEFITS IN CHRONIC HEART FAILURE BY AN ACTION ON HEMODYNAMIC VARIABLES
When first introduced into clinical use, SGLT2 inhibitors were believed to have immediate and clinically relevant hemodynamic effects. These drugs inhibit sodium reabsorption in the proximal renal tubule by an action to block glucose reuptake (via SGLT2) and possibly by an effect to interfere with sodium‐hydrogen exchanger isoform 3.15 In patients with acute heart failure, SGLT2 inhibitors promote an increase in urine volume that persists for several days,16 and this natriuretic action may lead to decreases in plasma and/or interstitial volume.17 In addition, SGLT2 inhibitors exert vasodilator effects that may underlie their action to lower blood pressure.18 Accordingly, it was suggested that SGLT2 inhibitors act primarily to lower cardiac filling pressures.
However, the effect of SGLT2 inhibitors on urinary sodium excretion in chronic heart failure appears to be modest and transient, and any reduction in plasma volume is often short lived.19, 20, 21 As a result, these drugs do not produce the immediate and striking decreases in circulating natriuretic peptides that are typically seen with loop diuretics, and treatment does not alleviate pulmonary or systemic congestion.22 In addition, the effects of loop diuretics have been associated with an increased risk of cardiovascular death,23 whereas SGLT2 inhibitors decrease cardiovascular mortality, both in patients with type 2 diabetes mellitus as well as those with chronic heart failure.5
Some have proposed that SGLT2 inhibitors may produce hemodynamic benefits by enhancing the production of ketone bodies.24 The short‐term infusion of β‐hydroxybutyrate in supraphysiological doses increases both cardiac contractility and heart rate in patients with a reduced ejection fraction.25 However, the clinical significance of this finding remains uncertain because SGLT2 inhibitors do not appear to produce positive inotropic and chronotropic effects in clinical trials. The failing heart already preferentially consumes ketone bodies as a fuel,26 and in experimental models, SGLT2 inhibition does not consistently improve myocardial ketone body use.27, 28, 29 Importantly, any improvement in cardiac performance that might result from the increase in myocardial adenosine triphosphate produced by SGLT2 inhibitors is not related to enhanced ketone body metabolism.28, 29 In hemodynamic studies, the cardiovascular benefits of these drugs are not related to changes in cardiac contractility or ventricular loading conditions.30
The cardioprotective effects of SGLT2 inhibitors evolve slowly, and their actions to reduce left ventricular mass and cardiac volumes are seen during prolonged therapy.31, 32 Furthermore, in a manner akin to conventional neurohormonal antagonists, SGLT2 inhibitors do not act rapidly to improve symptoms and show little effect on exercise tolerance or quality of life in trials of 3 months’ duration (Table).22 Any benefits of SGLT2 inhibitors on functional capacity33 may result from (rather than precede) the effects of these drugs to favorably influence the underlying cardiomyopathy.
Table 1.
Clinical, Cellular, and Molecular Features That Distinguish Hemodynamically Active Drugs, Established Neurohormonal Antagonists, and SGLT2 Inhibitors
| Diuretics, Systemic Vasodilators, and Positive Inotropic Drugs | Established Neurohormonal Antagonists | SGLT2 Inhibitors | |
|---|---|---|---|
| Immediate effects on cardiac output, filling pressures, and natriuretic peptides | Present and desirable | Often absent and frequently undesirable | Generally absent in clinically stable patients |
| Ability to rapidly improve symptoms, exercise tolerance, and quality of life | Frequently present | Frequently absent | Generally absent |
| Effect to reduce the risk of cardiovascular death | Usually absent | Characteristically present | Usually present |
| Effect to ameliorate oxidative stress, organellar dysfunction, and cellular inflammation | Usually absent | Characteristically present | Characteristically present |
| Enhancement of SIRT1/AMPK and attenuation of Akt/mTOR signaling | Inconsistent and not characteristic | Present with several drug classes | Characteristic of members of the drug class |
| Augmentation of autophagic flux | Inconsistent and not characteristic | Reported with several drug classes | Noted with several members of the drug class |
Akt indicates Akt/protein kinase B; AMPK, adenosine monophosphate‐activated protein kinase; mTOR, mammalian target of rapamycin; SGLT2, sodium‐glucose cotransporter 2; and SIRT1, sirtuin‐1.
SGLT2 INHIBITORS EXERT CARDIOPROTECTIVE EFFECTS IN EXPERIMENTAL MODELS THAT ARE SIMILAR TO THOSE PRODUCED BY CONVENTIONAL NEUROHORMONAL ANTAGONISTS
SGLT2 inhibitors exert cardioprotective effects in numerous models of diabetic and nondiabetic cardiac injury. Regardless of the inciting cause, treatment leads to the amelioration of oxidative stress, mitigation of mitochondrial dysfunction, attenuation of proinflammatory pathways, and a reduction in myocardial fibrosis.34, 35, 36, 37, 38, 39 The pattern of these effects is similar to that produced by ACE inhibitors, β‐blockers, mineralocorticoid receptor antagonists, and neprilysin inhibitors. ACE inhibitors reduce oxidative stress, preserve mitochondrial function, and mitigate proinflammatory pathways, effects that are superior to those seen with angiotensin receptor blockers.40, 41, 42 β‐blockers attenuate the generation of reactive oxygen species, inhibit mechanisms that trigger cellular inflammation, and promote mitochondrial biogenesis.43, 44, 45, 46 Mineralocorticoid receptor antagonists alleviate oxidative stress and reduce the inflammatory responses that lead to fibrosis.47, 48, 49, 50 Neprilysin inhibitors stabilize mitochondrial function and protect the heart from the injurious effects of reactive oxygen species and inflammation.51, 52, 53 The striking parallelism in the cellular effects of SGTL2 inhibitors and established neurohormonal antagonists support the hypothesis that the actions of SGLT2 inhibitors are akin to those produced by drugs that interfere with endogenous neurohormonal systems (Table).
The ability of SGLT2 inhibitors to exert cardioprotective effects on a cellular level is intriguing because the healthy or failing heart does not express SGLT2,54 and SGLT2 inhibitors do not bind to cardiac tissue.55 In contrast, cardiomyocytes have identifiable receptors for angiotensin II, norepinephrine, aldosterone, and natriuretic peptides, and interaction with these receptors is generally required for the effect of neurohormonal antagonists to ameliorate cellular stress and organellar dysfunction. Although SGLT2 inhibitors may modestly attenuate the sympathetic response to volume depletion,56 the direct measurements of sympathetic activity have demonstrated little inhibitory action of these drugs.57 Furthermore, SGLT2 inhibitors activate the renin‐angiotensin‐aldosterone system,58 suggesting that these drugs do not inhibit the mechanisms that are targeted by conventional neurohormonal antagonists.
Therefore, the benefits of SGLT2 inhibitors to alleviate cellular stress and organellar dysfunction result from interference with endogenous mechanisms that adversely affect the heart, but these are not the conventional pathways that are targeted by established drugs, and they are not mediated by identifiable receptor in the heart. What is the identity of the injurious mechanisms that are blocked by SGLT2 inhibitors?
IMPORTANCE OF AUTOPHAGIC FLUX IN ATTENUATING CELLULAR STRESS AND MITIGATING THE DEVELOPMENT OF CARDIOMYOPATHY
In chronic heart failure (regardless of etiology), glucose and lipid intermediates accumulate in cardiomyocytes59, 60 and play a critical role in causing oxidative and endoplasmic reticulum stress.61, 62 These stresses directly impair the structural integrity and normal functioning of mitochondria and peroxisomes,63, 64 thus promoting the generation of reactive oxygen species and activating proinflammatory pathways, further enhancing cellular stress. These cellular stresses and organellar derangements are normally constrained by a cellular housekeeping pathway known as autophagy.
Autophagy is a lysosome‐mediated degradative process that allows cardiomyocytes to clear the accumulation of intracellular glucose and lipid pools, and it also promotes the disposal of dysfunctional and damaged mitochondria and peroxisomes, thus muting oxidative stress and proinflammatory mechanisms.65, 66 The autophagic capacity of cardiomyocytes is markedly impaired in human heart failure67, 68; yet pharmacological stimulation of autophagic flux can directly ameliorate oxidative stress and organellar dysfunction, thereby preventing or reversing cardiomyocyte dysfunction and demise, and mitigating the development of cardiomyopathy.69, 70, 71, 72 Activation of autophagy represents a major shift in the priorities of cardiomyocytes away from growth toward the preservation of cellular homeostasis and survival.73 Importantly, the intensity of autophagic flux in cardiomyocytes is finely regulated by the balance of enzymes and transcription factors that are exquisitely sensitive to environmental conditions, particularly states of nutrient and energy deprivation and overabundance.
Transcription Factors That Modulate Autophagic Flux in Cardiomyocytes and Influence the Evolution of Progression of Myocardial Dysfunction
Autophagy is stimulated by nutrient depletion because any reduction in environmental fuel requires cells to curtail growth and direct their efforts to support organellar function and cellular homeostasis. The major sensors of glucose deprivation are SIRT1 (sirtuin‐1) and AMPK (adenosine monophosphate‐activated protein kinase). SIRT1 is a redox‐sensitive nicotinamide adenine dinucleotide–dependent deacetylase that functions to maintain blood glucose.74 AMPK is sensitive to the balance between adenosine triphosphate and adenosine diphosphate or adenosine monophosphate in the cytosol; its activation promotes the generation of adenosine triphosphate.75 Both SIRT1 and AMPK are enzymes that regulate the activity of hundreds of genes and proteins that are involved in metabolism and cellular homeostasis; their functions are intertwined, and they suppress energy storage and promote catabolic pathways. Both SIRT1 and AMPK act to stimulate autophagy and thus promote the clearance of dysfunctional organelles and glucose/lipid intermediates, which are the primary sources of cellular stress. In addition, they act directly to preserve organellar integrity and mute inflammasome activation.76, 77
In states of energy overabundance, cells prioritize growth over cellular stability and survival, and during such times, autophagic flux is constrained by activation of Akt (protein kinase B) and mTOR (mammalian target of rapamycin). Both Akt and mTOR are serine/threonine protein kinases that function as critical promoters of cell proliferation; mTOR is expressed as 2 protein complexes: mTORC1 and mTORC2. Both Akt and mTORC1 are upregulated by nutrient surplus and by growth factors (eg, insulin), and Akt potentiates the activation of mTORC1.78, 79 When activated by nutrient surplus, both Akt and mTORC1 enhance glucose and lipid storage, oxidative metabolism, and mitochondrial oxygen consumption, and they promote the anabolic pathways that are required for cellular hypertrophy and replication.80, 81
Therefore, whereas SIRT1 and AMPK signaling promotes autophagy and cellular homeostasis and survival during nutrient deprivation, Akt and mTORC1 act to suppress autophagy and facilitate cellular expansion during nutrient overabundance. The actions of the Akt and mTORC1 pathway counterbalance those of SIRT1 and AMPK.82
Abnormalities of Nutrient‐Sensitive Transcription Factor Signaling in Experimental and Clinical Heart Failure and Their Modulation by Conventional Neurohormonal Antagonists
Both experimentally and clinically, heart failure is characterized by the simultaneous impairment of signaling through SIRT1 and AMPK83, 84, 85 and by enhanced activation of the Akt/mTORC1 pathway in cardiomyocytes.67, 70, 86
Akt/mTORC2 signaling promotes normal and adaptive cardiac growth during development to manage physiological hemodynamic stresses,87, 88 and it has short‐term protective effects during acute myocardial ischemia.89 However, prolonged activation of mTORC1 in adulthood priorities growth over survival, and in doing so, it causes pathological hypertrophy, inflammasome activation, and impaired mitochondrial bioenergetics, leading to adverse remodeling and cardiac dysfunction.90, 91 Enhanced mTORC1 signaling in the human heart promotes cellular stress and myocardial fibrosis, and it portends a poor prognosis in patients with nonischemic cardiomyopathy92; mTORC1 upregulation impairs cardiac function in obesity‐related heart failure.93 Conversely, agents that inhibit the AkT/mTORC1 pathway (ie, rapamycin) ameliorate the evolution and progression of experimental cardiomyopathy.70, 93, 94
In contrast with the actions of the Akt/mTOR pathway, activation of SIRT1/AMPK prioritizes cellular survival over growth. Signaling through SIRT1/AMPK prevents adverse hypertrophy, mutes inflammation, promotes mitochondrial health, and preserves cardiac function during diverse forms of cardiac stress.95, 96, 97, 98 The activity of SIRT1 and AMPKα2 (the primary isoform that mitigates cardiac stress) is suppressed in experimental cardiomyopathy and is accompanied by increased oxidative stress and adverse structural and functional changes in the myocardium.99, 100, 101 Conversely, interventions that promote AMPK/SIRT1 signaling ameliorate the severity of cardiac injury and development of experimental cardiomyopathy, regardless of its cause.72, 102, 103, 104, 105 The combined effect of SIRT1/AMPK downregulation and Akt/mTOR activation that is seen in chronic heart failure is responsible for the suppression of autophagy in cardiomyopathic hearts.106
Interestingly, activation of the renin‐angiotensin system, sympathetic nervous system, and aldosterone as well as downregulation of natriuretic peptide signaling also cause SIRT1/AMPK suppression and Akt/mTOR activation. ACE inhibitors can counteract the adverse effects of angiotensin II by signaling through SIRT1107, 108 because SIRT1 upregulation may interfere with the injurious actions of angiotensin II on heart.109 The benefits of angiotensin receptor blockers may be mediated by activation of AMPK and inhibition of Akt/mTOR100, 110; the latter effect underlies the action of these drugs to promote autophagy.111 β‐adrenergic receptor stimulation leads to the suppression of AMPK,112 and β‐blockade is accompanied by the upregulation of AMPK45, 113; additionally, carvedilol exerts anti‐inflammatory effects by an action to promote autophagy through a stimulatory effect on SIRT1114 and by the inhibition of mTOR.115 Spironolactone activates SIRT1/AMPK in the heart,116 and AMPK activation interferes with aldosterone‐mediated cardiac fibrosis.117 In addition, the action of spironolactone to inhibit Akt/mTOR signaling may explain its ability to promote autophagic flux.118, 119 Natriuretic peptides promote activation of AMPK,120, 121 and intriguingly, neprilysin can directly activate Akt/mTOR signaling independent of its effect on natriuretic peptides.122, 123 Therefore, the ability of established neurohormonal antagonists to influence the interplay of SIRT1/AMPK and Akt/mTOR so as to promote autophagy may contribute to the benefits of these drugs.
SGLT2 Inhibitors Modulate the Effects of Transcription Factors That Regulate Cellular Stresses and Autophagy and Can Influence the Development of Cardiomyopathy
SGLT2 acts as a sensor of nutrient overabundance,124 and thus there is an inverse relationship between SGLT2 and SIRT1125 (which functions as the principal sensor of nutrient deprivation126); simultaneously, there is a direct relationship between the intensity of proximal tubular sodium‐glucose reabsorption and the activation of Akt/mTOR.54 When the actions of SGLT2 are inhibited, the resulting urinary loss of calories triggers systemic transcriptional reprogramming that closely mimics that seen during states of nutrient deprivation.127 The depletion of tissue nutrients that follows glycosuria leads to the activation of SIRT1 and AMPK and the suppression of kinases that are normally activated by nutrient excess (Akt and mTOR) (Figure).127, 128 Several SGLT2 inhibitors have been shown to upregulate SIRT1 and AMPK while suppressing the Akt/mTOR pathway,38, 127, 128, 129, 130, 131, 132 thus explaining the ability of these drugs to promote autophagy in diverse organs, including the heart.36 The induction of autophagy underlies the ability of SGLT2 inhibitors to mute oxidative stress, promote organellar integrity, suppress proinflammatory pathways, and ameliorate the course of cardiomyopathy.34, 35, 36, 37, 38, 39 Importantly, because nutrient deprivation signaling is a system‐wide response, these benefits do not depend on the expression of SGLT2 in the heart.
Figure 1. Effect of SGLT2 inhibitors on nutrient‐deprivation and nutrient‐excess sensor signaling and the development of cardiomyopathy.
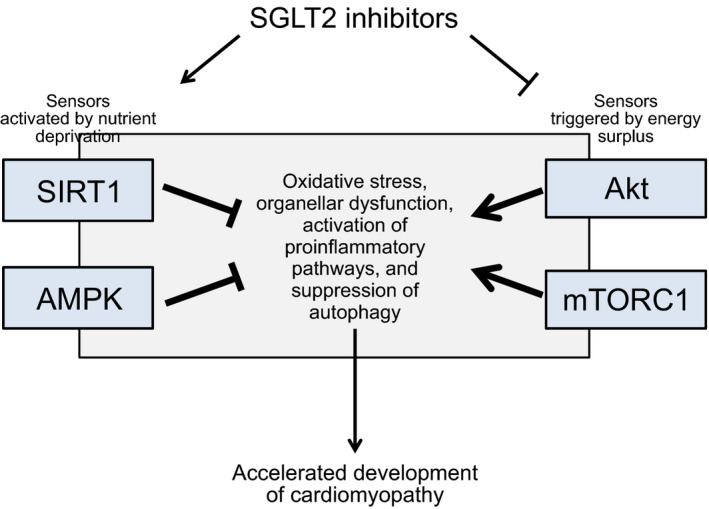
Akt indicates Akt/protein kinase B; AMPK, adenosine monophosphate‐activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; SGLT2, sodium‐glucose cotransporter 2 inhibitors; and SIRT1, sirtuin‐1.
CONCLUSIONS
Drugs that act primarily as diuretics or on hemodynamic variables can exert rapid effects on cardiac output, cardiac filling pressures, systemic vascular tone, or natriuretic peptides, and they can produce immediate effects to alleviate symptoms of congestion. However, long‐term treatment with these drugs often produces little benefit on the progression of heart failure and may increase the risk of death. In contrast, neurohormonal antagonists typically exert little immediate effect on symptoms or quality of life, but over time, they act to reduce morbidity and mortality because they shield the myocardium from the effects of endogenous mechanisms that can cause cellular stress and injury.
The pattern of responses to SGLT2 inhibitors closely mimics that of established neurohormonal antagonists. These drugs have limited immediate benefits on symptoms, quality of life, or natriuretic peptides, but sustained therapy reduces the risk of serious heart failure events. Experimentally, SGLT2 inhibitors ameliorate cellular stress, preserve mitochondrial integrity, and attenuate proinflammatory pathways, a profile of effects similar to that seen with inhibitors of the renin‐angiotensin system, β‐blockers, mineralocorticoid receptor antagonists, and neprilysin inhibitors. In addition, SGLT2 inhibitors promote the activation of SIRT1/AMPK and inhibit signaling through Akt/mTOR, thereby enhancing the cellular housekeeping process of autophagy and its effects to ameliorate cytosolic stress, enhance cellular survival, and ameliorate the development of cardiomyopathy. Similar effects on SIRT1/AMPK and Akt/mTOR as well as on autophagy have been noted with conventional neurohormonal antagonists. The striking parallelism of these molecular, cellular, and clinical profiles suggests that SGLT2 inhibitors should be regarded as neurohormonal antagonists when they are prescribed for the treatment of chronic heart failure with a reduced ejection fraction.
Sources of Funding
None.
Disclosures
Dr Packer has recently consulted for Abbvie, Actavis, Akcea, Amgen, AstraZeneca, Boehringer Ingelheim, Cardiorentis, Daiichi Sankyo, Johnson & Johnson, NovoNordisk, Pfizer, Sanofi, Synthetic Biologics, and Theravance.
(J Am Heart Assoc. 2020;9:e016270 DOI: 10.1161/JAHA.120.016270.)
For Sources of Funding and Disclosures, see pages 6 and 7.
References
- 1. Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol. 1992;20:248–254. [DOI] [PubMed] [Google Scholar]
- 2. Komajda M, Böhm M, Borer JS, Ford I, Tavazzi L, Pannaux M, Swedberg K. Incremental benefit of drug therapies for chronic heart failure with reduced ejection fraction: a network meta‐analysis. Eur J Heart Fail. 2018;20:1315–1322. [DOI] [PubMed] [Google Scholar]
- 3. Packer M. Double vision: replicating a trial showing a survival benefit. JACC Heart Fail. 2017;5:232–235. [DOI] [PubMed] [Google Scholar]
- 4. Thomsen MM, Lewinter C, Køber L. Varying effects of recommended treatments for heart failure with reduced ejection fraction: meta‐analysis of randomized controlled trials in the ESC and ACCF/AHA guidelines. ESC Heart Fail. 2016;3:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McMurray JJV, Solomon SD, Docherty KF, Jhund PS. The Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure Trial (DAPA‐HF) in context [published online ahead of print January 3, 2020]. Eur Heart J. DOI: 10.2337/dc14-1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Packer M, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, Kimura K, Zeller C, George J, Brueckmann M, et al. Evaluation of the effect of sodium‐glucose co‐transporter 2 inhibition with empagliflozin on morbidity and mortality of patients with chronic heart failure and a reduced ejection fraction: rationale for and design of the EMPEROR‐Reduced trial. Eur J Heart Fail. 2019;21:1270–1278. [DOI] [PubMed] [Google Scholar]
- 7. Packer M. How should physicians view heart failure? The philosophical and physiological evolution of three conceptual models of the disease. Am J Cardiol. 1993;71:3C–11C. [DOI] [PubMed] [Google Scholar]
- 8. Moe GW, Rouleau JL, Charbonneau L, Proulx G, Arnold JM, Hall C, de Champlain J, Barr A, Sirois P, Packer M. Neurohormonal activation in severe heart failure: relations to patient death and the effect of treatment with flosequinan. Am Heart J. 2000;139:587–595. [DOI] [PubMed] [Google Scholar]
- 9. Packer M, Medina N, Yushak M. Hemodynamic and clinical limitations of long‐term inotropic therapy with amrinone in patients with severe chronic heart failure. Circulation. 1984;70:1038–1047. [DOI] [PubMed] [Google Scholar]
- 10. Packer M, Pitt B, Rouleau JL, Swedberg K, DeMets DL, Fisher L. Long‐term effects of flosequinan on the morbidity and mortality of patients with severe chronic heart failure: primary results of the PROFILE trial after 24 years. JACC Heart Fail. 2017;5:399–407. [DOI] [PubMed] [Google Scholar]
- 11. Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325:1468–1475. [DOI] [PubMed] [Google Scholar]
- 12. Ikeda Y, Inomata T, Iida Y, Iwamoto‐Ishida M, Nabeta T, Ishii S, Sato T, Yanagisawa T, Mizutani T, Naruke T, et al. Time course of left ventricular reverse remodeling in response to pharmacotherapy: clinical implication for heart failure prognosis in patients with idiopathic dilated cardiomyopathy. Heart Vessels. 2016;31:545–554. [DOI] [PubMed] [Google Scholar]
- 13. Rogers WJ, Johnstone DE, Yusuf S, Weiner DH, Gallagher P, Bittner VA, Ahn S, Schron E, Shumaker SA, Sheffield LT. Quality of life among 5,025 patients with left ventricular dysfunction randomized between placebo and enalapril: the Studies of Left Ventricular Dysfunction. The SOLVD Investigators. J Am Coll Cardiol. 1994;23:393–400. [DOI] [PubMed] [Google Scholar]
- 14. Packer M, Colucci WS, Sackner‐Bernstein JD, Liang CS, Goldscher DA, Freeman I, Kukin ML, Kinhal V, Udelson JE, Klapholz M, et al. Double‐blind, placebo‐controlled study of the effects of carvedilol in patients with moderate to severe heart failure. The PRECISE Trial. Prospective Randomized Evaluation of Carvedilol on Symptoms and Exercise. Circulation. 1996;94:2793–2799. [DOI] [PubMed] [Google Scholar]
- 15. Pessoa TD, Campos LC, Carraro‐Lacroix L, Girardi AC, Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium‐glucose cotransporter isoform‐mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol. 2014;25:2028–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Damman K, Beusekamp JC, Boorsma EM, Swart HP, Smilde TDJ, Elvan A, van Eck JWM, Heerspink HJL, Voors AA. Randomized, double‐blind, placebo‐controlled, multicentre pilot study on the effects of empagliflozin on clinical outcomes in patients with acute decompensated heart failure (EMPA‐RESPONSE‐AHF). Eur J Heart Fail. 2020;22:713–722. [DOI] [PubMed] [Google Scholar]
- 17. Hallow KM, Helmlinger G, Greasley PJ, McMurray JJV, Boulton DW. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes Metab. 2018;20:479–487. [DOI] [PubMed] [Google Scholar]
- 18. Solini A, Giannini L, Seghieri M, Vitolo E, Taddei S, Ghiadoni L, Bruno RM. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: a pilot study. Cardiovasc Diabetol. 2017;16:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yasui A, Lee G, Hirase T, Kaneko T, Kaspers S, von Eynatten M, Okamura T. Empagliflozin induces transient diuresis without changing long‐term overall fluid balance in Japanese patients with type 2 diabetes. Diabetes Ther. 2018;9:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohara K, Masuda T, Murakami T, Imai T, Yoshizawa H, Nakagawa S, Okada M, Miki A, Myoga A, Sugase T, et al. Effects of the sodium‐glucose cotransporter 2 inhibitor dapagliflozin on fluid distribution: a comparison study with furosemide and tolvaptan. Nephrology. 2019;24:904–911. [DOI] [PubMed] [Google Scholar]
- 21. Sha S, Polidori D, Heise T, Natarajan J, Farrell K, Wang SS, Sica D, Rothenberg P, Plum‐Mörschel L. Effect of the sodium glucose co‐transporter 2 inhibitor canagliflozin on plasma volume in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2014;16:1087–1095. [DOI] [PubMed] [Google Scholar]
- 22. Nassif ME, Windsor S, Tang F, Khariton Y, Husain M, Inzucchi SE, McGuire D, Pitt B, Scirica BM, Austin B, et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fraction: the DEFINE‐HF trial. Circulation. 2019;140:1463–1476. [DOI] [PubMed] [Google Scholar]
- 23. Damman K, Kjekshus J, Wikstrand J, Cleland JG, Komajda M, Wedel H, Waagstein F, McMurray JJ. Loop diuretics, renal function and clinical outcome in patients with heart failure and reduced ejection fraction. Eur J Heart Fail. 2016;18:328–336. [DOI] [PubMed] [Google Scholar]
- 24. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA‐REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care. 2016;39:1108–1114. [DOI] [PubMed] [Google Scholar]
- 25. Nielsen R, Møller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frøkiær J, Eiskjaer H, Jespersen NR, et al. Cardiovascular effects of treatment with the ketone body 3‐hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abdurrachim D, Teo XQ, Woo CC, Chan WX, Lalic J, Lam CSP, Lee PTH. Empagliflozin reduces myocardial ketone utilization while preserving glucose utilization in diabetic hypertensive heart disease: a hyperpolarized 13C magnetic resonance spectroscopy study. Diabetes Obes Metab. 2019;21:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl Sci. 2018;3:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baker HE, Kiel AM, Luebbe ST, Simon BR, Earl CC, Regmi A, Roell WC, Mather KJ, Tune JD, Goodwill AG. Inhibition of sodium‐glucose cotransporter‐2 preserves cardiac function during regional myocardial ischemia independent of alterations in myocardial substrate utilization. Basic Res Cardiol. 2019;114:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Connelly KA, Zhang Y, Desjardins JF, Nghiem L, Visram A, Batchu SN, Yerra VG, Kabir G, Thai K, Advani A, et al. Load‐independent effects of empagliflozin contribute to improved cardiac function in experimental heart failure with reduced ejection fraction. Cardiovasc Diabetol. 2020;19:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soga F, Tanaka H, Tatsumi K, Mochizuki Y, Sano H, Toki H, Matsumoto K, Shite J, Takaoka H, Doi T, et al. Impact of dapagliflozin on left ventricular diastolic function of patients with type 2 diabetic mellitus with chronic heart failure. Cardiovasc Diabetol. 2018;17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verma S, Mazer CD, Yan AT, Mason T, Garg V, Teoh H, Zuo F, Quan A, Farkouh ME, Fitchett DH, et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes and coronary artery disease: the EMPA‐HEART CardioLink‐6 randomized clinical trial. Circulation. 2019;140:1693–1702. [DOI] [PubMed] [Google Scholar]
- 33. Kosiborod MN, Jhund PS, Docherty KF, Diez M, Petrie MC, Verma S, Nicolau JC, Merkely B, Kitakaze M, DeMets DL, et al. Effects of dapagliflozin on symptoms, function, and quality of life in patients with heart failure and reduced ejection fraction: results from the DAPA‐HF trial. Circulation. 2020;141:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li C, Zhang J, Xue M, Li X, Han F, Liu X, Xu L, Lu Y, Cheng Y, Li T, et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc Diabetol. 2019;18:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ye Y, Bajaj M, Yang HC, Perez‐Polo JR, Birnbaum Y. SGLT‐2 Inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther. 2017;31:119–132. [DOI] [PubMed] [Google Scholar]
- 36. Aragón‐Herrera A, Feijóo‐Bandín S, Otero Santiago M, Barral L, Campos‐Toimil M, Gil‐Longo J, Costa Pereira TM, García‐Caballero T, Rodríguez‐Segade S, Rodríguez J, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol. 2019;170:113677. [DOI] [PubMed] [Google Scholar]
- 37. Yurista SR, Silljé HHW, Oberdorf‐Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Sodium‐glucose co‐transporter 2 inhibition with empagliflozin improves cardiac function in non‐diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019;21:862–873. [DOI] [PubMed] [Google Scholar]
- 38. Mizuno M, Kuno A, Yano T, Miki T, Oshima H, Sato T, Nakata K, Kimura Y, Tanno M, Miura T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol Rep. 2018;6:e13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017;104:298–310. [DOI] [PubMed] [Google Scholar]
- 40. Abdel‐Wahab BA, Metwally ME, El‐khawanki MM, Hashim AM. Protective effect of captopril against clozapine‐induced myocarditis in rats: role of oxidative stress, proinflammatory cytokines and DNA damage. Chem Biol Interact. 2014;216:43–52. [DOI] [PubMed] [Google Scholar]
- 41. Hiona A, Lee AS, Nagendran J, Xie X, Connolly AJ, Robbins RC, Wu JC. Pretreatment with angiotensin‐converting enzyme inhibitor improves doxorubicin‐induced cardiomyopathy via preservation of mitochondrial function. J Thorac Cardiovasc Surg. 2011;142:396–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kanda T, Araki M, Nakano M, Imai S, Suzuki T, Murata K, Kobayashi I. Chronic effect of losartan in a murine model of dilated cardiomyopathy: comparison with captopril. J Pharmacol Exp Ther. 1995;273:955–958. [PubMed] [Google Scholar]
- 43. Le DE, Pascotto M, Leong‐Poi H, Sari I, Micari A, Kaul S. Anti‐inflammatory and pro‐angiogenic effects of beta blockers in a canine model of chronic ischemic cardiomyopathy: comparison between carvedilol and metoprolol. Basic Res Cardiol. 2013;108:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao K, Zhang WW, Yao L, Yang S, Nie W, Huang F. Carvedilol promotes mitochondrial biogenesis by regulating the PGC‐1/TFAM pathway in human umbilical vein endothelial cells (HUVECs). Biochem Biophys Res Commun. 2016;470:961–966. [DOI] [PubMed] [Google Scholar]
- 45. Ma L, Gul R, Habibi J, Yang M, Pulakat L, Whaley‐Connell A, Ferrario CM, Sowers JR. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the transgenic (mRen2) rat. Am J Physiol Heart Circ Physiol. 2012;302:H2341–H2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nishio M, Sakata Y, Mano T, Ohtani T, Takeda Y, Miwa T, Hori M, Masuyama T, Kondo T, Yamamoto K. Beneficial effects of bisoprolol on the survival of hypertensive diastolic heart failure model rats. Eur J Heart Fail. 2008;10:446–453. [DOI] [PubMed] [Google Scholar]
- 47. Nolly MB, Caldiz CI, Yeves AM, Villa‐Abrille MC, Morgan PE, Amado Mondaca N, Portiansky EL, Chiappe de Cingolani GE, Cingolani HE, Ennis IL. The signaling pathway for aldosterone‐induced mitochondrial production of superoxide anion in the myocardium. J Mol Cell Cardiol. 2014;67:60–68. [DOI] [PubMed] [Google Scholar]
- 48. Noda K, Kobara M, Hamada J, Yoshifuji Y, Shiraishi T, Tanaka T, Wang J, Toba H, Nakata T. Additive amelioration of oxidative stress and cardiac function by combined mineralocorticoid and angiotensin receptor blockers in postinfarct failing hearts. J Cardiovasc Pharmacol. 2012;60:140–149. [DOI] [PubMed] [Google Scholar]
- 49. Kobayashi N, Yoshida K, Nakano S, Ohno T, Honda T, Tsubokou Y, Matsuoka H. Cardioprotective mechanisms of eplerenone on cardiac performance and remodeling in failing rat hearts. Hypertension. 2006;47:671–679. [DOI] [PubMed] [Google Scholar]
- 50. Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, Sam F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005;111:420–427. [DOI] [PubMed] [Google Scholar]
- 51. Imran M, Hassan MQ, Akhtar MS, Rahman O, Akhtar M, Najmi AK. Sacubitril and valsartan protect from experimental myocardial infarction by ameliorating oxidative damage in Wistar rats. Clin Exp Hypertens. 2019;41:62–69. [DOI] [PubMed] [Google Scholar]
- 52. Kusaka H, Sueta D, Koibuchi N, Hasegawa Y, Nakagawa T, Lin B, Ogawa H, Kim‐Mitsuyama S. LCZ696, angiotensin II receptor‐neprilysin inhibitor, ameliorates high‐salt‐induced hypertension and cardiovascular injury more than valsartan alone. Am J Hypertens. 2015;28:1409–1417. [DOI] [PubMed] [Google Scholar]
- 53. Grois L, Hupf J, Reinders J, Schröder J, Dietl A, Schmid PM, Jungbauer C, Resch M, Maier LS, Luchner A, et al. Combined inhibition of the renin‐angiotensin system and neprilysin positively influences complex mitochondrial adaptations in progressive experimental heart failure. PLoS One. 2017;12:e0169743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Di Franco A, Cantini G, Tani A, Coppini R, Zecchi‐Orlandini S, Raimondi L, Luconi M, Mannucci E. Sodium‐dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol. 2017;243:86–90. [DOI] [PubMed] [Google Scholar]
- 55. Ghezzi C, Yu AS, Hirayama BA, Kepe V, Liu J, Scafoglio C, Powell DR, Huang SC, Satyamurthy N, Barrio JR, et al. Dapagliflozin binds specifically to sodium‐glucose cotransporter 2 in the proximal renal tubule. J Am Soc Nephrol. 2017;28:802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wan N, Rahman A, Hitomi H, Nishiyama A. The effects of sodium‐glucose cotransporter 2 inhibitors on sympathetic nervous activity. Front Endocrinol (Lausanne). 2018;9:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kusaka H, Koibuchi N, Hasegawa Y, Ogawa H, Kim‐Mitsuyama S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc Diabetol. 2016;15:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schork A, Saynisch J, Vosseler A, Jaghutriz BA, Heyne N, Peter A, Häring HU, Stefan N, Fritsche A, Artunc F. Effect of SGLT2 inhibitors on body composition, fluid status and renin‐angiotensin‐aldosterone system in type 2 diabetes: a prospective study using bioimpedance spectroscopy. Cardiovasc Diabetol. 2019;18:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. [DOI] [PubMed] [Google Scholar]
- 60. Nożyński J, Zakliczyński M, Konecka‐Mrowka D, Zielinska T, Zakliczynska H, Nikiel B, Mlynarczyk‐Liszka J, Mrowka A, Zembala‐Nozynska E, Pijet M, et al. Advanced glycation end product accumulation in the cardiomyocytes of heart failure patients with and without diabetes. Ann Transplant. 2012;17:53–61. [DOI] [PubMed] [Google Scholar]
- 61. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–H2190. [DOI] [PubMed] [Google Scholar]
- 62. Yao Y, Lu Q, Hu Z, Yu Y, Chen Q, Wang QK. A non‐canonical pathway regulates ER stress signaling and blocks ER stress‐induced apoptosis and heart failure. Nat Commun. 2017;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qiu Z, Wei Y, Song Q, Du B, Wang H, Chu Y, Hu Y. The role of myocardial mitochondrial quality control in heart failure. Front Pharmacol. 2019;10:1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Colasante C, Chen J, Ahlemeyer B, Baumgart‐Vogt E. Peroxisomes in cardiomyocytes and the peroxisome/peroxisome proliferator‐activated receptor‐loop. Thromb Haemost. 2015;113:452–463. [DOI] [PubMed] [Google Scholar]
- 65. Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caragnano A, Aleksova A, Bulfoni M, Cervellin C, Rolle IG, Veneziano C, Barchiesi A, Mimmi MC, Vascotto C, Finato N, et al. Autophagy and inflammasome activation in dilated cardiomyopathy. J Clin Med. 2019;8:1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, Takano H, Mizuno K, Shimizu W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Singh SR, Zech ATL, Geertz B, Reischmann‐Düsener S, Osinska H, Prondzynski M, Krämer E, Meng Q, Redwood C, van der Velden J, et al. Activation of autophagy ameliorates cardiomyopathy in Mybpc3‐targeted knockin mice. Circ Heart Fail. 2017;10:e004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, Ogino A, Tsujimoto A, Goto K, Maruyama R, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012;96:456–465. [DOI] [PubMed] [Google Scholar]
- 72. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age‐related disease. Nat Cell Biol. 2018;20:1338–1348. [DOI] [PubMed] [Google Scholar]
- 74. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature. 2005;434:113–118. [DOI] [PubMed] [Google Scholar]
- 75. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Salminen A, Hyttinen JM, Kaarniranta K. AMP‐activated protein kinase inhibits NF‐κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl). 2011;89:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. Clin Exp Pharmacol Physiol. 2014;41:127–133.24341361 [Google Scholar]
- 79. Yoon MS. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients. 2017;9:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4:a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Balasubramanian S, Johnston RK, Moschella PC, Mani SK, Tuxworth WJ Jr, Kuppuswamy D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc Hematol Agents Med Chem. 2009;7:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhao Y, Hu X, Liu Y, Dong S, Wen Z, He W, Zhang S, Huang Q, Shi M. ROS signaling under metabolic stress: cross‐talk between AMPK and AKT pathway. Mol Cancer. 2017;16:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Akkafa F, Halil Altiparmak I, Erkus ME, Aksoy N, Kaya C, Ozer A, Sezen H, Oztuzcu S, Koyuncu I, Umurhan B. Reduced SIRT1 expression correlates with enhanced oxidative stress in compensated and decompensated heart failure. Redox Biol. 2015;6:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu TM, Tsai JY, Chen YC, Huang CY, Hsu HL, Weng CF, Shih CC, Hsu CP. Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci. 2014;21:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sung MM, Zordoky BN, Bujak AL, Lally JS, Fung D, Young ME, Horman S, Miller EJ, Light PE, Kemp BE, et al. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc Res. 2015;107:235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ramos FJ, Kaeberlein M, Kennedy BK. Elevated MTORC1 signaling and impaired autophagy. Autophagy. 2013;9:108–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. [DOI] [PubMed] [Google Scholar]
- 88. Shende P, Xu L, Morandi C, Pentassuglia L, Heim P, Lebboukh S, Berthonneche C, Pedrazzini T, Kaufmann BA, Hall MN, et al. Cardiac mTOR complex 2 preserves ventricular function in pressure‐overload hypertrophy. Cardiovasc Res. 2016;109:103–114. [DOI] [PubMed] [Google Scholar]
- 89. Völkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ, et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128:2132–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wende AR, O'Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, et al. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion‐targeted nuclear genes. Mol Cell Biol. 2015;35:831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation. 2015;131:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yano T, Shimoshige S, Miki T, Tanno M, Mochizuki A, Fujito T, Yuda S, Muranaka A, Ogasawara M, Hashimoto A, et al. Clinical impact of myocardial mTORC1 activation in nonischemic dilated cardiomyopathy. J Mol Cell Cardiol. 2016;91:6–9. [DOI] [PubMed] [Google Scholar]
- 93. Völkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S, Ornelas L, Thuerauf DJ, Gude N, Friedrich K, et al. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 2014;6:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Choi JC, Worman HJ. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy. 2013;9:110–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Marín‐Aguilar F, Lechuga‐Vieco AV, Alcocer‐Gómez E, Castejón‐Vega B, Lucas J, Garrido C, Peralta‐Garcia A, Pérez‐Pulido AJ, Varela‐López A, Quiles JL, et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2020;19:e13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3‐LKB1-AMP‐activated kinase pathway. J Biol Chem. 2010;285:3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang Y, Mi SL, Hu N, Doser TA, Sun A, Ge J, Ren J. Mitochondrial aldehyde dehydrogenase 2 accentuates aging‐induced cardiac remodeling and contractile dysfunction: role of AMPK, Sirt1, and mitochondrial function. Free Radic Biol Med. 2014;71:208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang L, Quan N, Sun W, Chen X, Cates C, Rousselle T, Zhou X, Zhao X, Li J. Cardiomyocyte‐specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc Res. 2018;114:805–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, Zou MH, Chen C, Wang DW. AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res. 2018;122:712–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hernández JS, Barreto‐Torres G, Kuznetsov AV, Khuchua Z, Javadov S. Crosstalk between AMPK activation and angiotensin II‐induced hypertrophy in cardiomyocytes: the role of mitochondria. J Cell Mol Med. 2014;18:709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chen K, Kobayashi S, Xu X, Viollet B, Liang Q. AMP activated protein kinase is indispensable for myocardial adaptation to caloric restriction in mice. PLoS One. 2013;8:e59682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ma S, Feng J, Zhang R, Chen J, Han D, Li X, Yang B, Li X, Fan M, Li C, et al. SIRT1 activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid Med Cell Longev. 2017;2017:4602715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wan X, Wen JJ, Koo SJ, Liang LY, Garg NJ. SIRT1‐PGC1α‐NFκB pathway of oxidative and inflammatory stress during Trypanosoma cruzi infection: benefits of SIRT1‐targeted therapy in improving heart function in Chagas disease. PLoS Pathog. 2016;12:e1005954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ruan Y, Dong C, Patel J, Duan C, Wang X, Wu X, Cao Y, Pu L, Lu D, Shen T, et al. SIRT1 suppresses doxorubicin‐induced cardiotoxicity by regulating the oxide‐tive stress and p38MAPK pathways. Cell Physiol Biochem. 2015;35:1116–1124. [DOI] [PubMed] [Google Scholar]
- 105. Zilinyi R, Czompa A, Czegledi A, Gajtko A, Pituk D, Lekli I, Tosaki A. The cardioprotective effect of metformin in doxorubicin‐induced cardiotoxicity: the role of autophagy. Molecules. 2018;23:1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bartlett JJ, Trivedi PC, Pulinilkunnil T. Autophagic dysregulation in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2017;104:1–8. [DOI] [PubMed] [Google Scholar]
- 107. Marampon F, Gravina GL, Scarsella L, Festuccia C, Lovat F, Ciccarelli C, Zani BM, Polidoro L, Grassi D, Desideri G, et al. Angiotensin‐converting‐enzyme inhibition counteracts angiotensin II‐mediated endothelial cell dysfunction by modulating the p38/SirT1 axis. J Hypertens. 2013;31:1972–1983. [DOI] [PubMed] [Google Scholar]
- 108. Clarke NE, Belyaev ND, Lambert DW, Turner AJ. Epigenetic regulation of angiotensin‐converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin Sci (Lond). 2014;126:507–516. [DOI] [PubMed] [Google Scholar]
- 109. Shen T, Ding L, Ruan Y, Qin W, Lin Y, Xi C, Lu Y, Dou L, Zhu Y, Cao Y, et al. SIRT1 functions as an important regulator of estrogen‐mediated cardiomyocyte protection in angiotensin II‐induced heart hypertrophy. Oxid Med Cell Longev. 2014;2014:713894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mavroeidi V, Petrakis I, Stylianou K, Katsarou T, Giannakakis K, Perakis K, Vardaki E, Stratigis S, Ganotakis E, Papavasiliou S, et al. Losartan affects glomerular AKT and mTOR phosphorylation in an experimental model of type 1 diabetic nephropathy. J Histochem Cytochem. 2013;61:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wu X, He L, Cai Y, Zhang G, He Y, Zhang Z, He X, He Y, Zhang G, Luo J. Induction of autophagy contributes to the myocardial protection of valsartan against ischemia‐reperfusion injury. Mol Med Rep. 2013;8:1824–1830. [DOI] [PubMed] [Google Scholar]
- 112. Zhuo XZ, Wu Y, Ni YJ, Liu JH, Gong M, Wang XH, Wei F, Wang TZ, Yuan Z, Ma AQ, et al. Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation‐mediated endoplasmic reticulum stress. Apoptosis. 2013;18:800–810. [DOI] [PubMed] [Google Scholar]
- 113. Hu H, Li X, Ren D, Tan Y, Chen J, Yang L, Chen R, Li J, Zhu P. The cardioprotective effects of carvedilol on ischemia and reperfusion injury by AMPK signaling pathway. Biomed Pharmacother. 2019;117:109106. [DOI] [PubMed] [Google Scholar]
- 114. Wong WT, Li LH, Rao YK, Yang SP, Cheng SM, Lin WY, Cheng CC, Chen A, Hua KF. Repositioning of the β‐blocker carvedilol as a novel autophagy inducer that inhibits the NLRP3 inflammasome. Front Immunol. 2018;9:1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Allen SA, Tomilov A, Cortopassi GA. Small molecules bind human mTOR protein and inhibit mTORC1 specifically. Biochem Pharmacol. 2018;155:298–304. [DOI] [PubMed] [Google Scholar]
- 116. Liu GZ, Zhang S, Li YY, Liu YW, Zhang Y, Zhao XB, Yuan Y, Zhang JW, Khannanova Z, Li Y. Aldosterone stimulation mediates cardiac metabolism remodeling via Sirt1/AMPK signaling in canine model. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:851–863. [DOI] [PubMed] [Google Scholar]
- 117. Mummidi S, Das NA, Carpenter AJ, Kandikattu H, Krenz M, Siebenlist U, Valente AJ, Chandrasekar B. Metformin inhibits aldosterone‐induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt‐induced cardiac fibrosis in vivo. J Mol Cell Cardiol. 2016;98:95–102. [DOI] [PubMed] [Google Scholar]
- 118. Long HD, Lin YE, Liu MJ, Liang LY, Zeng ZH. Spironolactone prevents dietary‐induced metabolic syndrome by inhibiting PI3‐K/Akt and p38MAPK signaling pathways. J Endocrinol Invest. 2013;36:923–930. [DOI] [PubMed] [Google Scholar]
- 119. Li D, Lu Z, Xu Z, Ji J, Zheng Z, Lin S, Yan T. Spironolactone promotes autophagy via inhibiting PI3K/AKT/mTOR signalling pathway and reduce adhesive capacity damage in podocytes under mechanical stress. Biosci Rep. 2016;36:e00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Plante E, Menaouar A, Danalache BA, Broderick TL, Jankowski M, Gutkowska J. Treatment with brain natriuretic peptide prevents the development of cardiac dysfunction in obese diabetic db/db mice. Diabetologia. 2014;57:1257–1267. [DOI] [PubMed] [Google Scholar]
- 121. Ruiz‐Ojeda FJ, Aguilera CM, Rupérez AI, Gil Á, Gomez‐Llorente C. An analogue of atrial natriuretic peptide (C‐ANP4-23) modulates glucose metabolism in human differentiated adipocytes. Mol Cell Endocrinol. 2016;431:101–108. [DOI] [PubMed] [Google Scholar]
- 122. Kim J, Han D, Byun SH, Kwon M, Cho SJ, Koh YH, Yoon K. Neprilysin facilitates adipogenesis through potentiation of the phosphatidylinositol 3‐kinase (PI3K) signaling pathway. Mol Cell Biochem. 2017;430:1–9. [DOI] [PubMed] [Google Scholar]
- 123. Siepmann M, Kumar S, Mayer G, Walter J. Casein kinase 2 dependent phosphorylation of neprilysin regulates receptor tyrosine kinase signaling to Akt. PLoS One. 2010;5:e13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wakisaka M, Nagao T, Yoshinari M. Sodium glucose cotransporter 2 (SGLT2) plays as a physiological glucose sensor and regulates cellular contractility in rat mesangial cells. PLoS One. 2016;11:e0151585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Umino H, Hasegawa K, Minakuchi H, Muraoka H, Kawaguchi T, Kanda T, Tokuyama H, Wakino S, Itoh H. High basolateral glucose increases sodium‐glucose cotransporter 2 and reduces sirtuin‐1 in renal tubules through glucose transporter‐2 detection. Sci Rep. 2018;8:6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kanfi Y, Peshti V, Gozlan YM, Rathaus M, Gil R, Cohen HY. Regulation of SIRT1 protein levels by nutrient availability. FEBS Lett. 2008;582:2417–2423. [DOI] [PubMed] [Google Scholar]
- 127. Osataphan S, Macchi C, Singhal G, Chimene‐Weiss J, Sales V, Kozuka C, Dreyfuss JM, Pan H, Tangcharoenpaisan Y, Morningstar J, et al. SGLT2 inhibition reprograms systemic metabolism via FGF21‐dependent and ‐independent mechanisms. JCI Insight. 2019;4:e123130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Swe MT, Thongnak L, Jaikumkao K, Pongchaidecha A, Chatsudthipong V, Lungkaphin A. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin Sci (Lond). 2019;133:2415–2430. [DOI] [PubMed] [Google Scholar]
- 129. Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, Day EA, Salt IP, Steinberg GR, Hardie DG. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65:2784–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kim JW, Lee YJ, You YH, Moon MK, Yoon KH, Ahn YB, Ko SH. Effect of sodium‐glucose cotransporter 2 inhibitor, empagliflozin, and α‐glucosidase inhibitor, voglibose, on hepatic steatosis in an animal model of type 2 diabetes [published online ahead of print November 26, 2018]. J Cell Biochem. DOI: 10.1002/jcb.28141. [DOI] [PubMed] [Google Scholar]
- 131. Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK‐mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mohamed HE, Asker ME, Keshawy MM, Hasan RA, Mahmoud YK. Inhibition of tumor necrosis factor‐α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol Cell Biochem. 2020;466:45–54. [DOI] [PubMed] [Google Scholar]