

Abstract
Background
Mutations in the LMNA gene, encoding LMNA (lamin A/C), causes distinct disorders, including dilated cardiomyopathies, collectively referred to as laminopathies. The genes (coding and noncoding) and regulatory pathways controlled by LMNA in the heart are not completely defined.
Methods and Results
We analyzed cardiac transcriptome from wild‐type, loss‐of‐function (Lmna−/−), and gain‐of‐function (Lmna−/− injected with adeno‐associated virus serotype 9 expressing LMNA) mice with normal cardiac function. Deletion of Lmna (Lmna−/−) led to differential expression of 2193 coding and 629 long noncoding RNA genes in the heart (q<0.05). Re‐expression of LMNA in the Lmna−/− mouse heart, completely rescued 501 coding and 208 non‐coding and partially rescued 1862 coding and 607 lncRNA genes. Pathway analysis of differentially expressed genes predicted activation of transcriptional regulators lysine‐specific demethylase 5A, lysine‐specific demethylase 5B, tumor protein 53, and suppression of retinoblastoma 1, paired‐like homeodomain 2, and melanocyte‐inducing transcription factor, which were completely or partially rescued upon reexpression of LMNA. Furthermore, lysine‐specific demethylase 5A and 5B protein levels were increased in the Lmna−/− hearts and were partially rescued upon LMNA reexpression. Analysis of biological function for rescued genes identified activation of tumor necrosis factor‐α, epithelial to mesenchymal transition, and suppression of the oxidative phosphorylation pathway upon Lmna deletion and their restoration upon LMNA reintroduction in the heart. Restoration of the gene expression and transcriptional regulators in the heart was associated with improved cardiac function and increased survival of the Lmna−/− mice.
Conclusions
The findings identify LMNA‐regulated cardiac genes and their upstream transcriptional regulators in the heart and implicate lysine‐specific demethylase 5A and B as epigenetic regulators of a subset of the dysregulated genes in laminopathies.
Keywords: cardiomyopathies, KDM5, laminopathies, LMNA
Subject Categories: Cardiomyopathy, Basic Science Research, Myocardial Biology
Nonstandard Abbreviations and Acronyms
- AAV9
adeno‐associated virus serotype 9
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DCM
dilated cardiomyopathy
- GSEA
gene set enrichment analysis
- IPA
Ingenuity Pathway Analysis
- KDM5A
lysine‐specific demethylase 5A
- KDM5B
lysine‐specific demethylase 5B
- LADs
lamina‐associated domains
- limma
linear models for microarray data
- LMNA
lamin A
- MAPK4
mitogen‐activated protein kinase 4
- PCM1
pericentriolar material‐1
- PITX2
paired‐like homeodomain 2
- RB1
retinoblastoma 1
- TGF
transforming growth factor‐β1
- TP53
tumor protein 53
- TR
transcriptional regulator
- WT
wild‐type
Clinical Perspective
What Is New?
Genetic deletion of the Lmna gene, encoding LMNA (lamin A/C) protein, was associated with dysregulation of several thousand coding and noncoding genes in the heart and led to myocardial fibrosis, apoptosis, cardiac dysfunction, and premature death.
Genes whose expression levels were dysregulated predicted activation or suppression of over 2 dozen transcriptional regulators, including histone demethylases lysine‐specific demethylase 5A and B.
Reexpression of LMNA, predominantly in cardiac myocytes using recombinant adeno‐associated virus serotype 9 constructs, in the Lmna‐deficient mice normalized or partially restored expression levels of a few thousand genes, including targets of the lysine‐specific demethylase 5A and B, and restoration of gene expression in the heart was associated with improved cardiac function and prolonged survival.
What Are the Clinical Implications?
The findings by unraveling putative pathogenic transcriptional regulators of cardiac phenotypes in laminopathies, including lysine‐specific demethylase 5A and B, provide for potential therapeutic targets in heart failure caused by the LMNA mutations.
Mutations in the LMNA gene, encoding LMNA (lamin A/C), cause a diverse array of phenotypes involving multiple organs that are collectively referred to as laminopathies. 1 , 2 , 3 , 4 Notable among the laminopathies are dilated cardiomyopathy (DCM), cardiac conduction defects, arrhythmias, Hutchinson‐Gilford progeria syndrome, Emery‐Dreifuss muscular dystrophy, and familial partial lipodystrophy. 1 , 2 , 3 , 4 , 5 , 6 LMNA is among the most common causal genes for hereditary DCM, accounting for 6% to 8% of familial DCM and for a small fraction of arrhythmogenic cardiomyopathy. 2 , 5 , 6 DCM is the major cause of death in a subset of laminopathies involving striated muscles and typically manifests with conduction defects, arrhythmias, and cardiac dysfunction. 7 , 8 Accordingly, patients with LMNA mutations are at a high risk of sudden cardiac death, which typically necessitates implantation of a defibrillator/pacemaker. 7 , 8
LMNA is expressed in differentiated cells, including cardiac myocytes. Within the nucleus, lamin and associated proteins form a fibrous multiprotein network termed as nuclear lamina that lines the nucleoplasmic face of the inner nuclear membrane. Genomic regions that are in close contact with the nuclear lamina are known as lamina‐associated domains (LADs). 9 , 10 , 11 , 12 LMNA through LAD plays an important role in spatial chromatin organization and gene expression. Genes within LADs are expressed at low levels and are depleted for active histone marks like H3K4me3. 9 , 10 , 11 , 12 Recently, we identified over 300 LADs in human cardiac myocytes, encompassing several thousand coding and noncoding genomic regions. We further demonstrated that LADs are redistributed in DCM, and this redistribution affects gene expression either directly or indirectly. 13
The molecular pathogenesis of DCM in laminopathies is not well understood; however, a large number of genes and pathways are found to be dysregulated, correlating with the phenotypic diversity of the LMNA mutations. 9 , 11 , 13 , 14 , 15 , 16 Defects in several signaling pathways, such as those including forkhead box O and MAPK4 (mitogen‐activated protein kinase 4), have been implicated in the pathogenesis of DCM in laminopathies. 4 , 14 , 17 , 18 In addition, altered nuclear membrane mechanical integrity and DNA damage have emerged as putative mechanisms in the pathogenesis of laminopathies, including DCM. 19 , 20 , 21 , 22 However, the role of LMNA in regulating coding and noncoding genes, occurring before and independent of cardiac dysfunction, is not well understood. The present study is designed to bridge this knowledge gap by utilizing Lmna loss‐of‐function (Lmna−/−) and partial gain‐of‐function mouse models. The latter were generated upon reexpression of wild‐type (WT) LMNA in the heart in Lmna−/− mice using recombinant adeno‐associated viruses (henceforth referred to as Lmna−/−:AAV9‐Lmna WT).
We sequenced ribosomal‐depleted RNA samples, extracted from whole hearts of WT and Lmna−/− mice when cardiac function was normal to reduce potential confounding effects of cardiac dysfunction on gene expression. To complement the findings, we reexpressed LMNA in the Lmna−/− hearts using recombinant AAV9 viruses and determined the ensuing transcriptomic changes in the heart at the same age. Comparing gene expression in the WT, Lmna−/− and Lmna−/−:AAV9‐Lmna WT hearts, we identified coding and noncoding genes whose expression was regulated by LMNA in the absence of cardiac dysfunction. Differentially expressed genes (DEGs) were used to predict dysregulated activity of several transcriptional regulators (TRs), including lysine‐specific demethylase 5A (KDM5A) and B, implicating them in the pathogenesis of DCM in laminopathies.
METHODS
Further details of the material and methods are provided in Data S1.
Mice
The phenotype of the Lmna−/− mice has been published. 17 , 20 Lmna−/− and WT littermates were used in these experiments, the latter as controls. List of oligonucleotide primers used for genotype screening is listed in Table S1.
Anesthesia and Euthanasia
Anesthesia was induced with 3% inhaled isoflurane and was maintained at 0.5% to 1% isoflurane inhalation throughout the procedure. Mice were euthanized by CO2 inhalation followed by cervical dislocation. Animal studies were in accord with the NIH Guide for the Care and Use of Laboratory Animals published and approved by the Animal Care and Use Committee (AWC protocols: AWC‐15‐0052 and AWC‐17‐0146).
Echocardiography
Echocardiography was performed on mice anesthetized by intraperitoneal injection of sodium pentobarbital (60 mg/kg body weight). The mouse was positioned supine on a heating pad with embedded ECG leads. ECG and respiratory rate were recorded during the study. Wall thicknesses and left ventricular dimensions were measured from M‐mode images in at least 3 cardiac cycles and the mean values were used. Left ventricular fractional shortening and mass were calculated as previously described. 17 , 23 , 24
Immunoblotting
Immunoblotting was performed as published. 17 , 19 , 23 Nuclear protein isolation was performed using the nuclear extraction kit (Ab113474; Abcam, Cambridge, MA), and a 20‐ to 25‐μg aliquot of each nuclear protein extract was used for immunoblotting analysis. The primary and secondary antibodies and their titers are listed in Table S2.
Immunofluorescence
Immunofluorescence staining to detect transgene expression and localization was performed as published. 17 , 19 , 23 In brief, thin myocardial cryosections, fixed in optimal cutting temperature (OCT), were incubated with an anti‐Flag‐ LMNA or anti‐pericentriolar material‐1 (PCM1) antibody to detect expression of the transgenes within nuclei. The sections were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) to identify the nuclei. Details of antibodies are presented in Table S2.
RNA Sequencing
Total RNA depleted of ribosomal RNA was used. RNA fragmentation, first‐ and second‐strand cDNA synthesis (preserving strand information), and the addition of indexed adapters was carried out as previously described. 17 , 19 , 23 , 25 Paired‐end, 100‐bp reads were obtained on an HiSeq 4000 instrument (Illumina, San Diego, CA). Mapping of sequencing reads to the mouse genome GRCm38.91 (Ensembl) was performed using Tophat2. 26 The mean number of transcriptome‐aligned reads per heart was 27.6 million. Read quantitation on a per‐gene basis was performed with HTSeq 27 and the default gene annotation file (gtf) provided with the GRCm38.91 Ensembl genome distribution. Detectable RNAs were assessed as those present at or above 1 read per million (27 reads) in 50% of the samples, resulting in 15 096 RNAs for downstream analyses. Calculation of fold‐change and false discovery rate for differentially expressed RNAs were obtained by the R/Bioconductor packages linear models for microarray data (limma) and edgeR/variance modeling at the observational level. 28 A batch correction was performed by including batch as a blocking factor in the linear model used by limma, per a published protocol. 28 To obtain quantitative read information for lncRNAs HTSeq was used together with a customized gtf comprising the default Ensembl gtf together with annotation for an additional 87 774 lncRNAs that were present in the NONCODEv5 database (http://www.noncode.org/). The mean number of transcriptome‐aligned reads per heart, as determined by NONCODEv5, was 5.9×105. LncRNAs, identified by NONCODEv5, were defined at a detectable cutoff of 6 reads in at least 50% of the samples in either group, resulting in 15 695 lncRNAs. Fold‐change and false discovery rate of the differentially expressed lncRNAs were calculated using the R/Bioconductor packages edgeR and limma/variance modeling at the observational level.
Pathway Analysis
Gene set enrichment analysis (GSEA) was performed on normalized gene counts of DEGs in knockout as compared with control samples (q<0.05). Significance was assessed by analyzing signal‐to‐noise ratio and gene permutations based on 1000 permutations. Molecular signature database 3.0 curated gene sets for hallmark and canonical pathways were used in the GSEA. 29 , 30 Gene sets were also curated using the compute overlap, which is a hypergeometric distribution function of GSEA. The data were ordered based on enrichment score for gene set with afalse discovery rate cutoff of 0.05. When presenting results for specific gene sets, nominal enrichment score and false discovery rate values are shown.
Upstream Regulators Analysis
To identify likely TRs, the Upstream Regulator Analysis module of Ingenuity Pathway Analysis software (IPA, QIAGEN, Redwood City, CA) was used. DEGs (q<0.05) were used for this analysis. A Z score ≥+2 and ≤−2 was used to identify upstream regulators that were predicted to be altered and considered significantly changed. Targets of upstream regulators that overlap with DEGs and show expression pattern consistent with upstream regulator activation or suppression were obtained from IPA.
LncRNA‐mRNA Correlation Analysis
Correlation between LncRNA and mRNAs was performed on the count‐per‐million values for each lncRNA and mRNA in all samples using Pearson correlation analysis in R. Cis pairs for lncRNAs and mRNAs (located within 10 kb) were obtained using the “Nearest BEDTOOL” in local galaxy server platform (https://usegalaxy.org/). Heat plot and density plots that used the Pearson correlation coefficient r values were plotted in R.
Transduction of Mouse Heart With Recombinant AAV9 Constructs
Full‐length WT and N‐terminally Flag‐tagged murine Lmna cDNAs were cloned into a pTRUF11 plasmid downstream to a TATA‐less CMV promoter and rabbit beta‐globin intron at the StuI cloning site and were tested (Figure S1A through S1C). Expressions of the full‐length Flag‐tagged and nontagged LMNA proteins from the corresponding clones were analyzed by western blotting after transfection of the recombinant plasmids into HEK293 cells using transfectamine (Figure S1B). Approximately 1×1010 to 2×1010 vector genomes were injected subcutaneously at the nape of the back of Lmna−/− mice (Lmna−/−:AAV9‐Lmna WT) at postnatal days 2, 4, and 6. The early timing of injection was chosen to ensure adequate gene expression before the Lmna−/− mice exhibit cardiac dysfunction enabling an unconfounded transcriptomic analysis. 17 Three sequential injections were made to increase the likelihood of transducing newly formed cardiac myocytes during the early postnatal period, as a low level of proliferation continues this period. 31 , 32 , 33 As a control, an empty AAV9 vector construct was injected into Lmna−/− mice at the above time points and via the same subcutaneous route.
AAV9 Transduction Efficiency in Heart and Cardiac Myocytes
Transduction efficiency of the FLAG‐LMNA was determined at 1, 2, and 4 weeks after transduction. At least 2000 DAPI‐stained cells per each mouse heart were counted. Transduction efficiency was calculated as the percentage of FLAG‐stained cells to the total number of cells, identified by DAPI staining of the nuclei, assuming that one third of the DAPI‐stained cells in the heart were cardiac myocytes and that only myocytes are transduced with the AAV9 recombinant viruses in the heart.
To determine efficiency following transduction with the recombinant viruses expressing WT LMNA in the Lmna−/− mouse hearts, optimal cutting temperature–fixed thin myocardial cryosections were stained with an antibody against PCM1, and an antibody against LMNA and DAPI (nuclei). 31 , 32 , 33 The number of PCMI1 and LMNA costained cells (ie, myocytes) expressing LMNA was determined to confirm transduction efficiency of cardiac myocytes in the mouse heart. Data were analyzed without prior knowledge of the genotypes.
Statistical Analysis
Normality of data distribution was assessed by the Shapiro–Wilk test. Normally distributed data were analyzed by ordinary 1‐way ANOVA followed by Tukey’s multiple pairwise comparison test. Data deviating from normal distribution were analyzed by the Kruskal–Wallis test followed by Dunn’s multiple pairwise comparisons. The categorical data were analyzed by chi‐square test. The effect of AAV9‐Lmna WT on overall survival was evaluated by the Kaplan–Meier survival analysis, and the differences between subgroups were evaluated by log‐rank (Mantel‐Cox) test. Statistical analysis was performed by GraphPad Prism 8 (www.graphpad.com/scientific‐software/prism/).
Data Access
The Lmna−/− and WT heart RNA seq data have been submitted to NCBI GEO and are publicly available (GSE110341 and GSE123916). RNA seq data from the Lmna−/−:AAV9‐Lmna WT and the corresponding Lmna−/− have been submitted to GEO (GSE135288, reviewer token: gxylyyswhxopbsp) and will be released to the public upon manuscript publication.
RESULTS
Phenotype in the Lmna −/− Mice
The Lmna−/− mice develop progressive cardiac dilatation and dysfunction starting after 2 weeks of age, leading to premature death by 8 to 10 weeks of age. 17 Myocardial histopathology at 3 to 4 weeks of age is notable for increased fibrosis and apoptosis. The Lmna−/− mice have a median survival of 4 weeks, as described previously. 17
AAV9‐Mediated Reexpression of WT LMNA (LMNAWT) in Lmna−/− Mouse Hearts
To determine the role of LMNA in regulation of cardiac gene expression, we compared gene expression upon loss of LMNA (Lmna−/− or KO mice) and upon reexpression of LMNA in the LMNA null background (Lmna−/−:AAV9‐Lmna WT mice). A prerequisite to this approach is the ability to effectively express LMNA and to detect its expression and localization in the heart. Therefore, initial experiments were performed using recombinant AAV9 viruses expressing a FLAG‐tagged LMNA, which enabled detection of the transgene protein while avoiding the confounding effect of any potential residual expression of the endogenous LMNA protein in the heart of the Lmna−/− mice. Two factors were considered in determining timing of administration of the AAV9 viruses: (1) the short life span of the Lmna−/− mice, which was at most 8 to 10 weeks; and (2) a short delay in gene expression upon transduction with the AAV9 viruses. Therefore, in vivo gene transfer with the recombinant viruses started at postnatal day 2. To overcome the potential episomal loss of the viral genome caused by actively dividing myocytes during the few days after birth, injections were repeated at postnatal day 4 and postnatal day 6. A total dose of 2×1010 viral genome particle was delivered subcutaneously to each neonate.
To detect expression of the transgene in the hearts of Lmna−/−:AAV9‐LmnaFlag mice, thin myocardial sections were stained with an anti‐FLAG antibody at 1, 2, and 4 weeks after birth, which showed transgene protein expression and nuclear membrane localization in the heart (Figure 1A). Recombinant LMNA protein level was the lowest and regional heterogeneity was more prominent at 1 week after injection, while transgene expression was mostly homogenous and robust at 2 and 4 weeks following transduction with AAV9‐LmnaFlag. Approximately 19.3±3.2 % of the nuclei showed expression of the FLAG‐tagged LMNA, which corresponds to transduction of approximately 57% of myocytes (Figure 1B), assuming that about one third of the cells in the heart are cardiac myocytes and that AAV9 primarily transduces myocytes. 34 , 35 , 36
Figure 1. Expression and localization of LMNA in the heart of Lmna−/− mice upon AAV9‐mediated LMNA re‐expression.
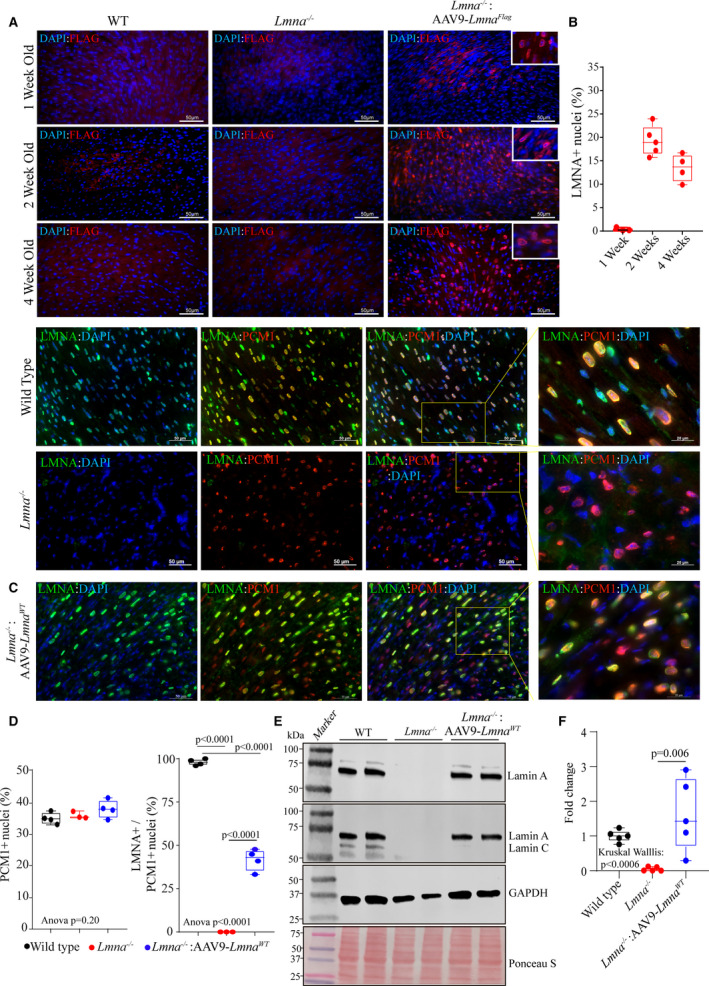
A, Immunofluorescence staining of thin myocardial sections showing FLAG‐LMNA (in red) expression in 1, 2, and 4‐weeks old wild type, Lmna−/− and Lmna−/−:AAV9–LmnaFlag mice. Nuclei were counterstained with DAPI (blue). B, Respective quantitation of LMNA+ nuclei as a fraction of total DAPI+ nuclei per heart (n=3–5). C, IF staining of myocardial sections in 2 weeks old wild type, Lmna−/− and Lmna−/−: AAV9‐Lmna WT LMNA (in green) and anti‐PCM1 antibody (in red). Higher magnification of the overlay of LMNA and PCM1 with DAPI. D, Respective quantitative data showing the percentage of PCM1 positive nuclei (marking cardiac myocytes) in the WT (n=4), Lmna−/− (n=3) and Lmna−/−:AAV9‐Lmna WT (n=4) and percentage of cardiac myocytes expressing LMNA (PCM1+/LMNA+nuclei). E, Immunoblot (IB) showing the expression of LMNA in whole heart lysates of 2‐week old mice in WT, Lmna−/− and Lmna−/−:AAV9‐Lmna WT mice using an anti‐LMNA antibody. Protein molecular weight markers are shown to the left, and GAPDH is used as a loading control together with total protein staining using Ponceau S. F, Quantitative data for LMNA protein expression fold change normalized to GAPDH and relative to WT mice. Data were obtained from N=5 mice heart/genotype and shows variability in the LMNA protein expression in the AAV9‐LMNA treated samples. Normality of data distribution was assessed by Shapiro–Wilks test. P values for Kruskal–Wallis test and Dunn’s multiple comparison are shown. Only P values that were significant (P<0.05) are shown. AAV9 indicates adeno‐associated virus serotype 9; DAPI, 4′,6‐diamidino‐2‐phenylindole; LMNA, lamin A; PCM1, pericentriolar material‐1; and WT, wild‐type.
Having established the feasibility of the approach, the experiments were repeated with an untagged LMNA, to eliminate potential confounding effects that might result from the Flag‐tagged LMNA. The Lmna−/− mice were injected subcutaneously with recombinant AAV9 viruses expressing nontagged LMNAWT (Lmna−/−:AAV9‐Lmna WT) protein, as described above. Thin myocardial sections obtained from 2‐week‐old Lmna−/− mice were stained with antibodies against LMNA and PCM1, the latter marking cardiac myocyte nuclei. The PCM1 along with LMNA staining of cardiac myocyte was used as a surrogate to determine the transduction and provide a quantitative measure of the overall experimental efficiency. The PCM1‐stained cells comprised approximately 35% of the nuclei in the myocardial sections, which is consistent with the previous data. 31 , 32 , 33 There were no differences in the percent PCM1‐positive nuclei among the experimental groups. At a dose of 2×1010 viral genome particle, 42±6.24% of cardiac myocytes, identified by PCM1 expression, were transduced and expressed LMNAWT, which was incorporated into the nuclear membrane (Figure 1C and 1D). Furthermore, the expression of LMNA protein in the hearts of the AAV9 treated Lmna−/− mice was confirmed by immunoblot analysis using an antibody that detects either LMNA or lamin A/C (Figure 1E). A degree of variability in the expression of LMNA was noted in the rescued group (Figure 1F).
Effects of LMNA on Expression of Coding Genes in the Heart
To identify Lmna regulated genes, RNA sequencing was performed from 2‐week old WT, Lmna−/− and Lmna−/−:AAV9‐Lmna WT mouse hearts. At 2 weeks of age, there was no discernible cardiac dysfunction or myocardial fibrosis (Table S3), as also reported previously. 17 Analysis of the cardiac gene expression showed differential expression of 2036 genes, comprised of 814 upregulated and 1222 downregulated genes (q<0.05) in the Lmna−/− as compared with WT mouse hearts (Figure 2A).
Figure 2. Differential coding and noncoding RNA expression in the WT, Lmna−/− and Lmna−/−: AAV9‐Lmna WT mouse hearts at 2 weeks of age.
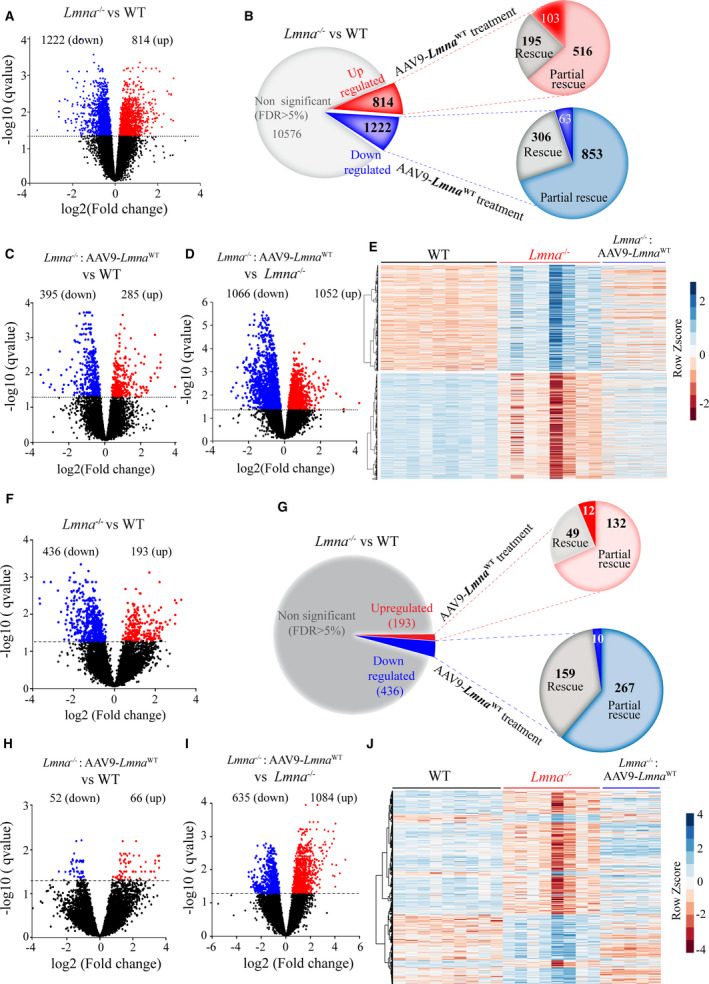
A, Volcano plots of RNA sequencing data showing the expression and significance levels of genes in Lmna−/− (n=8) vs WT (n=9) hearts. The dashed line denotes the significance level at a threshold of q<0.05 (or false discovery rate 5%). The up‐regulated and (are red), down regulated genes (blue) are shown. Number of upregulated and down regulated differentially expressed genes (DEGs) are indicated above each volcano plots. B, Pie charts illustrating the number of differentially upregulated (red), downregulated (blue), and unchanged (gray) genes in Lmna−/− hearts compared with the WT (q<0.05). A subset of genes that show complete and partial rescue upon AAV9 treatment (n=5) are shown along with the unaffected ones. C and D, Volcano plots of RNA‐seq data showing the expression and significance levels of genes in the Lmna−/−:AAV9‐Lmna WT vs Lmna−/− (C) and Lmna−/−:AAV9‐Lmna WT vs WT (D) heart. E, Heat map and hierarchical clustering of differentially expressed genes in the Lmna−/− compared with WT mouse hearts and the corresponding expression upon LMNA expression (Lmna−/−:AAV9‐Lmna WT). F, Volcano plots of the RNA sequencing data showing the expression and significance levels for lncRNAs in the Lmna−/− vs WT hearts. G, Pie charts illustrating the number of differentially upregulated (red), downregulated (blue), and unchanged (gray) lncRNAs in Lmna−/− hearts compared with the WT (q<0.05). A subset of lncRNAs that show complete and partial rescue upon AAV9 treatment are shown along with the ones that are unaffected. H and I, Volcano plots for lncRNA expression in the Lmna−/−: AAV9‐Lmna WT vs Lmna−/− (H) and in the Lmna−/−:AAV9‐Lmna WT− vs WT (I) mouse hearts. J, Heat map with hierarchical clustering of differentially expressed lncRNAs in Lmna−/− compared with WT mouse hearts and corresponding changes upon LMNA expression in the Lmna−/− mouse hearts. AAV9 indicates adeno‐associated virus serotype 9; LMNA, lamin A; lncRNA, long noncoding RNA; and WT, wild‐type.
Reexpression of LMNA led to the complete or partial reversal of a large fraction of the DEGs. Changes in the expression of the genes upon AAV9 administration were defined as follows:
Completely rescued genes: Genes whose expression levels were changed in the Lmna−/− hearts but were normalized upon AAV9‐Lmna WT injection. These genes were considered completely rescued and likely bonafide LMNA regulated genes. Accordingly, transcript levels of these genes were significantly different (q<0.05) between the Lmna−/− and WT mice but were not significantly different (q≥0.05) between the Lmna−/−:AAV9‐Lmna WT and WT groups. Therefore, transcript levels of these genes were considered normalized. The completely rescued genes comprised 23.9% of the upregulated (195/814) and 25.1% (306/1222) of the downregulated genes in the Lmna−/− hearts (Figure 2B and Figure S2A).
Partially rescued genes: Dysregulated genes in the Lmna−/− heart whose expression levels were significantly improved upon administration of the Lmna−/−:AAV9‐Lmna WT (q<0.05 as compared with their levels in the Lmna−/− hearts) but were not completely restored to normal levels and were not significantly different from the levels in the WT hearts. The majority of the DEGs were in this category, which included 63.3 % of the upregulated (516/814) and 69.8% of the downregulated (853/1222) genes (Figure 2B and Figure S2B).
Unchanged genes: Genes whose expression levels did not change upon injection of the AAV9‐Lmna WT viruses in the Lmna−/− hearts. Therefore, transcript levels of these genes remained altered in the Lmna−/−: AAV9‐Lmna WT hearts as compared with WT hearts. Thus, the reexpression of the LMNA did not affect the expression levels of this set of genes. This group of genes comprised 12.60% (103/814) of the upregulated and 5% (63/1222) of downregulated genes (Figure 2B and Figure S2C).
Thus, considering the above, cardiac gene expression profile in the Lmna−/−: AAV9‐Lmna WT mice closely resembled that of the WT mice, as only 680 genes remained differentially expressed between these 2 groups (Figure 2C). Compared with the Lmna−/− mice, the reintroduction of LMNA in the heart was associated with increased expression levels of 1052 genes and suppressed expression levels of 1066 genes (Figure 2D). Hierarchical clustering of DEGs showed that cardiac transcriptomic profile in the Lmna−/−: AAV9‐Lmna WT mice was closer to that in WT mice but was distinct from the transcript profile in Lmna−/− mouse hearts (Figure 2E).
Effects of LMNA on the Expression of Noncoding RNA Expression in the Heart
We have shown before that LADs were predominantly enriched at the noncoding and heterochromatic regions in human cardiac myocytes. 13 Therefore, effects of LMNA deficiency and reexpression on noncoding RNAs were analyzed. The noncoding RNAs (henceforth lncRNA), antisense RNAs, sense overlapping, and divergent transcripts, were included in the analysis, whereas snoRNAs, pre‐miRNA, and pseudogenes were excluded. A total of 629 lncRNAs were differentially expressed (193 upregulated and 436 downregulated) in Lmna−/− compared with WT mouse hearts (Figure 2F). Reexpression of LMNA in the heart completely rescued 49 of 193 (25.4%) of the upregulated and 159 of 436 (36.5%) of the downregulated lncRNAs in the heart (Figure 2G). The corresponding numbers for partial rescue were 132 of 193 (68.4%) for upregulated and 267 of 436 (61.2%) for downregulated lncRNAs (Figure 2G). In contrast, expression levels of only 118 lncRNAs were significantly different between the Lmna−/−: AAV9‐Lmna WT and WT groups (Figure 2H). Finally, as compared with Lmna−/− mouse heart, 1698 lncRNA were differentially expressed in the Lmna−/−: AAV9‐Lmna WT (Figure 2I). Consequently, hierarchical clustering analysis of the lncRNAs showed the Lmna−/−: AAV9‐Lmna WT group was clustering with the WT as compared with Lmna−/− mice (Figure 2J).
Effects of LMNA on Dysregulated Regulatory Network and Biological Pathways
GSEA was performed to identify biological pathways regulated by LMNA. Only genes that were classified as completely rescued were used for this analysis (Figure 3A). GSEA using the Hallmark Molecular Signature showed restoration of LMNA regulated pathways that are mainly involved in inflammation, cell death, and fibrosis through tumor necrosis factor‐α signaling, TP53 (tumor protein 53) and epithelial to mesenchymal transition. The pathways were predicted to be upregulated in the Lmna−/− compared with WT mouse hearts (Figure 3B) and suppressed in the Lmna−/−: AAV9‐Lmna WT as compared with Lmna−/− mouse hearts. Likewise, among the suppressed pathways genes involved in oxidative phosphorylation, E2F pathway, and G2M checkpoint (Figure 3C) were rescued as shown by suppression of these pathways in Lmna−/− (compared with WT) and reversal upon reexpression of LMNA in the heart (Lmna−/−: AAV9‐Lmna WT compared with Lmna−/−, Figure 3C).
Figure 3. Biological pathways in Lmna−/− and upon Lmna re‐expression.
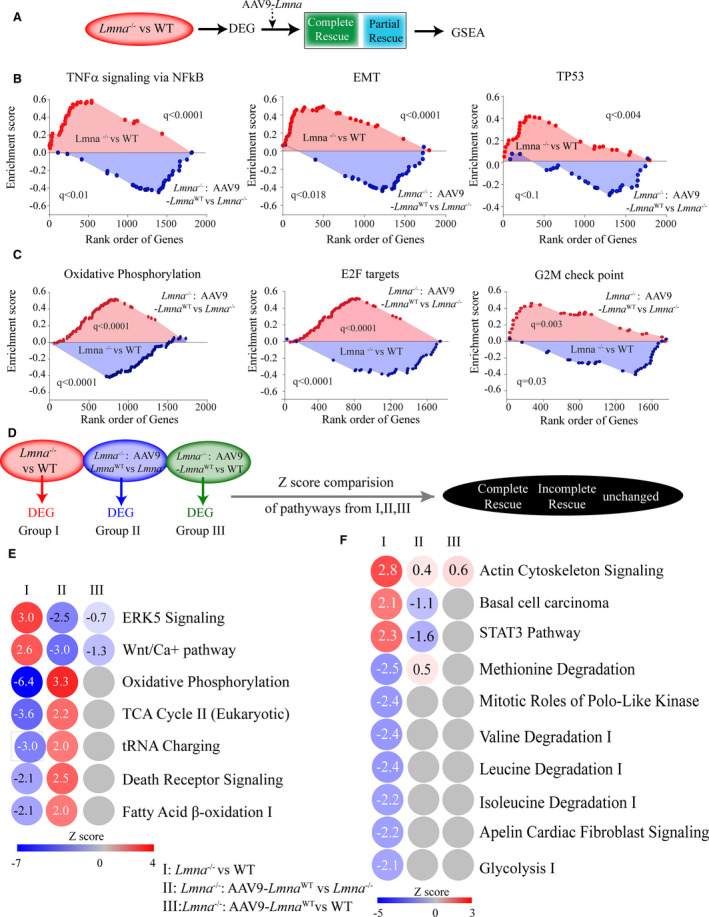
A, Schematic showing the genes selection for Gene Set Enrichment Analysis (GSEA). B and C, Top Hallmark signature upregulated (B) and downregulated (C) in Lmna−/− vs WT are shown and their corresponding status upon AAV9‐Lmna WT treatment (Lmna−/−:AAV9‐Lmna WT). D, Schematic showing the genes used for Ingenuity Pathway Analysis (IPA). Differentially expressed genes were obtained from Lmna−/− vs WT, Lmna−/−:AAV9‐Lmna WT vs Lmna−/−, and Lmna−/−:AAV9‐Lmna WT vs WT comparison and used to identify significantly altered pathways. Comparisons of pathways that significantly change in the Lmna−/− vs WT (Z score ≥2 and ≤−2 and P<0.05) were made to those in other groups. E and F, IPA analysis for enriched pathways comparisons in all three groups. Z scores are indicated for each pathway, activated pathways (red), and the inhibited (blue) are shown and the color of the circles is indexed to the Z score values. The comparison in Lmna−/− vs WT (first column), Lmna−/−:AAV9‐Lmna WT vs Lmna−/− (middle column), and Lmna−/−:AAV9‐Lmna WT vs WT (right column) are shown. E, Depicts the pathways that showed complete reversal defined as ones that are induced or suppressed in Lmna−/− vs WT group (Z‐score greater than or equal to 2 and less than or equal to −2 and P<0.05) and their reversal in Lmna−/−:AAV9‐Lmna WT group (Z score ≥2 and ≤−2 and P<0.05) and no change in Lmna−/−:AAV9‐Lmna WT vs WT group. F, Partially rescued pathways defined as ones that are induced or suppressed in Lmna−/− vs WT group but is not different in the Lmna−/−:AAV9‐Lmna WT vs Lmna−/− and Lmna−/− vs WT group. Z score was obtained from IPA. AAV9 indicates adeno‐associated virus serotype 9; DEG, differentially expressed gene; EMT, epithelial to mesenchymal transition; ERK5, extracellular signal‐regulated kinase 5; GSEA, gene set enrichment analysis; NFκB, nuclear factor kappa‐light‐chain enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3; TCA, tricarboxylic acid; TNFα, tumor necrosis factor‐α; TP53, tumor protein 53; and WT, wild‐type.
However, several pathways did not show rescue upon Lmna−/−: AAV9‐Lmna WT treatment (Table S4). Noteworthy among them was the gene set for apoptosis that showed induction in Lmna−/− versus WT group (q=0.013, Table S4) but remained unchanged upon AAV9‐Lmna WT treatment (q=0.8, Table S4).
IPA was performed to identify activated (Z score ≥2.0) and suppressed (Z score ≤2.0) biological pathways, as predicted from the DEGs among the 3 groups used as input (Figure 3D). IPA identified a subset of pathways and thereby reversal of Z score values upon AAV9‐Lmna WT treatment (Figure 3E). Likewise, some pathways showed only incomplete reversal of gene signature (Figure 3F) and their Z score was closer to WT upon AAV9‐Lmna treatment and did not reach the threshold cutoff (activated [Z score ≥2.0] and suppressed [Z score ≤−2.0]). Finally, the pathways that were not affected upon AAV9 treatment showed Z score value similar to the Lmna−/− group and were characterized as unchanged. Notable were those involved in cardiac hypertrophy/function (extracellular signal‐regulated kinase 5 and Wnt), as indicated by their induction (Z score ≥2.0) in the Lmna−/− mouse hearts and their reversal upon AAV9‐mediated LMNA expression (Z score ≤−2.0) and thereby, normalization toward WT (Figure 3E). Likewise, pathways involved in cardiac metabolism, mainly in mitochondrial function (oxidative phosphorylation, glycolysis, and fatty acid β‐oxidation I), were suppressed in the Lmna−/− mouse hearts (Figure 3E) and restored upon LMNA reexpression in Lmna−/− mouse hearts (Figure 3D and 3E). Taken together, these findings identify LMNA‐regulated pathways and implicate the role for LMNA in cardiac metabolism, cell death, and inflammation.
To identify TRs that were responsible for the dysregulated gene expression and were LMNA dependent, only genes that showed complete rescue and were therefore considered bona fide LMNA‐regulated genes were analyzed using the Upstream Regulators Analysis function of IPA. Over a dozen TRs that control this gene dysregulation in Lmna−/− were identified, including TP53, KDM5A (as the top candidate TR for activation). Retinoblastoma 1 (RB1) and paired‐like homeodomain 2 (PITX2) were identified as most likely TRs to be suppressed in the Lmna−/− mouse heart (Table S5).
DEGs were also analyzed to infer the TRs whose activity was restored after reexpression of the LMNA in the heart (Figure 4A). Significantly changed TRs that were identified in each group and then compared with the status of these TRs. According to changes in the DEGs upon administration of the AAV9 viruses, activated TP53, KDM5A, and nuclear receptor–interacting protein 1 were fully rescued, whereas KDM5B, forkhead box O, SMAD3 (Mothers Against Decapentaplegic Homolog 3), and several others were partially rescued (Figure 4B). Similarly, among the suppressed TRs, RB1, PITX2, and melanocyte‐inducing transcription factor were completely rescued, and several others were partially rescued (Figure 4C). The complete list of dysregulated TRs and inferred changes in their transcriptional activities upon AAV9 administration are shown in Figure 4B and 4C. A similar analysis of other regulators using IPA for DEG showed rescue of rapamycin‐insensitive companion of mTOR, insulin receptor, and serine‐threonine kinase 11, and an incomplete rescue of several others (Table S6). Finally, a subset of upstream regulators was inferred as unchanged, including TGF (transforming growth factor‐β1) (Table S6), RELA proto‐oncogene, NF‐kB subunit, signal transducer and activator of transcription 3, CAMP responsive element‐binding protein 1 and sex‐determining region Y‐box 2 (Figure 4D and 4E). Of the upstream TRs identified in this analysis, we have previously implicated TP53 and forkhead box O3 in the pathogenesis of LMNA‐associated DCM. 17 , 19 Notable among the unchanged regulator was TGF, a well‐characterized mediator of cardiac fibrosis (see below).
Figure 4. Transcriptional regulators (TR) perturbed in the Lmna−/− mice and their rescue in AAV9‐LmnaWT treated hearts.
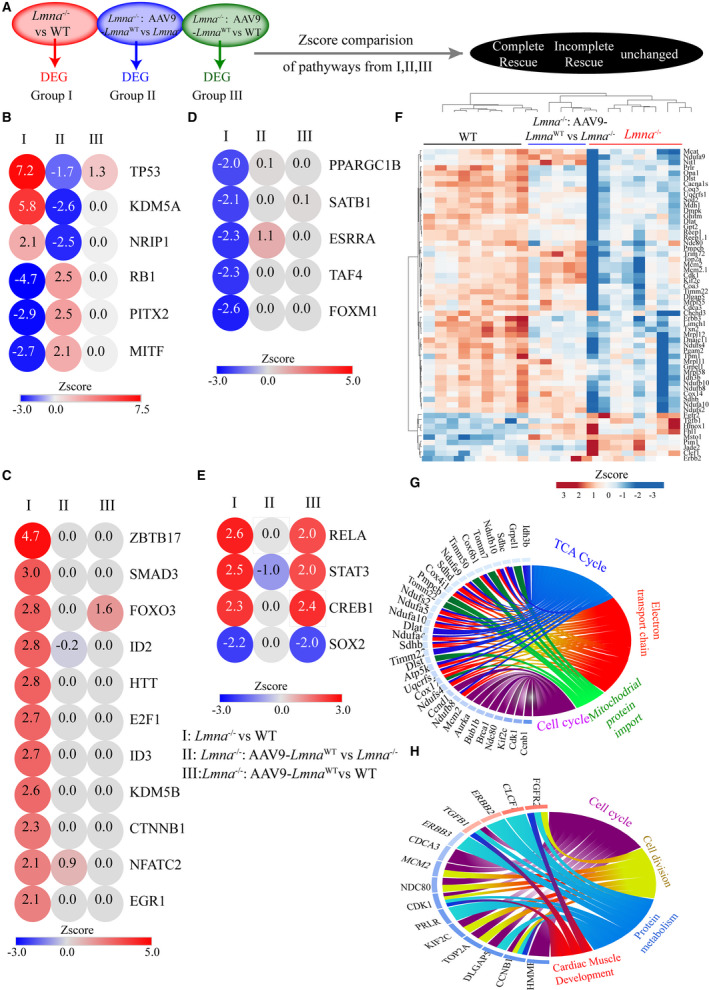
A, Schematic showing the genes used for Ingenuity Pathway Analysis (IPA). B through E, IPA analysis for upstream regulator showing top dysregulated TRs. Z‐Score comparisons for indicated TRs in 3 experimental groups are plotted. Activated ones are shown in red, and the inhibited in blue. The color of the circles is indexed to the Z score values. The experimental group used for comparison are marked as follows (I) Lmna−/− vs WT, (II) Lmna−/−:AAV9‐Lmna WT vs Lmna−/− (III) Lmna−/−:AAV9‐Lmna WT vs WT. B, Denotes complete reversal, (C) shows incompletely rescued upregulated and (D) shows incompletely rescued downregulated TRs. E, TR not affected by LMNA reexpression. F, Heat plot of the lysine‐specific demethylase 5A (KDM5A) and B targets genes that show overlap with the IPA curated database and show expression pattern consistent with KDM5A and B activation. Hierarchical clustering of the genes shows that WT and AAV9‐LMNAWT injected Lmna−/− cluster together and are distinct from the Lmna−/− group. G and H, Circos plot of gene ontology analysis of KDM5A (G) and KDM5B (H) targets that are differentially expressed in Lmna−/− mouse hearts compared with WT. AAV9 indicates adeno‐associated virus serotype 9; LMNA, lamin A; and WT, wild‐type.
The involvement of KDM5A and B in the laminopathies or cardiac dysfunction were novel findings. Therefore, we curated the KDM5A and B predicted targets from IPA and found that transcript levels of 37 of 42 KDM5A and 15 of 19 KDM5B target genes, were altered in the Lmna−/− mouse hearts (Figure 4F). KDM5A targets were enriched for genes involved in mitochondrial biogenesis and function/cardiac energy metabolism (Figure 4G), whereas KDM5B targets showed enrichment of genes involved in cell cycle and cell division (Figure 4H).
To validate activation of the KDM5A in the hearts in the Lmna−/− mice, transcript levels of a selected number of KDM5A target genes were analyzed by quantitative polymerase chain reaction, which confirmed decreased transcript levels of KDM5A target genes (Figure 5B). Evaluation of Kdm5a and Kdm5b mRNAs in these groups did not show any changes in the RNA levels among the experimental groups (Figure 5C). However, immunoblotting performed on myocardial nuclear protein extracts using antibodies that detect KDM5A and B showed increased nuclear levels of KDM5A by 18.3±8.9‐fold (n=3; P=0.02) and KDM5B by 7.5±2.7‐fold (n=3; P=0.008) in the Lmna−/− as compared with WT mouse hearts (Figure 5D). AAV9‐mediated LMNA reexpression partially rescued KDM5A and B protein levels in the Lmna−/− mouse hearts (Figure 5E). The data provided additional evidence of KDM5 dysregulation in the Lmna−/− mouse hearts.
Figure 5. l KDM5A and B are activated in Lmna−/− and partially rescued in AAV9‐LmnaWT treated hearts.
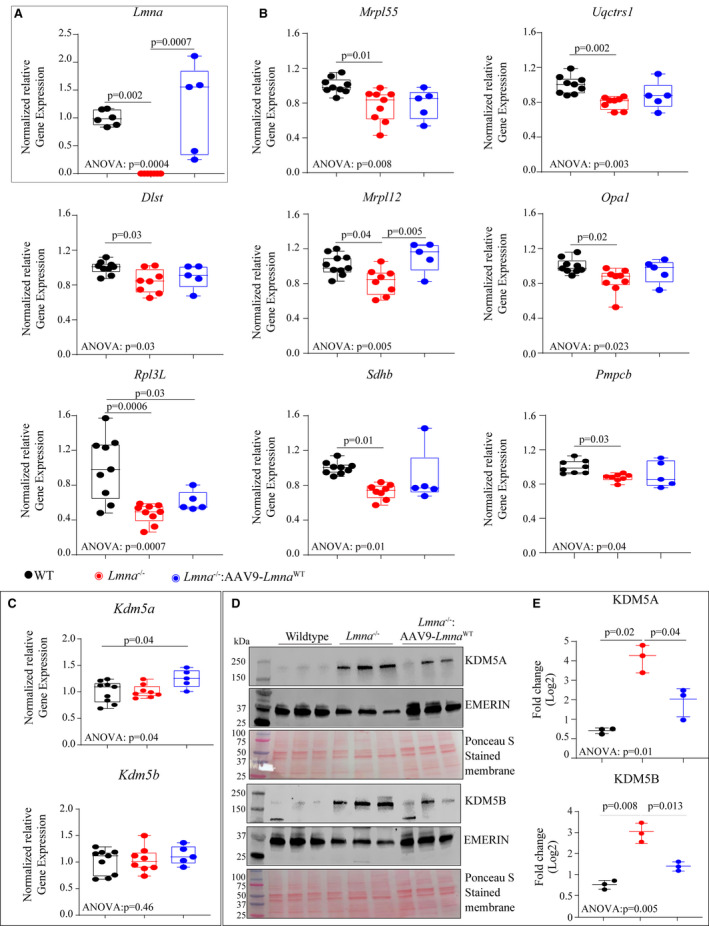
Quantitative PCR (qPCR) analysis of Lmna (A) and targets of KDM5A and KDM5B (B) in the WT, Lmna−/− and AAV9‐LMNAWT injected mice. C, qPCR analysis of Kdm5a and Kdm5b mRNAs showed no differential expression in the indicated groups. D, Immunoblot of nuclear extract from WT, Lmna−/− and AAV9‐LMNAWT injected Lmna−/− mouse hearts showing KDM5A (top panel) and KDM5B (lower panel) using anti‐KDM5A and anti‐KDM5B antibodies. Emerin was used as a nuclear loading control and Ponceau S stained membranes are shown to assess similar loading patterns. E, Respective quantitative data of KDM5A and KDM5B in the nuclear fraction in the heart of WT, Lmna−/−, and Lmna−/−: AAV9‐LMNAWT mice (n=3). For normally distributed dataset, 1‐way ANOVA followed by Tukey’s multiple pairwise comparison P values are shown and for once not normally distributed Kruskal–Wallis followed by Dunn’s multiple pairwise comparison P‐values are shown. Only P‐values that were significant (P<0.05) are shown. AAV9 indicates adeno‐associated virus serotype 9; KMD5, lysine‐specific demethylase 5; LMNA, lamin A; and WT, wild‐type.
LncRNA, mRNA Coexpression Network
Unlike protein‐coding genes, the majority of lncRNAs do not yet have defined biological functions. However, lncRNAs have been observed to regulate expression of mRNAs by acting in cis or trans, which enables the prediction of lncRNAs functions based upon co‐regulated mRNAs as proxies. 35 , 36 Pairwise analysis of completely rescued 207 lncRNA and 501 mRNAs revealed multiple strong correlations for lncRNA: mRNA pair, as indicated by a high r‐value (Figure S3A). Analysis of partially rescued 399 lncRNAs and 1369 mRNAs also showed a similar pairwise correlation (Figure S3A and S3B). Next, we reasoned that expressions of lncRNAs and mRNAs belonging to the similar biological pathways are likely to be coordinated and, thereby, are expected to show a high correlation in their expression pattern. To ascertain the biological roles of differentially expressed lncRNAs, we obtained highly correlated lncRNA: mRNA pairs that showed a Pearson correlation coefficient (r) >0.9 or −0.9 (Figure S3C). IPA for biological pathways involving these mRNAs (as a surrogate for the correlated lncRNAs), showed enrichment for genes involved in oxidative phosphorylation (Figure S3D). Further analysis for the transcriptional regulators of the correlated mRNAs showed enrichment for targets of TP53, KDM5A, NUPR1, predicting their activation in the Lmna−/− mouse hearts. In contrast, transcriptional regulators RB1, peroxisome proliferator–activated receptor gamma coactivator 1‐α, peroxisome proliferator–activated receptor gamma coactivator 1‐β, and PITX2 were predicted to be suppressed (Figure S3E and S3F).
Phenotypic Consequences of Reexpression of LMNAWT in Lmna−/− Mouse Hearts
The Lmna−/− mice exhibited cardiac dilatation and dysfunction at 4 weeks of age in accordance with previously published data, as indicated by a marked increase in left ventricular end‐diastolic and systolic diameters, and reduced left ventricular fractional shortening (Table S7). Consistent with the partial rescue of differentially expressed coding and noncoding genes, AAV9 mediated reexpression of LMNAWT improved cardiac function in the Lmna−/− mice, as noted by improvement in the echocardiographic indices of cardiac size (left ventricular end‐diastolic diameter and left ventricular end‐systolic diameter, Figure 6A) and function (Figure 6A and Tables S3 and S7). The improvement in cardiac function was associated with prolonged median survival (Figure 6B), from 28 days in the Lmna−/− mice to 52 days in the Lmna−/−:AAV9Lmna WT mice. Treatment of Lmna−/− mice with control viruses comprised of the AAV9 viral genome but without the Lmna gene did not affect survival (Figure 6B and Figure S4) or cardiac function (Figure 6A and Table S7). Likewise, treatment of Lmna−/−:AAV9Lmna Flag had a similar improvement in survival and cardiac function (Figure S4 and Table S8), further indicating that the FLAG epitope did not interfere with the beneficial effect of Lmna reexpression.
Figure 6. Phenotypic consequences of AAV9 mediated LmnaWT expression in the Lmna−/− mice.
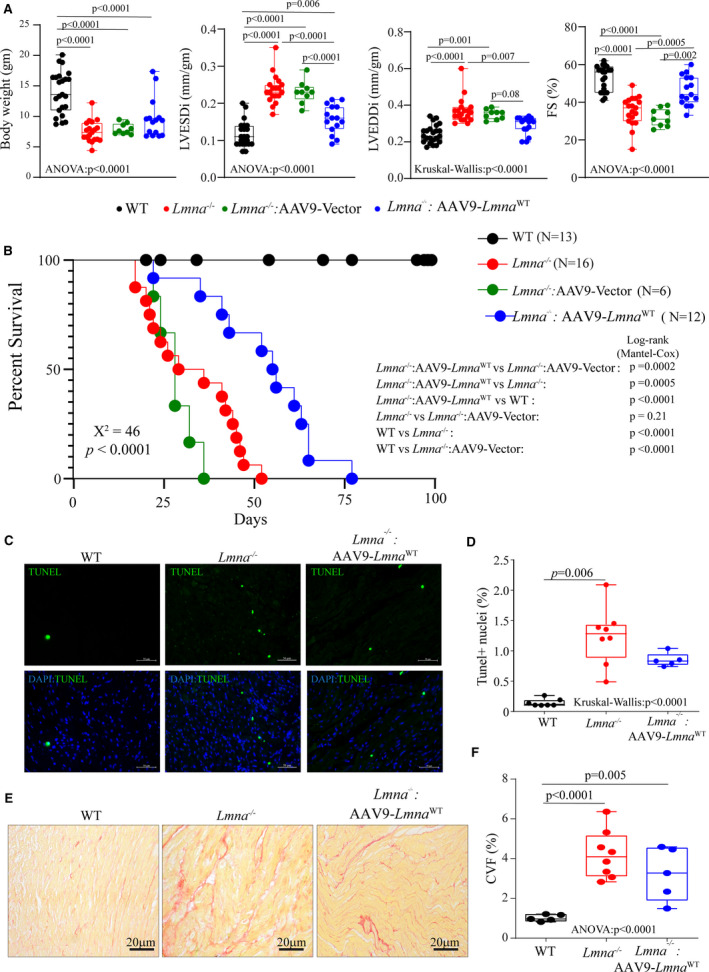
A, Selected echocardiographic indices of cardiac structure and function showing improvement of cardiac function upon vector or AAV9‐Lmna WT injection of Lmna−/− mice at 4 weeks after birth. LVEDDi: Left Ventricular End‐Diastolic Diameter indexed to the bodyweight; LVESDi, left ventricular end‐systolic diameter indexed to the bodyweight; FS, fractional shortening (age, sex, and number of mice used in each group are listed in Tables S3 and S7). B, Kaplan–Meier survival plots of WT, Lmna−/−, and Lmna−/− mice injected with vector or AAV9 expressing LmnaWT. Chi‐square and P value for the overall Kaplan–Meier survival analysis are indicated in bold. Log‐rank (Mantel‐Cox) pairwise analysis P values for each subgroup analysis are shown. C, Representative terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) stained thin myocardial cross‐section from 4‐week‐old WT, Lmna−/−, and Lmna−/−: AAV9‐Lmna WT are shown. The lower panel shows overlay images of TUNEL staining in green and nuclei (blue). D, Respective quantitative data of the TUNEL‐positive stained nuclei in WT (n=7), Lmna−/− (n=8), and Lmna−/− mice injected with AAV9‐Lmna WT (n=5). E, Representative Picrosirius red‐stained thin myocardial sections from 4‐week‐old WT, Lmna−/−, and Lmna−/−: AAV9‐Lmna WT. F, Respective quantitative data on collagen volume fraction in WT (n=5), Lmna−/− (n=8), and Lmna−/− mice injected with AAV9‐Lmna WT (n=5). One‐way ANOVA followed by Tukey’s multiple pairwise comparison P values is shown. Only P values that were significant (P<0.05) are shown. AAV9 indicates adeno‐associated virus serotype 9; KMD5, lysine‐specific demethylase 5; and WT, wild‐type.
Given that myocardial apoptosis is a phenotypic feature of the Lmna−/− mice and in view of activation of TP53 pathway, 19 the effect of reexpression of LMNA on cell death was evaluated by terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling TUNEL assay in heart sections. Consistent with the previous data, 17 the Lmna−/− mice showed increased TUNEL positive cells (1.24±0.47% in Lmna−/− versus 0.14±0.06% in WT control mice, P<0.001). Upon AAV9 treatment the number of TUNEL positive cells showed a trend toward a reduction in the Lmna−/−:AAV9Lmna WT hearts (0.85±0.11% in AAV9‐treated versus 1.24±0.47% in Lmna−/− mice, P=0.09, Figure 6C and 6D). Gene expression analysis of cell death genes with quantitative polymerase chain reaction also showed a similar trend with partial or no rescue upon AAV9 treatment (Figure S5A). Myocardial fibrosis quantitated and represented as collagen volume fraction was increased in Lmna−/− mice (3.59±1.32% in Lmna−/− versus 1.02±0.15% in WT, P=0.0003). However, the re‐expression of the LMNAWT did not have a discernible effect on collagen volume fraction in the Lmna−/− mice (3.23±1.34% in Lmna−/−: AAV9‐Lmna WT versus 3.59±1.32% in Lmna−/−, P=0.9, Figure 6E and 6F). Consistent with this observation AAV9‐Lmna WT treatment did not rescue the molecular signature of cardiac fibrosis, as indicated by the unchanged status of TGF, a key regulator of fibrosis in the treated group (Table S6). Likewise, quantitative polymerase chain reaction analysis of selected fibrosis markers showed partial or no rescue in the treated group (Figure S5B).
DISCUSSION
LMNA, a ubiquitously expressed protein in differentiated cells, interacts with numerous genomic regions through LADs, and influences chromatin structure. 12 , 13 , 17 , 37 , 38 We determined early transcriptomic changes (before the onset of cardiac dysfunction) in the hearts of Lmna−/− mice, compared with WT mice and following reexpression of LMNA in the Lmna−/− mouse heart. The approach was taken to reduce potential confounding effects of cardiac dysfunction on gene expression and identify DEGs and pathways that are likely pathogenic in these hearts (and not secondary to cardiac dysfunction). This methodology led us to identify 501 coding and 208 noncoding as direct LMNA‐regulated genes since their expression was completely rescued upon LMNA expression in the Lmna−/− mice. This study also identified 1862 coding and 607 lncRNA genes as partially rescued and dependent on LMNA for their expression and regulation.
Our study identified key upstream regulators of dysregulated transcripts namely KDM5A, TP53, and rapamycin‐insensitive companion of mTOR, which were activated, and RB1, PITX2, and melanocyte‐inducing transcription factor, which were suppressed. Of these, activation of the KDM5 family of proteins in the heart in laminopathies is a novel finding and was verified at the protein level. The induction of KDM5A expression was associated with suppression of its downstream targets, which included genes involved in mitochondrial biogenesis and function. These studies are also consistent with our previous data showing reduced mitochondrial electron transport complex I gene expression and enzymatic activity in the Lmna−/− mouse hearts. 17 Recent studies have also shown induction of KDM5A in other forms of cardiomyopathies associated with suppression of mitochondrial activity and oxidative phosphorylation, thereby underscoring the role of KDM5 in cardiac energy homeostasis in the context of heart failure. 39
Furthermore, our analysis identified several transcriptional regulators, including E2F, TP53, and forkhead box Os that were previously implicated in the laminopathies. 13 , 14 , 17 , 19 These altered regulators identified in the present study collectively regulate numerous biological processes relevant to laminopathies, including cell proliferation, cell death, inflammation, and metabolism, among others. These transcriptional factors were regulated by LMNA, as reexpression of LMNA in the Lmna−/− mice partially or reverted the regulators toward normal WT levels.
Gene expression analysis also showed that the cell death/apoptosis signature, which was induced upon LMNA loss, could not be rescued upon AAV9 treatment. These gene expression data were consistent with increased TUNEL positive cells in the Lmna−/− hearts that could not be rescued by AAV9 treatment. Expression of the differentially expressed lncRNA correlated with those of the coding RNAs. A subset of these lncRNAs correlated highly with the expression of mRNAs that are targeted by the upstream regulators such as TP53 and KDM5A. Additional studies would be required to delineate the role of TP53 and KDM5A in the regulation of lncRNAs and their biological functions in the heart and to substantiate their pathogenic roles in cardiac dysfunction associated with laminopathies.
The study has several limitations. Notable among them is that RNA sequencing was performed on whole heart RNA rather than in isolated cardiac myocyte RNA. Therefore, the observed transcriptomic changes in the Lmna−/− mouse hearts originated from multiple cell types in the heart that are deficient in the LMNA protein. However, the rescue experiments with the recombinant AAV9 viruses results in re‐expression of the LMNA protein mainly in cardiac myocytes and to a lesser extent, if at all, in other cell types. Whole heart RNA sequencing was adopted, as opposed to cardiac myocyte RNA sequencing, to determine global changes in the transcript levels, comprised of changes in the transduced cardiac myocytes and those in nonmyocyte cells, the latter encompassing the paracrine effects originating from transduced cardiac myocytes. The approach is not expected to rescue differentially expressed genes originating from nontransduced cardiac cells which likely are non‐cardiac myocyte cells in the heart. This limitation might in part explain the incomplete phenotypic rescue in the Lmna−/− mice. Consistent with this notion, cardiac fibrosis in the Lmna−/− mice was not affected upon AAV9 gene transfer, reflective of the ineffective transduction of cardiac fibroblasts with the AAV9 viruses. Similarly, apoptosis, as assessed by the TUNEL assay, was not rescued, likely indicative of cell death occurring in non‐myocyte LMNA‐deficient cells in the heart. Partial rescue of cardiac function despite the lack of an effect on apoptosis, points to a direct role of the LMNA in cardiac myocyte contractile function. While we surmise that apoptosis involved non‐myocyte cells, accurate identification of the cell type undergoing apoptosis remains unknown. Therefore, contributions of various cell types to the pathogenesis of cardiac phenotype in LMNA‐deficiency could not be accurately ascertained in the present study, but it is reasonable to conclude that cardiac myocytes are the major contributors to cardiac dysfunction and impaired survival in the LMNA‐deficient mice. Finally, the AAV9 virus is known to be highly cardiotrophic especially at the dose used in the present study. Nevertheless, other cell types such as skeletal muscles might be transduced as well and contribute to phenotypic rescue.
RNA sequencing data showed a preponderance of partially rescued genes upon AAV9‐LmnaWT treatment. This was also associated with variability in the gene expression as observed in the RNA sequencing and subsequent quantitative polymerase chain reaction experiments. Several factors may account for these observations, among them is that even though AAV9 has a well‐established tropism toward cardiac myocytes, the transduction efficiency was incomplete and to some extent heterogeneous among myocytes and in myocardial regions. Consequently, transduction with the recombinant AAV9 viruses leads to variable expression of the transgene among hearts and animals, and maybe one reason for the partial rescue of the phenotype. Furthermore, since LMNA regulates gene expression through multiple mechanisms the threshold of LMNA expression required to achieve complete reversal might be variable, dependent on a gene by gene basis, and may account for the partial rescue in gene expression pattern and ensuing phenotypes. In this study the numbers of the animals in the experimental group were not sufficiently large to conclusively determine the correlation between the expression level of the transgene and the partially rescued phenotypes such as survival or cardiac function.
The mechanism, by which LMNA influences gene expression, including expression of lncRNAs, is not fully understood and seemingly involves multiple mechanisms. Our study identifies KDM5A and KDM5B, and TP53 as the potential regulators. Future studies would be required to delineate the mechanism(s) responsible for the upregulation of KDM5A and KDM5B in the heart in DCM and determine their pathogenic roles in DCM associated with laminopathies. Nevertheless, transcriptomic restoration was sufficient to improve survival and cardiac function and to some extent reduce cell death. Collectively, the approach enabled the identification of the dysregulated transcriptome and the upstream regulators that are regulated by LMNA in the heart, independent of cardiac dysfunction in the Lmna−/− mice.
Taken together, our data point to the role of LMNA in regulating expression of coding and non‐coding RNAs in the heart and identify activation of KDM5A and KDM5B, TP53, and rapamycin‐insensitive companion of mTOR and suppression of RB1, PITX2 and melanocyte‐inducing transcription factor in the LMNA deficient hearts. These findings could serve as a platform for gaining mechanistic insights into the pathogenesis of the cardiac phenotypes in laminopathies, including cardiomyopathy, cardiac arrhythmias, and cardiac conduction defects.
Sources of Funding
This work was supported in part by grants from NIH (NIA, R21 AG060413‐01, NHLBI, R01 HL088498 and 1R01HL132401), Leducq Foundation (14 CVD 03), The Welch Foundation, The Ewing Halsell Foundation, and George and Mary Josephine Hamman Foundation.
Disclosures
None.
Supporting information
(J Am Heart Assoc. 2020;9:e015690 DOI: 10.1161/JAHA.119.015690.)
Raffaella Lombardi is currently located at the Division of Cardiology, Department of Advanced Biomedical Science, University of Naples Federico II, Italy.
Suet Nee Chen is currently located at the CU‐Cardiovascular Institute and Adult Medical Genetics Program, University of Colorado Anschutz Medical Campus, Aurora, CO.
For Sources of Funding and Disclosures, see page 16.
References
- 1. Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7:940–952. [DOI] [PubMed] [Google Scholar]
- 2. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction‐system disease. N Engl J Med. 1999;341:1715–1724. [DOI] [PubMed] [Google Scholar]
- 3. Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152:1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Worman HJ, Bonne G. "Laminopathies": a wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–1136. [DOI] [PubMed] [Google Scholar]
- 6. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–780. [DOI] [PubMed] [Google Scholar]
- 7. Anselme F, Moubarak G, Savoure A, Godin B, Borz B, Drouin‐Garraud V, Gay A. Implantable cardioverter‐defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. 2013;10:1492–1498. [DOI] [PubMed] [Google Scholar]
- 8. van Berlo JH, Duboc D, Pinto YM. Often seen but rarely recognised: cardiac complications of lamin A/C mutations. Eur Heart J. 2004;25:812–814. [DOI] [PubMed] [Google Scholar]
- 9. Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. [DOI] [PubMed] [Google Scholar]
- 10. Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. Single‐cell dynamics of genome‐nuclear lamina interactions. Cell. 2013;153:178–192. [DOI] [PubMed] [Google Scholar]
- 11. Perovanovic J, Dell'Orso S, Gnochi VF, Jaiswal JK, Sartorelli V, Vigouroux C, Mamchaoui K, Mouly V, Bonne G, Hoffman EP. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci Transl Med. 2016;8:335ra358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Steensel B, Belmont AS. Lamina‐associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, et al. Genomic reorganization of lamin‐associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. 2019;124:1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brayson D, Shanahan CM. Current insights into lmna cardiomyopathies: Existing models and missing lincs. Nucleus. 2017;8:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lund EG, Duband‐Goulet I, Oldenburg A, Buendia B, Collas P. Distinct features of lamin A‐interacting chromatin domains mapped by ChIP‐sequencing from sonicated or micrococcal nuclease‐digested chromatin. Nucleus. 2015;6:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paulsen J, Sekelja M, Oldenburg AR, Barateau A, Briand N, Delbarre E, Shah A, Sorensen AL, Vigouroux C, Buendia B, et al. Chrom3D: three‐dimensional genome modeling from Hi‐C and nuclear lamin‐genome contacts. Genome Biol. 2017;18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Auguste G, Gurha P, Lombardi R, Coarfa C, Willerson JT, Marian AJ. Suppression of activated FOXO transcription factors in the heart prolongs survival in a mouse model of laminopathies. Circ Res. 2018;122:678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226:316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen SN, Lombardi R, Karmouch J, Tsai JY, Czernuszewicz G, Taylor MRG, Mestroni L, Coarfa C, Gurha P, Marian AJ. DNA damage response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (lamin A/C) mutations. Circ Res. 2019;124:856–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C‐deficient mice. J Clin Invest. 2004;113:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikolova‐Krstevski V, Leimena C, Xiao XH, Kesteven S, Tan JC, Yeo LS, Yu ZY, Zhang Q, Carlton A, Head S, et al. Nesprin‐1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C‐deficient cardiomyopathy. J Mol Cell Cardiol. 2011;50:479–486. [DOI] [PubMed] [Google Scholar]
- 22. Sullivan T, Escalante‐Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of a‐type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheedipudi SM, Hu J, Fan S, Yuan P, Karmouch J, Czernuszewicz G, Robertson MJ, Coarfa C, Hong K, Yao Y, et al. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc Res. 2020;116:1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. [DOI] [PubMed] [Google Scholar]
- 25. Gurha P, Chen X, Lombardi R, Willerson JT, Marian AJ. Knockdown of plakophilin 2 downregulates miR‐184 through CpG hypermethylation and suppression of the E2F1 pathway and leads to enhanced adipogenesis in vitro. Circ Res. 2016;119:731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics. 2009;25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Law CW, Alhamdoosh M, Su S, Smyth GK, Ritchie ME. RNA‐seq analysis is easy as 1‐2‐3 with limma, Glimma and edgeR. F1000Res. 2016;5:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bergmann O, Zdunek S, Alkass K, Druid H, Bernard S, Frisen J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp Cell Res. 2011;317:188–194. [DOI] [PubMed] [Google Scholar]
- 32. Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, Wurch A, Bonisch U, Gunther S, Backofen R, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Preissl S, Schwaderer M, Raulf A, Hesse M, Gruning BA, Kobele C, Backofen R, Fleischmann BK, Hein L, Gilsbach R. Deciphering the epigenetic code of cardiac myocyte transcription. Circ Res. 2015;117:413–423. [DOI] [PubMed] [Google Scholar]
- 34. Ishikawa K, Tilemann L, Fish K, Hajjar RJ. Gene delivery methods in cardiac gene therapy. J Gene Med. 2011;13:566–572. [DOI] [PubMed] [Google Scholar]
- 35. Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte‐specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, Zolotukhin I, Tarantal AF, Byrne BJ. Recombinant adeno‐associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. [DOI] [PubMed] [Google Scholar]
- 37. de Leeuw R, Gruenbaum Y, Medalia O. Nuclear lamins: thin filaments with major functions. Trends Cell Biol. 2018;28:34–45. [DOI] [PubMed] [Google Scholar]
- 38. Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Mol Genet Metab. 2006;87:289–302. [DOI] [PubMed] [Google Scholar]
- 39. Chen L, Yang F, Chen X, Rao M, Zhang NN, Chen K, Deng H, Song JP, Hu SS. Comprehensive myocardial proteogenomics profiling reveals C/EBPalpha as the key factor in the lipid storage of ARVC. J Proteome Res. 2017;16:2863–2876. [DOI] [PubMed] [Google Scholar]
- 40. Kohlbrenner E, Henckaerts E, Rapti K, Gordon RE, Linden RM, Hajjar RJ, Weber T. Quantification of AAV particle titers by infrared fluorescence scanning of coomassie‐stained sodium dodecyl sulfate‐polyacrylamide gels. Hum Gene Ther Methods. 2012;23:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.