

Abstract
Background
Variants of the desmosomal protein desmoplakin are associated with arrhythmogenic cardiomyopathy, an important cause of ventricular arrhythmias in children and young adults. Disease penetrance of desmoplakin variants is incomplete and variant carriers may display noncardiac, dermatologic phenotypes. We describe a novel cardiac phenotype associated with a truncating desmoplakin variant, likely causing mechanical instability of myocardial desmosomes.
Methods and Results
In 2 young brothers with recurrent myocarditis triggered by physical exercise, screening of 218 cardiomyopathy‐related genes identified the heterozygous truncating variant p.Arg1458Ter in desmoplakin. Screening for infections yielded no evidence of viral or nonviral infections. Myosin and troponin I autoantibodies were detected at high titers. Immunohistology failed to detect any residual DSP protein in endomyocardial biopsies, and none of the histologic criteria of arrhythmogenic cardiomyopathy were fulfilled. Cardiac magnetic resonance imaging revealed no features associated with right ventricular arrhythmogenic cardiomyopathy, but multifocal subepicardial late gadolinium enhancement was present in the left ventricles of both brothers. Screening of adult cardiomyopathy cohorts for truncating variants identified the rare genetic variants p.Gln307Ter, p.Tyr1391Ter, and p.Tyr1512Ter, suggesting that over subsequent decades critical genetic/exogenous modifiers drive pathogenesis from desmoplakin truncations toward different end points.
Conclusions
The described novel phenotype of familial recurrent myocarditis associated with a desmoplakin truncation in adolescents likely represents a serendipitously revealed subtype of arrhythmogenic cardiomyopathy. It may be caused by a distinctive adverse effect of the variant desmoplakin upon the mechanical stability of myocardial desmosomes. Variant screening is advisable to allow early detection of patients with similar phenotypes.
Keywords: arrhythmogenic cardiomyopathy, cardiovascular genetics, desmoplakin, genome‐environment interactions, myocarditis
Subject Categories: Cardiomyopathy, Genetics, Arrhythmias, Cell Biology/Structural Biology, Mechanisms
Nonstandard Abbreviations and Acronyms
- AC
arrhythmogenic cardiomyopathy
- AM
acute myocarditis
- DCM
dilated cardiomyopathy
- DIFC
desmosome‐intermediate filament complex
- EMB
endomyocardial biopsy
- FACS
fluorescence‐activated cell sorting
- LV
left ventricular
- MRI
magnetic resonance imaging
- PBMC
peripheral blood mononuclear cell
Clinical Perspective
What Is New?
We describe a novel cardiac phenotype associated with a truncating desmoplakin variant, likely causing mechanical instability of desmosome‐intermediate filament complex as the primary, and myocarditis as a secondary phenomenon.
Association between desmosomal protein variants and recurrent myocarditis triggered by exercise has implications for adult and pediatric cardiology.
Testing of the “mechanical failure” hypothesis in experimental models is warranted.
What Are the Clinical Implications?
Recurrent myocarditis should serve as a possible indicator of genetic cardiomyopathy and implicates state‐of‐the‐art genetic screening, unless other myocarditis triggers are unequivocally established.
The association of recurrent myocarditis with competitive sports illustrates the need to consider genome‐environment interactions, even if a pathogenic genetic variant has been established.
Limitation of peaks of physical exercise may be highly effective before secondary pathomechanisms (eg, myocardial inflammation) ensue.
Introduction
Arrhythmogenic cardiomyopathy (AC) is considered an autosomal dominant inherited disease linked to variants in genes encoding desmosomes or desmosome‐related proteins. There is no single gold standard test for AC patients, but diagnosis is based on a score of major and minor criteria evaluating data from 6 different diagnostic task force criteria.1 Characteristic histopathological features include fibrofatty replacement of right ventricular myocardium, corresponding to an advanced disease stage. Importantly, inflammation of the myocardium is often observed in AC, and differential diagnosis of early AC from myocarditis may be challenging.2 The primary treatment goal is prevention of sudden cardiac death. AC patients are often diagnosed in adolescence or young adulthood and the clinical decision for implantable cardioverter‐defibrillator implantation is a particularly challenging problem.3 Due to uncertainties about the molecular and cellular pathogenesis of AC,4 there are no established strategies to not only prevent sudden cardiac death but to also slow down myocardial degeneration and progressive heart failure.
Genetic screening in AC has identified a host of variants5 in desmosomal or desmosome‐related proteins (junction plakoglobin, desmoplakin, plakophilin 2, desmoglein‐2, desmocollin‐2), sarcomeric or intermediate filaments (desmin, titin), and immunoregulatory proteins (transforming growth factor‐β3).
Here, we describe 2 brothers presenting with recurrent myocarditis associated with a desmoplakin truncation. These unusual cases point to genetic predisposition within the myocarditis spectrum and the relevance of early genetic testing in myocarditis in a much broader sense. Moreover, we report a striking discrepancy between essentially complete loss of desmoplakin protein from the myocardium of these patients despite their only heterozygous desmoplakindefect at the genome level. It appears indispensable to analyze the transcript and protein level within the tissue actually affected in patients, to recognize their individual pathogenesis. This is of particular importance since desmoplakin variant carriers may display dermatologic phenotypes without cardiac involvement.6, 7 Haploinsufficiency of the truncated desmoplakin protein, leading to dysfunctional desmosomes, may be responsible for the severe familial phenotype of the 2 brothers.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Patients and Variant Screening
The family depicted in Figure 1 was identified during routine clinical patient management including genetic testing for cardiomyopathy‐related variants, to which all investigated family members gave informed written consent. Screening was performed using targeted next‐generation sequencing combined with the gold standard Sanger technique as described.8, 9
Figure 1. Genetic analysis of the family.
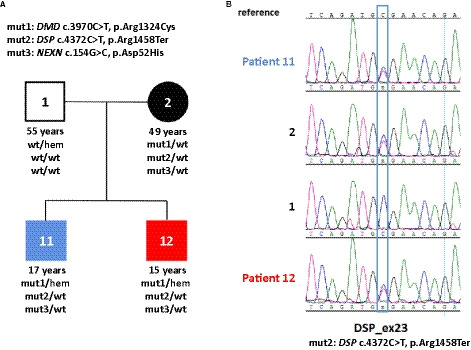
A, Large‐scale variant screening by sequencing of 218 cardiomyopathy‐related genes was conducted in both brothers to identify a possible genetic basis of their disease. Subsequent filtering with a minor allele frequency of 0.001 revealed three variants in cardiomyopathy‐associated genes: Dystrophin c.3970C>T (p.Arg1324Cys), Desmoplakin c.4372C>T (p.Arg1458Ter) and nexilin c.154G>C (p.Asp52His). The desmoplakin variant leads to truncation of the central rod domain and complete loss of the C‐terminal plakin repeat domains of desmoplakin involved in the transmission of mechanical signals. The relevance of 2 other rare variants of cardiac structural proteins (nexilin, dystrophin) detected in both brothers is uncertain. B, Sanger sequencing of the desmoplakin variantSanger sequencing of the desmoplakin variant in patient 11, patient 12, and individuals 1 and 2.
Other patients with truncating desmoplakin variants were identified by analysis of genetic databases generated during a project conducted at Heidelberg Medical University. This project was approved by the university's institutional ethics committee (S‐390/2011) with written informed consent from each proband. In total, 384 cardiomyopathy patients from Heidelberg were tested for desmoplakin variants. Preparation of sequencing libraries and data analysis was done using well‐established protocols for either panel, whole‐exome, or genome sequencing, as published.10, 11, 12
Desmoplakin Expression in Myocardium and Immune Cells
Desmoplakin transcription was evaluated in left ventricular (LV) myocardium and blood from patients with dilated cardiomyopathy (DCM), and in fluorescence‐activated cell sorting (FACS)‐sorted peripheral blood mononuclear cell (PBMC) subpopulations, by use of RNA sequencing as described.10, 11, 12, 13, 14
Cardiac Magnetic Resonance Imaging and Analyses
The cardiac magnetic resonance imaging (MRI) studies were performed and evaluated by standard procedures as described.15
Histological, Immunohistological, and Virological Analyses
Endomyocardial biopsies (EMBs) and PBMCs were investigated for the presence of intramyocardial or intracellular viral genomes, and desmoplakin and other structural myocardial proteins were detected as described.16, 17, 18, 19 Specifically, immunohistochemical detection of desmoplakin was performed using polyclonal rabbit anti‐desmoplakin antibody (Novus) at 1:50 dilution, dystrophin using polyclonal rabbit anti‐dystrophin antibody (Zytomed) at 1:70 dilution, and dysferlin20, 21, 22 using polyclonal rabbit anti‐dysferlin antibody (Invitrogen) at 1:100 dilution.
Serum Autoantibody Detection and FACS Analyses of PBMCs
Autoantibodies against cardiac myosin, troponin I or Z, were assayed as described.23, 24, 25 Specifically, serum blood samples were collected and antibody titers were determined using sandwich ELISA technique.25 To measure the titer of anti–troponin I or anti–major histocompatibility complex autoantibodies, plates were coated with 50 μL/well anti–troponin I antibody (produced in hybridoma cell culture) or anti–major histocompatibility complex antibody (GeneTex GTX20015, Biozol) (1 μg/mL) in coating buffer (0.1 mol/L NaHCO3; 34 mmol/L Na2CO3; pH 9.5) and incubated overnight. The GTX20015 antibody is myosin α and β heavy‐chain specific (affinity constants for human ventricular myosin heavy chains—3.33×108 M−1, human atrial myosin heavy chains—1.48×108 M−1, human skeletal muscle myosin—1.06×108 M−1); 50 μL/well of human cardiac troponin I (3 lg/mL) or porcine major histocompatibility complex (Sigma) (3 μg/mL) were added to complete coating. Dilution series of serum samples were 1:40, 1:80, 1:160, and 1:320. Anti‐human secondary antibody for IgG (1:10,000 dilution, BD) was used for detection. Anti–troponin I antibody or immunoadsorption‐eluate positive for major histocompatibility complex was used as a positive control. We determined the optical density at a wavelength of 450 nm. Antibody end point titer for each individual serum was calculated as the highest positive dilution of antibody. FACS studies of PBMCs (Table S1) were conducted using published methods13, 26, 27 that are generally employed for diagnostics and research in clinical immunology and related disciplines.
Results
Two brothers were admitted to our clinics with massive chest pain. The younger brother (patient 12) at age 15 was first diagnosed as having acute myocarditis with a massive increase in troponin T (Figure 2) and ST elevations in the inferior and lateral leads on ECG (Figure 3). He reported dyspnea accompanied by vomiting and 3 days of intermittent left arm pain. During hospitalization he had 2 episodes of nonsustained ventricular tachycardia (Figure 3A) and 1850 ventricular extrasystoles per 24 hours on Holter, and 3 weeks after the initial event his ECG showed repolarization abnormalities with T‐wave inversions (Figure 3B and 3C). Two weeks later the 17‐year‐old elder brother (patient 11) suffered a similar, although less severe, event with ST elevations (Figure S1) interpreted as acute myocarditis. There was no prior medication or recent vaccination in either brother, but their mother reported recurrent events of “chest pains” during their childhood related to physical exercise. Six months later the elder brother (patient 11) was readmitted, this time with an episode as severe as initially in his brother (Figure 2). Recovery was slow after this recurrence, and troponin T returned to normal after 6 months. Over a period of 16 months, the younger brother (patient 12) was entirely asymptomatic and had resumed normal activities including sports. Unexpectedly, however, he suffered a second myocarditis event with sudden massive increase of troponin T (Figure 2). Of note, his mother spontaneously remarked that before his first as well as this second event he had performed competitive sports. Both brothers performed sports with frequent high‐intensity training (American football, basketball).
Figure 2. Clinical and laboratory course.
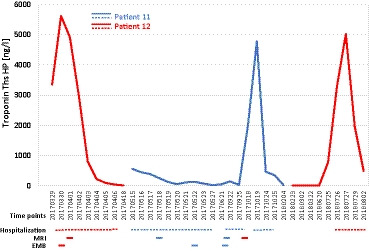
Repetitive high levels of cardiac troponin T were observed in 2 brothers (patient 11 and 12) carrying a truncating desmoplakin variant p.Arg1458Ter. In both patients, the initial clinical diagnosis was “myocarditis” on the basis of their symptoms, ECG changes, and laboratory findings. EMB indicates endomyocardial biopsy; and MRI,cardiac magnetic resonance imaging.
Figure 3. Electrocardiographic findings.
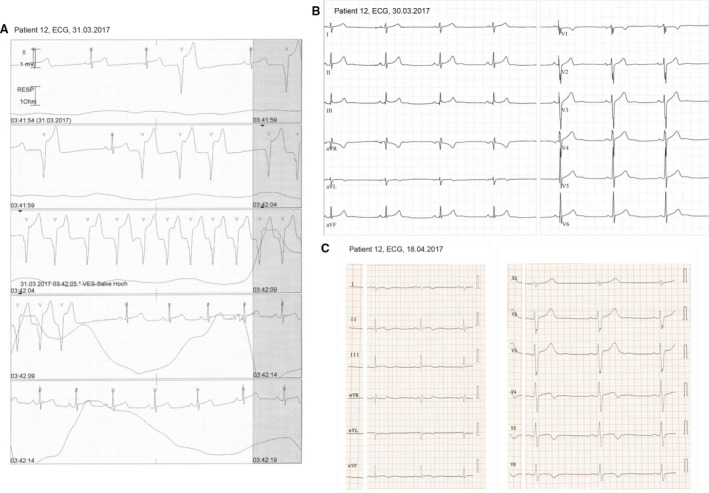
A, Nonsustained ventricular tachycardia of patient 12 during his first day of hospitalization. B, Twelve‐lead ECG of this patient on admission with acute chest pain and high troponin T. C, ECG at follow‐up when symptoms had subsided and Ths HP had returned to near normal values. Ths HP indicates cardiac troponin T.
Both brothers underwent echocardiographic and cardiac MRI on admission and during follow‐up. Echocardiographic examinations revealed a decreased LV ejection fraction in patient 12 at initial diagnosis and at follow‐up after 18 months. Results and images of initial and follow‐up MRI investigations are shown in Figure 4 and Table S2. Multifocal subepicardial late gadolinium enhancement was present in both brothers at initial and follow‐up studies. Patient 12 showed reduced LV ejection fraction only at follow‐up, while patient 11 demonstrated normal LV function at all time points. Focal wall abnormalities and myocardial edema were absent, implicating that no acute myocarditis was present. Cardiac catheterization was performed in both brothers at initial presentation and at follow‐up, and EMBs revealed low‐level immune cell infiltration in the absence of intramyocardial virus genomes (Table S3). There was no evidence of myocyte death supporting the Dallas criteria and only low‐level myocardial inflammation without detection of perforin‐positive cytotoxic T cells.19 In addition, circulating blood mononuclear cell analyses, as well as serologic studies yielded no evidence of viral or nonviral infections (Table S4). Immunologic screening identified circulating cardiac autoantibodies against myosin (IgM) and troponin I (IgG and IgM) in both brothers, consistent with autoimmune involvement. FACS studies of PBMCs to identify possible immune cell defects revealed only mild anomalies, which were, however, essentially identical in both cases (Table S1). Both displayed relative a reduction of α/β and an increase of γ/δ T cells, as well a discrete CD4/8 T‐cell inversion.
Figure 4. Cardiac magnetic resonance imaging studies. Late gadolinium enhancement images of cardiac magnetic resonance imaging (MRI) short‐axis views (left to right: basal, midventricular, apical).
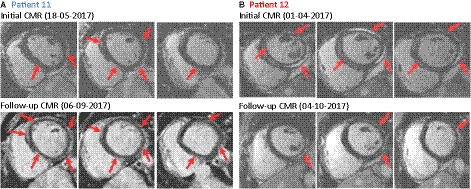
A, Initial and follow‐up cardiac MRI of patient 11 with multifocal subepicardial late gadolinium enhancement anterior, posterior, and septal. Initial left ventricular ejection fraction (LVEF) was 65%, at FU 64%. B, Initial and follow‐up cardiac MRI of patient 12 with multifocal subepicardial LGE posteroseptal and lateral. Initial LVEF was 59%, at follow‐up 47%. Regions of positive LGE are highlighted with red arrows. For further cardiac MRI data, see Table S2.
Based on the familial occurrence of a condition presenting as acute myocarditis the patients were referred for genetic analysis. Screening of 218 cardiomyopathy‐related genes was conducted in both brothers to identify a possible genetic defect. Three rare variants were identified in patients 11 and 12: dystrophin c.3970C>T, p.Arg1324Cys, desmoplakin c.4372C>T, p.Arg1458Ter, and nexilin F‐actin–binding protein c.154G>C, p.Asp52His (Figure 1). The heterozygous variant desmoplakin p.Arg1458Ter was graded as likely pathogenic according to the American College of Medical Genetics guidelines.28 The desmoplakin variant is reported in one individual in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), where it is classified as pathogenic for “dilated cardiomyopathy with woolly hair and keratoderma, arrhythmogenic right ventricular cardiomyopathy, type 8.” It occurs at low frequency (0.000003980) in gnomAD (https://gnomad.broadinstitute.org), and it is predicted to truncate approximately half of the protein, inducing loss of the C‐terminal plakin repeat domains of desmoplakin that are involved in the mechanical signal transmission.29 The hemizygous missense variant dystrophin p.Arg1324Cys and the heterozygous variant nexilin F‐actin–binding protein p.Asp52His were classified as variants of unknown significance
The detection of the desmoplakin variant also led us to inspect a possible cutaneous phenotype. Both brothers have wooly hair; their mother originates from a Latino ethnic background and has curly hair; and the father is European with straight hair. Any specific recognizable cutaneous phenotype such as keratoderma was not visible in the brothers or the parents.
Segregation analysis revealed that the heterozygous desmoplakin variant p.Arg1458Ter was inherited from the 49‐year‐old mother (Figure 1), who reported no history or symptoms of cardiovascular or other diseases. Clinical examination and cardiac imaging including cardiac MRI showed no cardiac abnormalities. Ventricular size and function were normal, and there was no regional akinesia or dyskinesia (Figure S2). The 55‐year‐old father (patient 1) carried none of these variants, had suffered a myocardial infarction several years ago, and had no signs of cardiomyopathy from medical records.
Only after this detection of the predictably truncating nonsense desmoplakin variant in both brothers, a number of additional histologic and immunohistologic studies were conducted on their EMBs. The heterozygous desmoplakin variant was expected to result in either an abnormal truncated protein product or approximately 50% loss of protein from the variant allele through nonsense‐mediated RNA decay. Unexpectedly, there was complete loss of desmoplakin protein in the myocardium of patients, as far as detectable by immunohistochemistry (Figure 5A). Of note, no such loss of desmoplakin was observed in patients with “classical” arrhythmogenic right ventricular cardiomyopathy, even when all other histologic criteria of the disease were fully evolved (Figure 5A). In contrast to these findings regarding desmoplakin protein, there were no detectable differences in dystrophin immunoreactivity between p.Arg1458Ter patients or “classical” arrythmogenic right ventricular cardiomyopathy patients and normal controls (Figure 5B). Furthermore, immunostaining for the sarcolemmal cell surface protein dysferlin 20, 21, 22 revealed no differences between patients and controls (Figure 5C). Desmoplakin truncating variant p.Arg1458Ter did not affect myocardial structure as assessed by hematoxylin and eosin and trichrome stain (Figure 5D and 5E), and there was only low level inflammatory activation (Figure 5F). Interestingly, histology could not detect fibrofatty replacement of right ventricular myocardium at biopsy.
Figure 5. Protein expression in endomyocardial biopsies. Immunhistochemistry detecting desmoplakin and dystrophin was conducted in endomyocardial biopsies of patient 11.
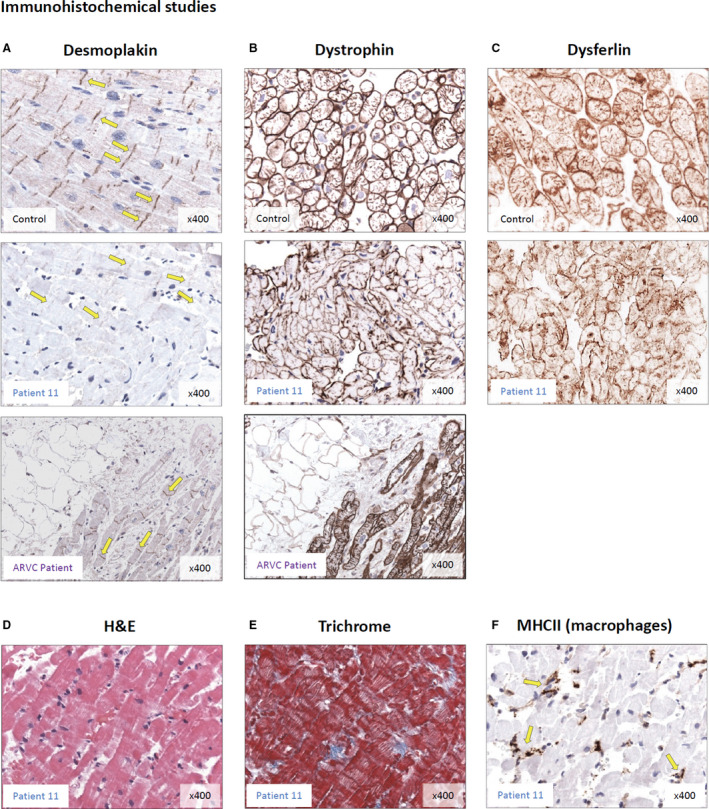
A, Immunodetection reveals strong desmoplakin signals in desmosomes (yellow arrows) of the control biopsy. In the biopsy of patient 11 almost no significant desmoplakin signal was detectable. Of note, in a right heart biopsy from a patient with “classical” arrhythmogenic right ventricular cardiomyopathy, normal desmoplakin expression was observed. The anti‐desmoplakin antibody recognizes an epitope within the N‐terminal part of desmoplakin. Nuclei are shown in light blue. Magnification is ×400. B, Immunodetection of dystrophin appears unaffected in the biopsy of patient 11 compared with the control and arrhythmogenicright ventricular cardiomyopathy patient. C, Immunostaining for the sarcolemmal cell surface protein dysferlin revealed no differences between patients and controls. D and E, H&E and trichrome stainings identified no alterations of myocardial structure. F, MHCII staining showed low level inflammation with infiltrating macrophages (for further EMBx data see Table S3). H&E indicates hematoxylin and eosin; and MHCII, major histocompatibility complex class II.
We detected desmoplakin transcript in PBMCs (Figure S3), which raises the question to what extent desmoplakin variants affect immune functions. Of note, in a cohort of DCM and in control patients, we were able to detect desmoplakin transcripts in both PBMCs and left‐ventricular heart tissue (Figure S3A).30 In PBMCs from the p.Arg1458Ter carriers, desmoplakin transcript was likewise detected (Figure S3B). To our knowledge, the functions of desmoplakin in immunity have not been addressed so far. Unpublished data from a previous RNA‐sequencing study on sorted human PBMC subpopulations13 (Figure S3C) suggest that desmoplakin transcription is restricted to certain immune cell subtypes (CD3+, CD4+, CD19+), further supporting the hypothesis of immune functions of DSP.
To understand the phenotypic consequences of truncating desmoplakin variants, we screened additional cohorts of adult cardiomyopathy patients for such variants. We identified the nonsense variant Gln307Ter in a 37‐year‐old woman with “classical” arrythmogenic right ventricular cardiomyopathy phenotype. The sister of this patient died of heart failure at the age of 39. A 59‐year‐old woman with LV noncompaction phenotype carried the nonsense variant Tyr1391Ter. Here, family history was unclear; the brother probably died of coronary artery disease. In a 71‐year‐old woman with DCM phenotype, with no family history, the nonsense variant Tyr1512Ter was identified. These examples underline the phenotypic heterogeneity of cardiomyopathies associated with unique severe gene defects and indicate the critical role of modifiers (genetic or exogenous) in driving pathogenesis either way.
Overall, neither of the 2 brothers fulfilled the 2010 Task Force Criteria for the diagnosis of arrhythmogenic cardiomyopathy.1 Patient 12 met 1 major (identification of a pathogenic variant categorized as probably associated with AC) and 1 minor criterion (nonsustained ventricular tachycardia of right ventricular outflow tract configuration), therefore resulting in a borderline diagnosis of AC. Patient 11 fulfilled only 1 major criterion (identification of a pathogenic variant categorized as probably associated with AC) that results in a possible diagnosis of AC.
Discussion
Myocardial Inflammation in AC Pathogenesis
It is well established that desmoplakin variant–associated arrythmogenic right ventricular cardiomyopathy can present as myocarditis. An increasingly discussed and clinically important aspect of AC in general is its relationship with and differentiation from myocarditis.31 Myocarditis is often used synonymously with virus‐induced myocardial inflammation, whereas de facto immune cell infiltration of the myocardium is a common phenomenon triggered by multiple types of myocardial injury. These injuries range from ischemia to pressure or volume overload to storage diseases (eg, amyloidosis). It is indispensable to avoid a priori association of the term myocarditis with infectious etiology, unless a recent or current infectious trigger is clearly established. Otherwise, stepwise exclusion of the multiple alternative causes of myocardial inflammation is required before a definitive diagnosis is made. Regarding AC pathogenesis in patients with monogenic defects, this is critical since essential pathogenic cofactors obviously exist, but their nature has not been clarified yet. In the current study, we made an extensive effort to decide whether the observed recurrent phenotype in 2 brothers carrying a truncating desmoplakin variant might have been triggered by an infectious agent. After extensive screening for intramyocardial infectious genomes, recent or current systemic infection with a broad spectrum of viral or nonviral agents, or unspecific laboratory signs of infection, in combination with the absence of clinical signs or symptoms of infection, we had to exclude an infectious trigger as entirely speculative. Further, noninvasive and invasive (EMB) diagnostics excluded ischemia, pressure or volume overload, and myocardial storage disorders as cofactors. In addition, FACS screening exluded major cellular or humoral immune disturbances as possible cofactors of disease pathogenesis. In the described 2 particular cases with a mechanically highly relevant desmoplakin variant, all clinical and other findings are instead fully consistent with the alternative model proposed in Figure 6. To our knowledge, there is no prior report encompassing comprehensive exclusion of intramyocardial or systemic infections, immune disturbances, and the above‐mentioned possible alternative cofactors, in AC patients with desmoplakin variants.
Figure 6. Mechanical failure hypothesis.
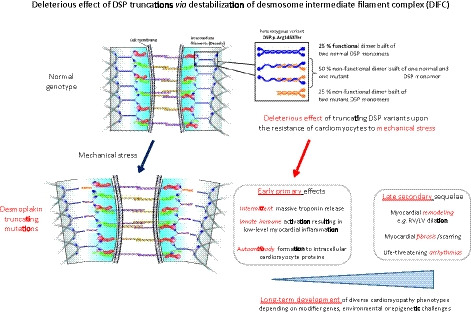
Ultrastructural studies of myocardial desmosomes suggest a possible consequence of haploinsufficiency due to the p.Arg1458Ter truncation. Desmosome‐intermediate filament complex (DIFC) denotes a multiprotein complex supporting mechanical stability. Desmoplakin needs to forms homodimers (white arrow), and midregion coiled‐coil rod domain of desmoplakin with amino acids 1057 to 1945 (red arrows) is essential for homodimerization. Variants p.Tyr1391Ter, p.Arg1458Ter, and p.Tyr1512Ter delete major parts of this domain. Glc307Ter deletes the domain entirely. If normal and pathogenic variant allele were translated with ≈1:1 ratio into protein, the cell should harbor one‐fourth normal, one‐fourth pathogenic variant, and one‐half mixed desmoplakin dimer, which is likely to significantly impair coiled‐coil domain‐dependent mechanical function within the DIFC. Loss of the C‐terminal “head” of desmoplakin (yellow arrow) that is involved in mechanical signal transmission may further impair mechanical robustness of the heart. LV indicates left ventricular; and RV, right ventricular.
Important previous studies32, 33 suggested that previously silent recessive defects of the myocardium may predispose to acute heart failure presenting as “acute myocarditis” (AM), notably after common viral infections in children. While this model is of high clinical interest and may be relevant in a fraction of “genetically predisposed” individuals, it obviously does not apply to our cases. Another study investigated the genetic basis of AM in AC and tested for possible association with poorer prognosis and higher risk of ventricular arrhythmias.31, 34 Among 131 patients with AC, 7 presented with a clinical diagnosis of AM, but this study did not screen for viral or other infectious genomes in EMBs, serologic or other evidence of past or current infections, or immune disturbances. In one paper reporting desmoplakin variant–associated AC presenting as AM,35 no EMB data on viral or other infectious genomes, histology, or immunohistochemistry were presented. Assumption of an infectious trigger in these studies therefore remains speculative.
Beyond this lack of evidence of infection, the “single event” clinical phenotype of AM in these reports is strikingly distinct from the “recurrent” life‐threatening phenotype of the sibling cases described here. Of note, detection of “inflammation” per se by cardiac MRI36 or EMB has low differential diagnostic and thus clinical potential, since it occurs unspecifically in multiple etiologically unrelated types of injury. Of course, the cardiac MRI findings cannot be invoked to suggest an infectious trigger without further direct evidence.
“Mechanical Failure” Model
The described brothers with severe myocarditis‐like events had episodes of massive chest pain and significant transient troponin elevation that were remarkably associated with physical exercise. The lack of any evidence of infections or other trigger events suggests that these episodes may represent early active phases of a pathogenic process primarily enabled by instability of the desmosome‐intermediate filament complex (DIFC) attributable to the truncating defect of DSP, which constitutes 1 key component of the DIFC (Figure 6).
Important in‐depth studies of myocardial desmosome structure down to the electron microscopic level37, 38, 39, 40, 41 have provided insight into the functions of desmoplakin within the desmosome and suggest a straightforward mechanistic hypothesis to explain the adverse clinical effect of the p.Arg1458Ter variant (hypothesis, Figure 6). Desmoplakin haploinsufficiency may lead to dysfunctional desmosomes with loss of numerous proteins from the DIFC.42, 43, 44 Whereas gap junctions allow electrical coupling between cardiomyocytes, desmosomes are junctions that link to the intermediate filament cytoskeleton, thus providing mechanical strength to the myocardium.37 Regarding factors that trigger the disease, our cases suggest that mechanical overload per se may suffice to elicit disease. Troponin peaks above 5.000 ng/mL in both cases dropped rapidly after the acute events. This is consistent with high‐level physical activity as trigger events and reminiscent of the peaklike elevations of troponin upon fulminant pulmonary embolism resulting in acute mechanical overload.45
Of note, any dimeric protein component of the DIFC could in principle exert a similar adverse effect upon DIFC function if the encoding gene carries a truncating variant as exemplified by desmoplakin. We suggest that the described phenotype of familial recurrent myocarditis represents a serendipitously revealed molecular subtype of AC, primarily triggered by distinctive adverse effect of the trunctated DSP upon the mechanical stability of myocardial desmosomes. Subsequent secondary events would include “unspecific” activation of innate and adaptive immunity and determine the long‐term clinical course dependent on exogenous and epigenetic cofactors (Figure 6).
Primary Versus Secondary Pathogenic Factors
In our report, the detection of high‐titer autoantibody against cardiac myosin and troponin I23, 24 is consistent with disturbed autoimmune control as a contributing factor aggravating a primarily mechanical defect. While this justifies the term autoimmune myocarditis, recurrent massive bouts of troponin release cannot be linked to stably elevated autoantibody titers. Thus, mechanical instability of the DIFC appears as the primary, and autoantibody formation as a secondary phenomenon.
One may speculate that the anomalous truncated desmoplakin protein may have initiated this disturbance, thus not only acting directly by its dysfunctionality, but also as an innate immunity trigger. In fact, desmoplakin protein is targeted in a number of dermatologic autoimmune diseases.46 Desmoplakin autoantibodies may bind to the surface of keratinocytes, become internalized via plasmalemmal vesicles, and may then be found within cutaneous desmosomal plaques, providing a possible mechanism for how the autoantibodies exert pathogenic effects. Similar processes may occur in the myocardium when pathogenic variant desmoplakin becomes exposed to the immune system.
Variable Phenotypic Expression of Desmoplakin Variants
Disease mechanisms vary according to the specific types of desmoplakin variant and include haploinsufficiency, dominant negative effects, or a combination of both.47 In the cases reported here, definitive resolution of the molecular pathomechanism cannot possibly be achieved because of lack of sufficient myocardial biopsy material. Of note, the biopsy‐based myocardial findings in the affected brothers yield information about a far earlier and therefore clinically more relevant phase of the disease process than what can be learned from postmortem hearts, which often are the sole source of information in genetic cardiomyopathies. As desmosomal proteins are expressed in myocardium and epidermis, AC and DCM are occasionally associated with cutaneous manifestations like palmoplantar keratoderma and woolly hair.47 The protein exists in 2 isoforms, DSP1 and DSP2, produced by alternative splicing, and DSP1 is the only isoform expressed in the myocardium, whereas both DSP1 and DSP2 are expressed in the epidermis. The variant p.Arg1458Ter is located in DSP1, leading to premature truncation affecting the DSP1 isoform only. Our study also shows, for the first time, desmoplakin transcription restricted to immune cell subtypes CD3+, CD4+, and CD19+, suggesting unknown immune functions of desmoplakin, which warrant further investigation.
The truncating variants Gln307Ter, Tyr1391Ter, and Tyr1512Ter were detected in adult cardiomyopathy patients at 37, 59, and 71 years of age, respectively. In these patients, variant‐dependent myocardial pathomechanisms had 2 to 5 decades more to evolve. Therefore, their divergent clinical phenotypes—at time of diagnosis—may well be a consequence of decades of disease‐modifying exogenous or genetic background factors driving pathogenesis into different directions. In the young brothers, relevance of their heterozygosity for the rare variant p.Asp52His in the nexilin F‐actin–binding protein is uncertain in that regard (Figure S4). Of note, however, nexilin F‐actin–binding protein knockout in mice leads to severe cardiomyopathy and endomyocardial fibroelastosis.47 Further, nexilin F‐actin–binding protein was identified as causative and as a modifier gene of DCM,48, 49 and a new component of junctional membrane complexes required for cardiac T‐tubule formation.50, 51, 52
The course of AC is influenced by sex hormones, with elevated serum testosterone levels in men and decreased estradiol levels in women independently associated with major adverse cardiovascular events.53 Among patients with sporadic AC, men had a significantly higher risk of ventricular tachycardia/ventricular fibrillation, whereas women had a significantly higher risk of heart failure death or heart transplantation attributable to heart failure.54 Pediatric patients disproportionately presented with sudden cardiac death.54 Of note, our patients’ mother carried the desmoplakin variant but was clinically unaffected, possibly because of the known protective influence of female sex.
Implications for the Clinical Care of Patients at Risk
Earliest possible recognition of patients at genetic risk is of critical importance, since in these individuals the identification and avoidance of exogenous triggers may allow survival for decades. Since duplicate recurrent myocarditis in 1 family is a most unusual situation, variant screening even in isolated cases of “myocarditis” seems advisable to enable early genetic risk detection and preventive measures in these often young patients. The bouts of “chest pain” in our cases during their childhood, as reported by their mother, may reflect earlier similar events that went undiagnosed because of their unsuspicious nature.55, 56, 57 Whereas inhibition of pathogenic variant desmoplakin expression is not yet clinically feasible58 and there is no pharmacologic approach to compensate for desmoplakin deficiency,59 limitation of physical exercise may be highly effective if initiated early enough before secondary pathomechanisms (eg, myocardial inflammation) ensue. Both brothers carry event recorders, so far without detection of critical arrhythmias, so that in view of their currently normal cardiac function, there is no indication for implantable cardioverter‐defibrillator implantation.
Study Limitations
After extensive exclusion of alternative disease trigger factors, the proposed mechanical failure model is fully consistent with state‐of‐the‐art basic research and clinical knowledge. Limited availability of patient myocardial tissue for in‐depth ultrastructural and ex vivo functional studies in variant carriers prevents, however, direct proof of the model. Since any defective dimeric protein component of the DIFC could exert a similarly strong effect upon mechanical DIFC function as the desmoplakin pathogenic variant reported here, further testing of the mechanical failure hypothesis in experimental models appears warranted.
Conclusions
The described novel phenotype of familial recurrent myocarditis associated with a desmoplakin truncation likely represents a serendipitously revealed subtype of AC. After comprehensive exclusion of alternative known trigger factors, and considering recent ultrastructural knowledge on desmoplakin functions, this peculiar phenotype may be triggered by a distinct adverse of the variant desmoplakin upon the mechanical stability of myocardial desmosomes. Variant screening is advisable to allow early detection of patients with a similar phenotype, to possibly enable prophylactic measures targeting this type of pathomechanism.
Author Contributions
WP and SK conceived and designed the research. JH, KK, JK, MG, ZK, FE, EK, FD, BOR, FB, HCM, CS, and BH acquired the data. LM, JH, and MG performed statistical analyses. WP and SK handled funding and supervision, and drafted the manuscript. JH, LM, BM, and UL made critical revision of the manuscript for key intellectual content.
Sources of Funding
Human samples were from the biobank, Competence Network for Congenital Heart Defects, Germany, funded by the Federal Ministry of Education and Research (BMBF; grant no:1GI0601 until 2014, and the DZHK (German Centre for Cardiovascular Research; as of 2015). This work was supported by the DZHK (FKZ 81Z3100331).
Disclosures
None.
Supporting information
Tables S1–S4 Figures S1–S4
(J Am Heart Assoc. 2020;9:e015289 DOI: 10.1161/JAHA.119.015289.)
For Sources of Funding and Disclosures, see page 11.
References
- 1. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur Heart J. 2010;31:806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patriki D, Gresser E, Manka R, Emmert MY, Luscher TF, Heidecker B. Approximation of the incidence of myocarditis by systematic screening with cardiac magnetic resonance imaging. JACC Heart Fail. 2018;6:573–579. [DOI] [PubMed] [Google Scholar]
- 3. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, Bauce B, Basso C, Brunckhorst C, Tsatsopoulou A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36:3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Bortoli M, Calore C, Lorenzon A, Calore M, Poloni G, Mazzotti E, Rigato I, Marra MP, Melacini P, Iliceto S, et al. Co‐inheritance of mutations associated with arrhythmogenic cardiomyopathy and hypertrophic cardiomyopathy. Eur J Hum Genet. 2017;25:1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pilichou K, Lazzarini E, Rigato I, Celeghin R, De Bortoli M, Perazzolo Marra M, Cason M, Jongbloed J, Calore M, Rizzo S, et al. Large genomic rearrangements of desmosomal genes in italian arrhythmogenic cardiomyopathy patients. Circ Arrhythm Electrophysiol. 2017;10:e005324. [DOI] [PubMed] [Google Scholar]
- 6. Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, Aho H, Heikinheimo K, Bruckner‐Tuderman L, Fertala A, et al. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J Invest Dermatol. 2010;130:968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paller AS, Czarnowicki T, Renert‐Yuval Y, Holland K, Huynh T, Sadlier M, McAleer MA, Tran G, Geddes GC, Irvine AD, Guttman‐Yassky E. The spectrum of manifestations in desmoplakin gene (DSP) spectrin repeat 6 domain mutations: immunophenotyping and response to ustekinumab. J Am Acad Dermatol. 2018;78:498–505.e492. [DOI] [PubMed] [Google Scholar]
- 8. Ochoa JP, Sabater‐Molina M, Garcia‐Pinilla JM, Mogensen J, Restrepo‐Cordoba A, Palomino‐Doza J, Villacorta E, Martinez‐Moreno M, Ramos‐Maqueda J, Zorio E, et al. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72:2457–2467. [DOI] [PubMed] [Google Scholar]
- 9. Ortiz‐Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado‐Aranda R, Climent V, Padron‐Barthe L, Duro‐Aguado I, Jimenez‐Jaimez J, Hidalgo‐Olivares VM, et al. Truncating FLNC mutations are associated with high‐risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. [DOI] [PubMed] [Google Scholar]
- 10. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135a. [DOI] [PubMed] [Google Scholar]
- 11. Sedaghat‐Hamedani F, Haas J, Zhu F, Geier C, Kayvanpour E, Liss M, Lai A, Frese K, Pribe‐Wolferts R, Amr A, et al. Clinical genetics and outcome of left ventricular non‐compaction cardiomyopathy. Eur Heart J. 2017;38:3449–3460. [DOI] [PubMed] [Google Scholar]
- 12. Haas J, Mester S, Lai A, Frese KS, Sedaghat‐Hamedani F, Kayvanpour E, Rausch T, Nietsch R, Boeckel JN, Carstensen A, et al. Genomic structural variations lead to dysregulation of important coding and non‐coding RNA species in dilated cardiomyopathy. EMBO Mol Med. 2018;10:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gast M, Rauch B, Haghikia A, Nakagawa S, Haas J, Stroux A, Schmidt D, Schumann P, Weiss S, Jensen L, et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res. 2019;115:1886–1906. [DOI] [PubMed] [Google Scholar]
- 14. Gast M, Rauch BH, Nakagawa S, Haghikia A, Jasina A, Haas J, Nath N, Jensen L, Stroux A, Bohm A, et al. Immune system‐mediated atherosclerosis caused by deficiency of long non‐coding RNA MALAT1 in ApoE−/−mice. Cardiovasc Res. 2019;115:302–314. [DOI] [PubMed] [Google Scholar]
- 15. Achenbach S, Barkhausen J, Beer M, Beerbaum P, Dill T, Eichhorn J, Fratz S, Gutberlet M, Hoffmann M, Huber A, et al. Consensus recommendations of the German Radiology Society (DRG), the German Cardiac Society (DGK) and the German Society for Pediatric Cardiology (DGPK) on the use of cardiac imaging with computed tomography and magnetic resonance imaging. Rofo. 2012;184:345–368. [DOI] [PubMed] [Google Scholar]
- 16. Mueller K, Heinzmann D, Klingel K, Fallier‐Becker P, Kandolf R, Kilias A, Walker‐Allgaier B, Borst O, Kumbrink J, Kirchner T, et al. Histopathological and immunological characteristics of tachycardia‐induced cardiomyopathy. J Am Coll Cardiol. 2017;69:2160–2172. [DOI] [PubMed] [Google Scholar]
- 17. Pavlicek V, Kindermann I, Wintrich J, Mahfoud F, Klingel K, Bohm M, Ukena C. Ventricular arrhythmias and myocardial inflammation: long‐term follow‐up of patients with suspected myocarditis. Int J Cardiol. 2019;274:132–137. [DOI] [PubMed] [Google Scholar]
- 18. Akdis D, Medeiros‐Domingo A, Gaertner‐Rommel A, Kast JI, Enseleit F, Bode P, Klingel K, Kandolf R, Renois F, Andreoletti L, et al. Myocardial expression profiles of candidate molecules in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia compared to those with dilated cardiomyopathy and healthy controls. Heart Rhythm. 2016;13:731–741. [DOI] [PubMed] [Google Scholar]
- 19. Escher F, Kuhl U, Lassner D, Stroux A, Gross U, Westermann D, Pieske B, Poller W, Schultheiss HP. High perforin‐positive cardiac cell infiltration and male sex predict adverse long‐term mortality in patients with inflammatory cardiomyopathy. J Am Heart Assoc. 2017;6:e005352 DOI: 10.1161/JAHA.116.005352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cox D, Henderson M, Straub V, Barresi R. A simple and rapid immunoassay predicts dysferlinopathies in peripheral blood film. Neuromuscul Disord. 2019;29:874–880. [DOI] [PubMed] [Google Scholar]
- 21. Rekik S, Sakka S, Ben Romdhan S, Farhat N, Baba Amer Y, Lehkim L, Authier FJ, Mhiri C. Novel missense CAPN3 mutation responsible for adult‐onset limb girdle muscular dystrophy with calves hypertrophy. J Mol Neurosci. 2019;69:563–569. [DOI] [PubMed] [Google Scholar]
- 22. Dai Y, Liang S, Dong X, Zhao Y, Ren H, Guan Y, Yin H, Li C, Chen L, Cui L, Banerjee S. Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy‐dystroglycanopathy. J Cell Mol Med. 2019;23:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muller AM, Bockstahler M, Hristov G, Weiss C, Fischer A, Korkmaz‐Icoz S, Giannitsis E, Poller W, Schultheiss HP, Katus HA, Kaya Z. Identification of novel antigens contributing to autoimmunity in cardiovascular diseases. Clin Immunol. 2016;173:64–75. [DOI] [PubMed] [Google Scholar]
- 24. Haghikia A, Kaya Z, Schwab J, Westenfeld R, Ehlermann P, Bachelier K, Oettl R, von Kaisenberg CS, Katus HA, Bauersachs J, Hilfiker‐Kleiner D. Evidence of autoantibodies against cardiac troponin I and sarcomeric myosin in peripartum cardiomyopathy. Basic Res Cardiol. 2015;110:60. [DOI] [PubMed] [Google Scholar]
- 25. Doesch AO, Mueller S, Nelles M, Konstandin M, Celik S, Frankenstein L, Goeser S, Kaya Z, Koch A, Zugck C, Katus HA. Impact of troponin I‐autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic Res Cardiol. 2011;106:25–35. [DOI] [PubMed] [Google Scholar]
- 26. Loebel M, Holzhauser L, Hartwig JA, Shukla PC, Savvatis K, Jenke A, Gast M, Escher F, Becker SC, Bauer S, et al. The forkhead transcription factor FOXO3 negatively regulates natural killer cell function and viral clearance in myocarditis. Eur Heart J. 2018;39:876–887. [DOI] [PubMed] [Google Scholar]
- 27. Bobbert P, Scheibenbogen C, Jenke A, Kania G, Wilk S, Krohn S, Stehr J, Kuehl U, Rauch U, Eriksson U, et al. Adiponectin expression in patients with inflammatory cardiomyopathy indicates favourable outcome and inflammation control. Eur Heart J. 2011;32:1134–1147. [DOI] [PubMed] [Google Scholar]
- 28. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Albrecht LV, Zhang L, Shabanowitz J, Purevjav E, Towbin JA, Hunt DF, Green KJ. GSK3‐ and PRMT1‐dependent modifications of desmoplakin control desmoplakin‐cytoskeleton dynamics. J Cell Biol. 2015;208:597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meder B, Haas J, Sedaghat‐Hamedani F, Kayvanpour E, Frese K, Lai A, Nietsch R, Scheiner C, Mester S, Bordalo DM, et al. Epigenome‐wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation. 2017;136:1528–1544. [DOI] [PubMed] [Google Scholar]
- 31. Lopez‐Ayala JM, Pastor‐Quirante F, Gonzalez‐Carrillo J, Lopez‐Cuenca D, Sanchez‐Munoz JJ, Oliva‐Sandoval MJ, Gimeno JR. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12:766–773. [DOI] [PubMed] [Google Scholar]
- 32. Belkaya S, Kontorovich AR, Byun M, Mulero‐Navarro S, Bajolle F, Cobat A, Josowitz R, Itan Y, Quint R, Lorenzo L, et al. Autosomal recessive cardiomyopathy presenting as acute myocarditis. J Am Coll Cardiol. 2017;69:1653–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campuzano O, Fernandez‐Falgueras A, Sarquella‐Brugada G, Sanchez O, Cesar S, Mademont I, Allegue C, Mates J, Perez‐Serra A, Coll M, et al. A genetically vulnerable myocardium may predispose to myocarditis. J Am Coll Cardiol. 2015;66:2913–2914. [DOI] [PubMed] [Google Scholar]
- 34. Lopez‐Ayala JM, Oliva‐Sandoval MJ, Sanchez‐Munoz JJ, Gimeno JR. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2015;385:662. [DOI] [PubMed] [Google Scholar]
- 35. Reichl K, Kreykes SE, Martin CM, Shenoy C. Desmoplakin variant‐associated arrhythmogenic cardiomyopathy presenting as acute myocarditis. Circ Genom Precis Med. 2018;11:e002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martins D, Ovaert C, Khraiche D, Boddaert N, Bonnet D, Raimondi F. Myocardial inflammation detected by cardiac MRI in arrhythmogenic right ventricular cardiomyopathy: a paediatric case series. Int J Cardiol. 2018;271:81–86. [DOI] [PubMed] [Google Scholar]
- 37. Cerrone M, Agullo‐Pascual E, Delmar M. The intercalated disc: a molecular network that integrates electrical coupling, intercellular adhesion, and cell excitability In Zipes D, Jalife J, Stevenson WG, eds. Cardiac Electrophysiology: From Cell to Bedside. Elsevier: Philadelphia; 2018:198–211. [Google Scholar]
- 38. Goossens S, Janssens B, Bonne S, De Rycke R, Braet F, van Hengel J, van Roy F. A unique and specific interaction between alphaT‐catenin and plakophilin‐2 in the area composita, the mixed‐type junctional structure of cardiac intercalated discs. J Cell Sci. 2007;120:2126–2136. [DOI] [PubMed] [Google Scholar]
- 39. Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85:69–82. [DOI] [PubMed] [Google Scholar]
- 40. North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, Hatzfeld M, Green KJ, Magee AI, Garrod DR. Molecular map of the desmosomal plaque. J Cell Sci. 1999;112(Pt 23):4325–4336. [DOI] [PubMed] [Google Scholar]
- 41. Al‐Amoudi A, Castano‐Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three‐dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci USA. 2011;108:6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey‐Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chatterjee D, Fatah M, Akdis D, Spears DA, Koopmann TT, Mittal K, Rafiq MA, Cattanach BM, Zhao Q, Healey JS, et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur Heart J. 2018;39:3932–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Calkins H. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy: is this too good to be true? Eur Heart J. 2018;39:3945–3946. [DOI] [PubMed] [Google Scholar]
- 45. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- 46. Foedinger D, Anhalt GJ, Boecskoer B, Elbe A, Wolff K, Rappersberger K. Autoantibodies to desmoplakin I and II in patients with erythema multiforme. J Exp Med. 1995;181:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rasmussen TB, Hansen J, Nissen PH, Palmfeldt J, Dalager S, Jensen UB, Kim WY, Heickendorff L, Molgaard H, Jensen HK, et al. Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms. Clin Genet. 2013;84:20–30. [DOI] [PubMed] [Google Scholar]
- 48. Wang H, Li Z, Wang J, Sun K, Cui Q, Song L, Zou Y, Wang X, Liu X, Hui R, Fan Y. Mutations in NEXN, a Z‐disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2010;87:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, Just S, Hess A, Ehlermann P, Weichenhan D, et al. Nexilin mutations destabilize cardiac Z‐disks and lead to dilated cardiomyopathy. Nat Med. 2009;15:1281–1288. [DOI] [PubMed] [Google Scholar]
- 50. Liu C, Spinozzi S, Chen JY, Fang X, Feng W, Perkins G, Cattaneo P, Guimaraes‐Camboa N, Dalton ND, Peterson KL, et al. Nexilin is a new component of junctional membrane complexes required for cardiac T‐tubule formation. Circulation. 2019;140:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu YW, Guo FX, Xu YJ, Li P, Lu ZF, McVey DG, Zheng L, Wang Q, Ye JH, Kang CM, et al. Long noncoding RNA NEXN‐AS1 mitigates atherosclerosis by regulating the actin‐binding protein NEXN. J Clin Invest. 2019;129:1115–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soni S, Raaijmakers AJ, Raaijmakers LM, Damen JM, van Stuijvenberg L, Vos MA, Heck AJ, van Veen TA, Scholten A. A proteomics approach to identify new putative cardiac intercalated disk proteins. PLoS ONE. 2016;11:e0152231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akdis D, Saguner AM, Shah K, Wei C, Medeiros‐Domingo A, von Eckardstein A, Luscher TF, Brunckhorst C, Chen HSV, Duru F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: from a stem cell derived cardiomyocyte‐based model to clinical biomarkers of disease outcome. Eur Heart J. 2017;38:1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kimura Y, Noda T, Otsuka Y, Wada M, Nakajima I, Ishibashi K, Miyamoto K, Okamura H, Aiba T, Kamakura S, et al. Potentially lethal ventricular arrhythmias and heart failure in arrhythmogenic right ventricular cardiomyopathy: what are the differences between men and women? JACC Clin Electrophysiol. 2016;2:546–555. [DOI] [PubMed] [Google Scholar]
- 55. Te Riele A, James CA, Sawant AC, Bhonsale A, Groeneweg JA, Mast TP, Murray B, Tichnell C, Dooijes D, van Tintelen JP, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy in the pediatric population: clinical characterization and comparison with adult‐onset disease. JACC Clin Electrophysiol. 2015;1:551–560. [DOI] [PubMed] [Google Scholar]
- 56. te Riele AS, James CA, Groeneweg JA, Sawant AC, Kammers K, Murray B, Tichnell C, van der Heijden JF, Judge DP, Dooijes D, et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J. 2016;37:755–763. [DOI] [PubMed] [Google Scholar]
- 57. Steinmetz M, Krause U, Lauerer P, Konietschke F, Aguayo R, Ritter CO, Schuster A, Lotz J, Paul T, Staab W. Diagnosing ARVC in pediatric patients applying the revised task force criteria: importance of imaging, 12‐lead ECG, and genetics. Pediatr Cardiol. 2018;39:1156–1164. [DOI] [PubMed] [Google Scholar]
- 58. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M, Leistner DM, Jakob P, Nakagawa S, Blankenberg S, et al. Non‐coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. 2018;39:2704–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Giuliodori A, Beffagna G, Marchetto G, Fornetto C, Vanzi F, Toppo S, Facchinello N, Santimaria M, Vettori A, Rizzo S, et al. Loss of cardiac Wnt/beta‐catenin signalling in desmoplakin‐deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovasc Res. 2018;114:1082–1097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4 Figures S1–S4