

Abstract
Efficient release of promoter-proximally paused Pol II into productive elongation is essential for gene expression. Recently, we reported that the Integrator complex can bind paused Pol II and drive premature transcription termination, potently attenuating the activity of target genes. Premature termination requires RNA cleavage by the endonuclease subunit of Integrator, but the roles of other Integrator subunits in gene regulation have yet to be elucidated. Here, we report that Integrator subunit 8 (IntS8) is critical for transcription repression and is required for association with Protein Phosphatase 2A (PP2A). We find that Integrator-bound PP2A dephosphorylates the Pol II C-terminal domain and Spt5 and prevents the transition to productive elongation. Blocking PP2A association with Integrator thus stimulates pause release and gene activation. These results reveal a second catalytic function associated with Integrator-mediated transcription termination and suggest a model for the control of productive elongation involving active competition between transcriptional kinases and phosphatases.
eTOC blurb
The Integrator Complex binds promoter-proximally paused Pol II and drives premature transcription termination. Huang et al. identified a motif within Integrator subunit 8 (INTS8) that recruits Protein Phosphatase 2A (PP2A) to Pol II. Integrator-PP2A then dephosphorylates residues within Spt5 and the Pol II CTD to inhibit pause release and facilitate termination.
Graphical Abstract
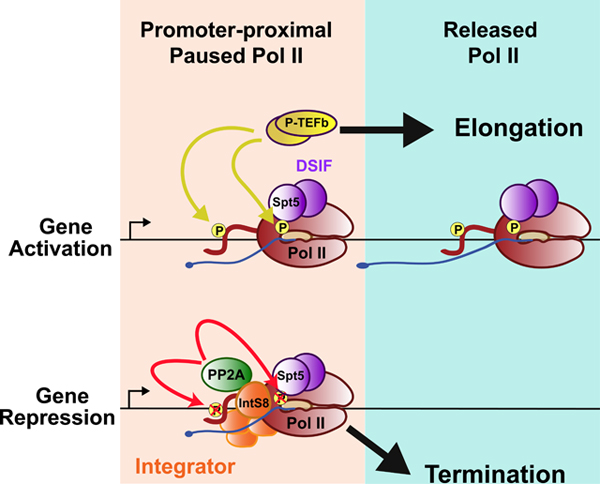
INTRODUCTION
The assembly of general transcription factors and recruitment of RNA polymerase II (Pol II) to promoters allows for the initiation of RNA synthesis (Haberle and Stark, 2018; Sainsbury et al., 2015). Pol II then pauses in early elongation, associated with a 25–60 nucleotide (nt) long RNA (Core and Adelman, 2019; Zhou et al., 2012). Central to Pol II pausing is the association of two protein complexes, Negative Elongation Factor (NELF) and DRB-sensitivity inducing factor (DSIF), a heterodimer of the Spt5 and Spt4 proteins (Wada et al., 1998a; Yamaguchi et al., 1999). These factors interact with Pol II and the nascent RNA, helping to maintain and stabilize the paused state (Vos et al., 2018b). During pausing, the C-terminal domain of the Pol II largest subunit, Rpb1, is phosphorylated within a heptameric repeat sequence (YS2PTS5PS7) by the Cdk7 kinase (Sainsbury et al., 2015). Cdk7 targets Serine residues at positions 5 and 7 within this repeat (hereafter referred to as Ser5P and Ser7P) (Akhtar et al., 2009), which stimulates association of the RNA capping enzymes that add a protective 7-methylguanosine cap to the nascent RNA 5’ end (Fabrega et al., 2004).
The lifetime of paused Pol II varies significantly from gene to gene (Henriques et al., 2013; Krebs et al., 2017). Notably, paused Pol II can either be released into productive elongation to form a functional mRNA, or it can be subjected to promoter-proximal termination, which prevents gene expression (Brannan et al., 2012; Ehrensberger et al., 2013; Henriques et al., 2013; Jonkers et al., 2014). Pol II pause release is mediated by the positive transcription elongation factor b (P-TEFb, a heterodimer of Cdk9 and Cyclin T). P-TEFb phosphorylates several proteins within the paused complex and the Pol II CTD at Serine 2 residues (Ser2P; (Czudnochowski et al., 2012; Lis et al., 2000; Peterlin and Price, 2006; Sanso et al., 2016). P-TEFb-mediated phosphorylation of the DSIF subunit Spt5 is particularly important for pause release and increases the rate of Pol II elongation (Cheng and Price, 2007; Wada et al., 1998b). Phosphorylation by P-TEFb occurs within the Spt5 C-terminal region (e.g. Thr806 in mammals, Thr847 in Drosophila) and the linker domain between conserved Kyrpides-Ouzounis-Woese (KOW) motifs 4 and 5 (Ser666 in humans, Ser707 in Drosophila) (Sanso et al., 2016; Yamada et al., 2006). Although the mechanisms by which these modifications of Spt5 enhance elongation are not yet known, Ser666/Ser707 is located near the channel through which RNA exits Pol II, suggesting a potential role in Spt5-RNA interactions (Vos et al., 2018b). Importantly, P-TEFb activity triggers the transition of Pol II into processive transcription across gene bodies (Ehara et al., 2017; Vos et al., 2018a).
As Pol II approaches the gene 3’ end, it undergoes a transition towards slower elongation that accompanies synthesis of the polyadenylation signal (PAS) (Cortazar et al., 2019; Logan et al., 1987; Whitelaw and Proudfoot, 1986). Recognition of the PAS element in nascent RNA by the cleavage and polyadenylation (CPA) complex triggers RNA cleavage by the cleavage and polyadenylation specificity factor 73 (CPSF73) subunit, using a metallo-β-lactamase/β-CASP domain (Mandel et al., 2006). Cleavage releases the mRNA from Pol II to enable polyadenylation, and creates a free, uncapped 5’ end on the nascent RNA that provides an accessible entry point for the 5’ to 3’ RNA exonuclease Xrn2 (Eaton et al., 2018; Eaton et al., 2020; Kim et al., 2004; West et al., 2004). Purification and proteomic analysis of the CPA machinery reveals the presence of multiple protein phosphatases (Shi et al., 2009) and recent work indicates that these enzymes play key roles in mediating termination. In organisms as diverse as yeast and humans, Protein Phosphatase 1 (PP1) or its orthologues were found to associate with Pol II and/or the CPA machinery near the PAS (Cortazar et al., 2019; Eaton et al., 2020; Kecman et al., 2018; Parua et al., 2018; Schreieck et al., 2014). Importantly, PP1 was shown to dephosphorylate Spt5 residue Thr806 as well as residues within the CTD, to slow down the Pol II complex and sensitize it to termination (Cortazar et al., 2019). This combination of slowed elongation and cleavage of nascent RNA are thought to destabilize Pol II, ultimately leading to Xrn2-mediated termination of transcription (Proudfoot, 2016). Thus, the emerging model is that kinases play a pivotal role in stimulating rapid and efficient Pol II elongation, and that phosphatases antagonize this activity to facilitate transcription termination.
Promoter-proximal Pol II is also susceptible to termination. We recently found that the association of the Integrator complex with paused Pol II bound by DSIF and NELF (Gardini et al., 2014; Skaar et al., 2015; Stadelmayer et al., 2014) can trigger premature transcription termination and repress gene activity (Elrod et al., 2019). Integrator is a metazoan-specific complex of at least 14 subunits, first described for its role in 3’ end formation of the non-coding small nuclear RNAs (snRNAs; (Baillat et al., 2005; Baillat and Wagner, 2015). The Integrator subunit 11 (IntS11) harbors an RNA endonuclease activity that is paralogous to CPSF73 (Albrecht and Wagner, 2012). Like CPSF73, IntS11 cleaves nascent RNA emanating from Pol II to enable transcription termination. Likewise, depletion of IntS11 causes transcriptional readthrough of snRNAs 3’ ends (Baillat et al., 2005). Similar findings have been reported for non-coding enhancer RNAs (eRNA), where loss of Integrator causes inappropriate eRNA elongation (Lai et al., 2015).
At protein-coding genes, depletion of IntS9, IntS11, or expression of a catalytically inactive IntS11 mutant leads to increased transcription of many genes (Elrod et al., 2019). Analysis of nascent RNA at Integrator target loci using Precision Run-on sequencing (PRO-seq) demonstrated that, in wild type cells, Pol II paused near Integrator-target promoters was unable to successfully transition into productive elongation, consistent with low RNA expression levels (Elrod et al., 2019). Depletion of Integrator increased productive elongation, and genes were markedly upregulated. We proposed, analogous to the situation at gene 3’ ends, that RNA cleavage by IntS11 stimulates termination by providing an uncapped RNA 5’ end that is susceptible to attack by exonucleases. However, the mechanisms underlying Integrator-mediated termination remained elusive, suggesting that other Integrator subunits or associated activities might facilitate termination following RNA cleavage.
Depletion of nearly any Integrator subunit leads to snRNA misprocessing (Chen et al., 2012; Ezzeddine et al., 2011) and increased transcription at protein coding genes (Tatomer et al., 2019) indicating that all known subunits contribute to the overall function and/or integrity of the Integrator complex. However, given the paucity of structural and functional information on Integrator, the role of most subunits remains unclear. Here, we show that IntS8 is critical for the association of protein phosphatase 2A (PP2A) with Integrator and that PP2A is required to repress transcription and to prevent elongation by paused Pol II. We further identify conserved substrates of Integrator-associated PP2A (Integrator-PP2A) in Drosophila and human cells, including Spt5 and the Pol II CTD. These results indicate that termination of paused Pol II by Integrator requires dephosphorylation of key pause-inducing factors and transcription regulators, and highlight that progression through the transcription cycle, long appreciated to be driven by transcriptional kinases, is also directed by protein phosphatases.
RESULTS
Loss of IntS8 Leads to Upregulation of Gene Expression
To probe the role of Integrator subunits outside the cleavage module in transcription termination, we first investigated IntS8, for two reasons: 1) In the genome-scale RNAi screen that identified Integrator subunits as repressors of reporter gene transcription, the knockdown of IntS8 caused the greatest level of transcriptional activation (Tatomer et al., 2019); 2) aberrant expression or mutation of IntS8 is associated with numerous human diseased states including neurodevelopment disorders and cancer (Cheng et al., 2013; Federico et al., 2017; Oegema et al., 2017; Simpson et al., 2015). These two observations suggest that IntS8 performs a vital yet undefined role within the Integrator complex, one that is critical to human physiology.
We used RNA interference (RNAi) in Drosophila DL1 cells to deplete IntS8 over 60 h (Figure S1A) and identified mRNA expression changes in polyA-selected RNA-seq (see STAR Methods). We previously established that this timecourse of RNAi is sufficient to detect transcriptional changes due to Integrator depletion but short enough so as to not perturb steady-state levels of snRNAs, thereby minimizing potential indirect effects (Elrod et al., 2019). Nevertheless, genes with any evidence of altered splicing in IntS8-depleted cells were removed from all further analyses, enabling us to focus on transcriptional targets of Integrator. We detected 1099 upregulated and 182 downregulated mRNAs upon IntS8 depletion (Figure 1A, Table S2). Notably, this analysis reveals a general trend for genes with low expression levels in control cells to exhibit upregulation upon loss of IntS8, with many exhibiting strong activation. Expression changes observed with RNA-seq were validated using RT-qPCR (Figure S1B). The large number of genes upregulated after IntS8 depletion is similar to that observed after depletion of subunits required for RNA cleavage (Elrod et al., 2019). Further, comparison of fold changes in gene expression after IntS8 knockdown to those after IntS9 or IntS11 knockdown revealed a strong correspondence at upregulated genes (Figures 1B and S1C). As reported previously, there was little correlation observed between downregulated genes upon depletion of different Integrator subunits (Figures 1B and S1C), suggesting that these genes are indirectly affected by Integrator depletion (Elrod et al., 2019).
Figure 1. Depletion of IntS8 causes widespread activation of genes bound by Integrator.

(A) DL1 cells were treated with dsRNA targeting IntS8 or ß-galactosidase (Control) for 60 h and RNA isolated for total RNA-seq (n=3 per condition). Normalized counts and fold changes are shown for mRNA genes with no alteration in splicing patterns under these conditions (N=9303). Affected genes show a fold-change > 1.5 and adjusted P value <0.0001.
(B) Comparison of fold changes in RNA-seq signals between cells depleted of IntS8 and IntS9. Pearson correlations are shown for genes upregulated (red) or downregulated (green) upon depletion of IntS8.
(C) Composite metagene analysis of IntS1 ChIP-seq reads around promoters of mRNA genes upregulated (N=1009) or unchanged (N=8022) upon IntS8-depletion. Data are shown as average reads per gene in 25-bp bins.
(D) Promoter-proximal read counts for IntS1 ChIP-seq were summed around snRNAs (N=31), randomly selected regions (N=5000) or promoters upregulated or unchanged by IntS8-depletion, as in C. Violin plots depict the range of values, with median indicated by a line. P value calculated using a Mann-Whitney test.
To test the hypothesis that genes upregulated after IntS8 depletion are direct targets of Integrator, we took advantage of our previous ChIP-seq datasets from DL1 cells using antibodies to Drosophila IntS1 or IntS12 (Elrod et al., 2019). We found that genes repressed by IntS8 were significantly enriched in promoter-proximal IntS1 and IntS12 ChIP-seq signals as compared to genes unaffected by IntS8 loss (Figures 1C and S1D). Moreover, similar to findings for IntS9-regulated genes, IntS1 and IntS12 levels observed at IntS8-repressed promoters were even higher than levels at snRNA genes (Figures 1D and S1E). Overall, these results show that depletion of IntS8 results in upregulation of >1000 genes that are also significantly enriched in Integrator occupancy. Moreover, the similarity between gene targets of IntS8 with those of the Integrator cleavage module suggests that IntS8 is critical for Integrator-mediated transcriptional repression.
PP2A Associates with Integrator through a Conserved Motif within IntS8
To gain insights into a role for IntS8, we analyzed the constituency of the Drosophila Integrator complex using affinity purification followed by LC-MS analysis of their tryptic peptides. We purified Drosophila Integrator from S2 cell nuclear extracts derived from four independent cell lines stably expressing FLAG-tagged IntS1, IntS5, IntS8, or IntS11. The rationale for choosing these subunits in addition to IntS8, is that: IntS1 is the largest Integrator subunit and likely functions as a scaffold, IntS5 has been found to directly interact with IntS8 (see below), and IntS11 is the endonuclease. As expected, we readily observed all 14 Integrator subunits in each purification derived from extracts containing FLAG-tagged Integrator subunits but not from control S2 nuclear extract, which lacks any FLAG-tagged proteins (Figure 2A). We also detected significant levels of the Drosophila PP2A subunits PR65 and PP2Ac in each Integrator purification (Figure 2A). Overall, we note that the stringency of our purifications reproducibly isolated the 14 Integrator subunits, PP2A subunits, and only a small number of other associated proteins (shown in Figure S2A) thus suggesting that PP2A is a stable component of Integrator complexes. The association of PP2A with the fly Integrator complex is consistent with purifications of human Integrator (Baillat et al., 2016; Herzog et al., 2012; Malovannaya et al., 2011; Malovannaya et al., 2010; Yadav et al., 2017) and provides previously unappreciated evidence that PP2A association with Integrator is evolutionarily conserved. To further explore the association of PP2A with Drosophila Integrator, we generated nuclear extract from S2 cells stably expressing FLAG-PR65 and purified associated proteins using anti-FLAG affinity resin followed by LC-MS analysis. Similar to Integrator purifications, we could detect high levels of all 14 Integrator subunits as well as PP2Ac associating with PR65 indicating that PR65 can reciprocally pull down the complete Drosophila Integrator complex (Figure 2A). These results were confirmed using independent anti-FLAG affinity purifications that were probed with antibodies raised against Drosophila Integrator subunits as well as PR65 and PP2Ac (Figure 2B).
Figure 2. PR65/PP2Ac require a discrete motif within IntS8 to associate with Integrator.

(A) Heatmap derived from IP LC-MS analysis of FLAG immunoprecipitations from S2 cells expressing the indicated FLAG-tagged proteins. Heatmaps reflect normalized spectral counts observed from analysis of samples performed in triplicate. Control cells lack any exogenously expressed FLAG-tagged protein.
(B) Western blot analysis of input nuclear extracts (left) and immunoprecipitations (right) from S2 cells stably expressing FLAG-tagged proteins as indicated. Immunoprecipitations conducted using anti-FLAG affinity resin were normalized to FLAG signals in each immunoprecipitation.
(C) Yeast two-hybrid analysis where PR65 was fused to the Gal4 DNA binding domain and 14 Integrator subunits were individually expressed as fusions with the Gal4 activation domain. Empty vector is the negative control. Permissive plates lack leucine and tryptophan (-L/-W) to allow growth of plasmid-containing yeast whereas selective plates also lack histidine to screen for interaction (-L/-W/-H). Yeast are plated in five-fold serial dilutions.
(D) Alignment of the N-terminus of IntS8 from multiple species. Scanning mutations are depicted where four consecutive amino acids are converted to alanine. Note the numbering is relative to Homo sapiens INTS8.
(E) Results of yeast two-hybrid analysis where either PR65 or IntS5 are fused to the Gal4 DNA binding domain and either wild-type or alanine mutant IntS8 constructs are fused to the Gal4 activation domain.
(F) Western blot analysis of nuclear extracts (left) and immunoprecipitations (right) from stable cell lines expressing FLAG-IntS8-WT or FLAG-IntS8-WFEF/A.
The canonical mode for PP2A binding to target proteins is through differential association of the PR65/PP2Ac catalytic heterodimer with members of a broad family of PP2A ‘B’ regulatory subunits (Seshacharyulu et al., 2013; Shi, 2009). However, despite high levels of both PR65 and PP2Ac associated with Integrator, no such B subunit was found in any of the four Integrator purifications (Figure 2A). The lack of observed B subunits associated with Integrator is unlikely due to evasion of detection in MS/MS because we could readily observe four different B subunits in the FLAG-PR65 pulldown. The significant levels of PR65/PP2Ac associated with Integrator in the absence of a PP2A regulatory subunit raised the possibility that one of the Integrator proteins could be functioning as a surrogate B subunit. To test this hypothesis, we conducted a directed yeast two-hybrid screen wherein we expressed PR65 fused to the Gal4 DNA binding domain and each of the 14 Drosophila Integrator subunits fused to the Gal4 activation domain. As shown in Figure 2C, yeast expressing Gal4 fusions of PR65 and IntS8 exhibited robust growth on media lacking histidine, indicative of an interaction between these proteins.
To better understand this interaction, we searched for predicted structural similarity between IntS8 and PP2A B subunits, but no similarities were readily apparent. The structure of PP2A has been determined and reveals that PR65 contains 15 HEAT repeats that function as a scaffold for association of both the PP2Ac and B subunits (Shi, 2009): PP2Ac binds HEAT repeats 11–15 while B subunit family members bind within HEAT repeats 1–10 (Cho and Xu, 2007; Xu et al., 2008). With this in mind, we further analyzed IntS8 association with PR65 and found PR65 HEAT repeats 1–7 are both necessary and sufficient to support growth on selective media lacking histidine, indicating that this is the site of PR65 interaction with IntS8 (Figure S2B). Thus, the same region of PR65 that binds B subunits is sufficient to mediate association with IntS8, despite no obvious homology between IntS8 and known PP2A B subunits.
To determine which region of IntS8 is required to associate with PR65, we created deletion mutants of IntS8 (Figure S2C). We found that removal of 18 N-terminal amino acids of IntS8 is sufficient to disrupt association with PR65 but does not impact interaction with IntS5. Alignment of the IntS8 N-terminus from diverse species revealed the presence of a small region of highly conserved amino acids, compelling us to test this region for PR65 interaction using alanine-scanning mutagenesis (Figure 2D). Notably, none of the alanine mutants disrupted binding of IntS8 to IntS5, indicating that the overall folding of each IntS8 mutant and assembly into the Integrator complex is likely not significantly impacted (Figure 2E). We found that mutants five, six, and to a lesser degree four, specifically disrupted IntS8 association with PR65. We focused on mutant five for further analyses because all four of the residues in this region (WFEF) are entirely conserved in the species aligned (mt. 5, Figure 2E).
To determine whether mutating the IntS8 WFEF residues to alanine, hereafter called ‘IntS8-WFEF/A’, also disrupts IntS8-PP2A association in cells, we created an S2 cell line expressing FLAG-tagged IntS8-WFEF/A. We isolated nuclear extract from FLAG-IntS8-WFEF/A cells and compared the associated proteins with that of FLAG-tagged IntS8-WT using anti-FLAG affinity purification. Western blot analysis of each purification revealed a striking reduction in PR65 and PP2Ac in FLAG-IntS8-WFEF/A compared to FLAG-IntS8-WT (Figure 2F). In contrast, there was little change in the level of associated Integrator subunits between FLAG-IntS8-WT and FLAG-IntS8-WFEF/A. Subjecting each purification to LC-MS analysis demonstrated that FLAG-IntS8-WFEF/A purification contained no enrichment of PR65 and PP2Ac, but displayed comparable levels of all Integrator subunits and associated proteins to that of FLAG-IntS8-WT (Figure S2A). We conclude that PP2A binding with Integrator is conserved from flies to humans, and that IntS8 bridges PP2A to Integrator, likely in the place of a canonical B subunit. Critically, we have identified a 4 amino acid motif within IntS8 that is required for PP2A association, but which is dispensable for assembly of IntS8 with the other Integrator subunits.
Association of PP2A with IntS8 is Required for Transcriptional Repression of Integrator Target Genes
To address the importance of PP2A association to Integrator function, we explored the transcriptional impact of the IntS8-WFEF/A mutant in DL1 cells. The advantage of utilizing the IntS8 mutant as opposed to PP2A knockdown or PP2A inhibitors, is that PP2A has hundreds of targets and broad cellular disruption of PP2A activity is certain to cause widespread indirect consequences. In contrast, the IntS8-WFEF/A mutant specifically abrogates PP2A phosphatase activity associated with Integrator without overtly disrupting other PP2A functions. Accordingly, we devised a method to induce expression of either IntS8-WT or IntS8-WFEF/A mutant in cells where endogenous IntS8 has been depleted using RNAi. We created an RNAi-resistant IntS8 cDNA harboring silent mutations throughout the region targeted by dsRNA (Figure 3A). Using this modified IntS8 cDNA, we created stable cell lines expressing either a FLAG-tagged IntS8-WT or IntS8-WFEF/A mutant cDNA under control of a copper-inducible promoter. We observed that treatment of S2 cells with IntS8 dsRNA resulted in effective depletion of endogenous IntS8 while induction of IntS8 transgenes allowed for expression of near endogenous levels of either IntS8-WT or IntS8-WFEF/A proteins (Figure 3B).
Figure 3. The interaction between IntS8 and PP2A is critical for proper gene expression profiles.

(A and B) DL1 cells were depleted of IntS8 using 60 h treatment with dsRNA. RNA was harvested for RNA-seq (n=3 per condition). Where indicated, IntS8-depleted cells were rescued using a transgene expressing wild-type IntS8 (WT) or IntS8 with mutations that disrupt its interaction with PP2A (WFEF/A). To specifically deplete endogenous IntS8 from rescue samples, silent mutations were introduced into the transgene to prevent loss by RNAi (region highlighted in orange). Representative Western blot is shown, indicating IntS8 protein levels in each condition. Tubulin was used as a loading control.
(C) SP1029 (CG11956) locus showing an upregulated gene whose expression is rescued by WT IntS8, but not IntS8-WFEF/A. RNA-seq tracks are shown in control cells and each of the treatments. IntS1 ChIP-seq is from Elrod et al., 2019.
(D) Heatmap representation of RNA-seq fold changes in IntS8-depleted cells, as compared to cells rescued with WT or WFEF/A mutant IntS8. Genes shown are those upregulated by IntS8 depletion (N=649), ranked by fold change.
(E) Fold change in RNA-seq signal upon IntS8 depletion at genes upregulated by IntS8 (N=649). Changes in RNA-seq levels as compared to the control cells are shown following IntS8-depletion, and with rescue by WT or WFEF/A mutant IntS8. Violin plots show range of values, with a line indicating median.
With the RNAi-rescue system established, we then analyzed gene expression using RNA-seq of the following four conditions: control, using a non-targeting dsRNA against β-galactosidase; IntS8 depletion; and IntS8 depletion with concurrent expression of either IntS8-WT or IntS8-WFEF/A. Additionally, because both IntS8 transgenes were induced by copper, we treated all four conditions with copper and also only considered genes to be upregulated after IntS8 depletion if they were upregulated in the absence (Figure 1) and presence of copper (see Star Methods). As anticipated, we observed significantly increased expression of previously defined Integrator target genes upon IntS8 depletion (e.g. Figure 3C). Importantly, repression of transcription was fully restored by IntS8-WT but not IntS8-WFEF/A (Figure 3C). Genome-wide, we observed that expression of IntS8-WT in cells depleted of endogenous IntS8 resulted in >95% restoration of transcriptional repression at Integrator target genes (Figures 3D and 3E). In contrast, expression of the IntS8-WFEF/A mutant was unable to rescue the IntS8 depletion phenotype, and instead these cells showed upregulation of Integrator targets that matched or exceeded that observed in IntS8-depleted cells (Figures 3D and 3E), suggestive that this mutant could have a dominant negative effect. These results were validated by RT-qPCR of selected targets (Figure S3A).
Finally, to attain direct evidence for PP2A involvement in Integrator-mediated gene repression, we conducted two independent experiments. First, we depleted PR65 or PP2Ac using RNAi (Figure S3B) and measured expression of Integrator-repressed genes using RT-qPCR. Depletion of either PP2A subunit resulted in significant activation of all Integrator targets tested, at levels that were comparable to those observed after depletion of IntS8 (Figure S3C). As a second approach, we treated DL1 cells with low doses of PP2A inhibitors Calyculin A or Phendione (Yue et al., 2020). We found that pharmacological reduction in PP2A activity also caused upregulation of Integrator-repressed genes (Figure S3D). We therefore conclude that PP2A association with Integrator is critical for transcriptional repression of Integrator-target genes.
Integrator-PP2A Interaction is Necessary for Promoter-Proximal Termination
Our results thus far suggest that Integrator-bound PP2A promotes termination. To investigate this possibility, we conducted Precision Run-On sequencing (PRO-seq) in control cells, IntS8-depleted cells, IntS8-depleted cells with exogenous IntS8-WT expression, or IntS8-depleted cells with exogenous expression of the IntS8-WFEF/A mutant. We then compared spike-normalized PRO-seq signals across these samples, focusing specifically on the genes upregulated in RNA-seq upon IntS8 knockdown (see STAR Methods). Consistent with our previous work on Integrator-affected genes, we found that IntS8 targets exhibit significantly lower RNA-seq expression levels in unperturbed cells as compared to unaffected genes (Figure S4A). Nonetheless, these genes efficiently recruit Pol II to their promoters, with significantly higher promoter-proximal PRO-seq signal than unaffected genes (Figure S4B). This indicates that Pol II associated with IntS8 target genes is engaged in early elongation but fails to produce full-length mRNAs. To quantify the amount of paused vs. elongating Pol II for all active genes (N=9303, see Figure 1A), we calculated the Pausing Index as the ratio of PRO-seq signal density near promoters (from TSS to +150 nt) over gene body signal (+250 to +2250 downstream of the TSS), using data from control cells. We then asked what percentage of highly paused genes were Integrator targets. Of genes in the top 10% of Pausing Index, nearly half were upregulated upon IntS8-depletion (47%, 441 of 930 genes in the top decile of Pausing Index; indicated in Table S2), indicating a broad effect of Integrator on genes at which paused Pol II does not efficiently enter productive elongation.
Upon depletion of IntS8, we observed significantly more elongating Pol II within the bodies of these genes, consistent with increased productive elongation (Figures 4A, 4B, 4C, and S4C). As expected, we observed no change in PRO-seq signals within the bodies of genes found to be unaffected by IntS8-depletion in RNA-seq (Figure 4C). Re-expression of IntS8-WT in IntS8-depleted cells significantly reduced Pol II elongation at these genes, rendering the PRO-seq profile nearly indistinguishable from that of control cells (Figures 4B and 4C). In contrast, expression of IntS8-WFEF/A in IntS8-depleted cells did not reinstate transcriptional repression and resulted in strong gene activation, as seen in IntS8-depleted cells (Figures 4A, 4B and 4C).
Figure 4. Integrator-mediated attenuation at coding and non-coding loci requires the IntS8-PP2A interaction.

(A) PRO-seq tracks are shown at the 5’ end of the upregulated oaf gene, with TSS designated by arrow.
(B) Heatmap representations of PRO-seq reads from indicated samples, treated as in Figure 3. The location of TSSs is indicated by an arrow. Genes that are upregulated upon IntS8 depletion in RNA-seq are shown (N=649), ranked from most to least upregulated. Color bar at bottom indicates range of read counts per 10-nt bin.
(C) Violin plots depict the fold change in gene body PRO-seq reads (+500 to +2500 nt from TSS) for upregulated (N=649) or unchanged genes (N=7,182) genes. Plots show the range of values, with a line indicating median. P values are from Kruskal-Wallis test, with Dunn’s multiple comparisons test performed against control. *** indicates P<0.0001; n.s. indicates P>0.05.
(D) The difference in PRO-seq signal at upregulated genes between IntS8-depleted samples and control samples is shown. Data are shown in 5-nt bins, smoothed over 2 nearest neighbors.
(E) Average distribution of PRO-seq signal is shown at enhancers previously shown to be targeted by Integrator (defined in Elrod et al., 2019). Data represent N=228 Integrator-target eRNAs in 25-nt bins.
To probe the mechanisms underlying gene activation in the absence of IntS8-PP2A, we calculated the difference in PRO-seq signal between control cells and each of the IntS8-depleted samples, focusing on the region just downstream of TSSs (Figure 4D). Despite loss of IntS8 causing an overall increase in PRO-seq signal near target promoters (Figure S4D), this high-resolution analysis revealed an interesting shift in Pol II location: we observed less Pol II paused promoter-proximally (<50 nt from the TSS) upon depletion of IntS8 or loss of Integrator-PP2A association, and more Pol II present downstream towards the gene body (Figures 4D and S4C). This effect is reminiscent of that observed upon recruitment of the kinase P-TEFb, which phosphorylates Spt5 and the Pol II CTD to drive release of paused Pol II (Lis et al., 2000). Our results thus suggest that loss of PP2A phosphatase activity associated with Integrator allows phosphorylation to accumulate on the paused elongation complex and stimulates pause release.
To determine whether IntS8-PP2A functions similarly at enhancers, we investigated PRO-seq signal around enhancer TSSs (eTSSs) previously shown to be Integrator targets (Elrod et al., 2019). Indeed, at these enhancers, like at protein-coding genes, loss of IntS8-PP2A resulted in significantly upregulated transcription elongation (Figures 4E and S4D), indicating that PP2A activity can also restrain eRNA transcription. Finally, we investigated PRO-seq signal at the Drosophila snRNA genes, where Integrator is required for proper 3’ end formation and termination. Accordingly, IntS8 depletion resulted in a significant increase in PRO-seq signal downstream of the 3’ processing site, indicative of transcriptional read through (Figure S4E). As expected, proper 3’ end processing was restored by expression of IntS8-WT, but not the IntS8-WFEF/A mutant (Figure S4E). We conclude that the interaction of Integrator with PP2A is required for activity at both coding and non-coding RNA loci, indicating a conserved function.
Integrator-PP2A Dephosphorylates Specific Residues within the Pol II CTD and Spt5
The above data suggest that one or more proteins within the paused elongation complex is subject to dephosphorylation mediated by Integrator-PP2A. To identify the substrate(s) of this activity we conducted parallel in vitro and cell-based assays. Since Integrator binds Pol II promoter-proximally, and phosphorylation of both the Pol II CTD and Spt5 regulates the release of paused Pol II (Core and Adelman, 2019), we focused our analyses of Integrator-PP2A activity on phosphorylated residues within these proteins. We created a collection of synthetic peptides with three copies of the CTD consensus repeat (Figures 5A and S5A). These peptides contained unphosphorylated CTD repeats as a control, or were phosphorylated within the heptamer at Ser2, Ser5, or Ser7. We also created CTD peptides with all three combinations of serine phosphorylation. Finally, we generated a peptide derived from Drosophila Spt5 containing phosphorylation at Ser707 (orthologous position to human Spt5-Ser666). To monitor dephosphorylation in vitro, we used a sensitive colorimetric assay wherein an indicator dye, malachite green, is converted from yellow to green upon orthophosphate release (Geladopoulos et al., 1991). As a control, we treated each synthetic peptide with non-selective alkaline phosphatase, which confirmed that each peptide was capable of discharging orthophosphate (Figure S5B).
Figure 5. The Pol II CTD and Spt5 are in vitro and in vivo substrates of Integrator-PP2A.

(A) Schematics of synthetic peptides used for in vitro phosphatase assays.
(B) Results of malachite green assay where peptides shown in panel A were incubated with Integrator-PP2A purified with anti-FLAG affinity resin from nuclear extracts expressing FLAG-PR65. Control is from S2 cells not expressing a FLAG-tagged protein. Representative colorimetric results are shown in the upper panel. Results from triplicate assays are quantified as picomoles of orthophosphate produced above negative control.
(C) Results of malachite green assay using Integrator-PP2A purified from cells expressing FLAG-IntS8-WT or FLAG-IntS8-WFEF/A mutant. The substrate used is CTD-Ser7P (n=3).
(D) Dose curves of Integrator-PP2A incubated with CTD-Ser7P and increasing amounts of PP2A inhibitors Calyculin A or Okadaic Acid (n=3).
(E) Western blot analysis of lysates from cells either mock-depleted or depleted of IntS8. Antibodies shown probe either endogenous proteins or are phospho-specific, as indicated.
(F) Western blot analysis of lysates from control or IntS8-depleted cells. Where indicated, cells were induced to express IntS8-WT or IntS8-WFEF/A mutant.
Using anti-FLAG affinity resin, we purified Integrator-PP2A from nuclear extracts containing FLAG-tagged PR65 protein, and used extracts lacking a FLAG-tagged protein as a control. Either purification was then incubated with each of the synthetic peptides. We observed a potent phosphatase activity that was significantly above the control when Integrator-PP2A was incubated with CTD peptides that contain either a singular mark at Ser7P or dual modifications at Ser5P/Ser7P or Ser2P/Ser7P. Moreover, we detected robust phosphatase activity of Integrator-PP2A with the Spt5-Ser707P peptide (Figure 5B). These results indicate that purified Integrator-PP2A can dephosphorylate residues associated with paused Pol II and release into productive elongation, with an in vitro preference for the CTD containing Ser7P and Spt5 containing Ser707P.
To confirm this result, we evaluated the phosphatase activity of Integrator-PP2A purified from nuclear extract containing either FLAG-IntS8-WT or FLAG-IntS8-WFEF/A. Importantly, when Integrator-PP2A was purified using the FLAG-IntS8-WFEF/A mutant, the level of released phosphate was reduced by >75%, consistent with reduced association of PP2A (Figure 5C). We also incubated Integrator-PP2A purified using FLAG-IntS8-WT with increasing amounts of PP2A inhibitors Calyculin A or Okadaic Acid, and observed greatly reduced dephosphorylation activity, at IC50 values consistent with those defined previously for inhibition of PP2A with these compounds (Figure 5D) (Cohen et al., 1989; Ishihara et al., 1989). Overall, these results confirm that the IntS8-WFEF/A mutations found to disrupt PP2A association also reduce in vitro phosphatase activity and provide additional support that the Integrator-associated phosphatase is indeed PP2A.
To investigate Integrator-PP2A substrates in Drosophila S2 cells, we depleted IntS8 using RNAi and measured phosphorylation changes within the Pol II CTD and Spt5 using phospho-specific antibodies (Figure 5E). We reasoned that loss of PP2A recruitment to Pol II upon IntS8 depletion would result in increased levels of phosphorylation on the cellular targets of Integrator-PP2A. We observed that depletion of IntS8 increased the level of CTD phosphorylation states associated with early elongation, particularly Ser7P and Ser5P. Notably, the effects of Integrator-PP2A on Ser5P in vivo is stronger than that observed in our in vitro assay, suggesting that Integrator-PP2A has additional targets in cells and/or that there is some cross-talk between Ser7P and Ser5P levels. Nonetheless, we observed no substantive change in phosphorylation of the CTD at Tyr1P, Ser2P or Thr4P nor in the total levels of Rpb1 (Figure 5E). We did however note an upwards shift in migration of the species recognized by the Tyr1P antibody, indicating that this antibody can recognize a CTD hyperphosphorylated at Ser5 and/or Ser7. Importantly, knockdown of IntS8 led to increased levels of Spt5-Ser707P, consistent with our in vitro findings (Figure 5E). IntS8 depletion does not alter total Spt5 expression or phosphorylation of Spt5 residue Thr847, in agreement with recent work indicating that the analogous residue to Thr847 in human Spt5 (Thr806) is dephosphorylated by the phosphatase PP1 at gene 3’ ends (Cortazar et al., 2019).
To confirm the specificity of these results, we probed lysates from cells depleted of IntS8 wherein IntS8-WT or IntS8-WFEF/A mutant was re-expressed. As anticipated, the increases in Ser5P, Ser7P, and Spt5-Ser707P observed after IntS8 depletion were reversed by expression of IntS8-WT but not by the IntS8-WFEF/A mutant protein (Figure 5F). Collectively, these in vitro and cellular data converge on a model wherein Integrator-PP2A removes CTD phosphorylation normally present on paused Pol II and reverses or prevents the phosphorylation of Spt5-Ser707 by P-TEFb, thereby constraining pause release.
Association of PP2A with Integrator is Conserved in Human Cells
Our results indicate that Drosophila PP2A assembly with Integrator is dependent on a small conserved motif within IntS8 that is critical to attenuate transcription. To determine whether human Integrator interacts similarly with PP2A, we introduced a single, N-terminal FLAG epitope into endogenous genes encoding INTS1, INTS5, INTS8, INTS11, and PR65 in 293T cells using CRISPR genome editing. From these individual cell lines, we generated nuclear extract and purified native human Integrator complexes with anti-FLAG affinity resin and analyzed the associated proteins using Western blot and MS/MS. As expected, each Integrator purification contained all other Integrator subunits as well as high levels of PR65 and PP2Ac (Figure 6A). Moreover, reciprocal purification of human FLAG-PR65 revealed the presence of associated Integrator subunits. Each FLAG-Integrator purification was also subjected to LC-MS analysis, indicating that, like in Drosophila, no PP2A B regulatory subunits were detectable within any Integrator purifications despite high levels of PR65 and PP2Ac (Figure S6).
Figure 6. Assembly and function of Integrator-PP2A is conserved in human cells.

(A) Western blot analysis of nuclear extracts (left) and immunoprecipitations (right) from 293T cells harboring FLAG epitope tags within human Integrator genes generated using CRISPR/Cas9 genome-editing. Immunoprecipitations were conducted using anti-FLAG affinity resin and were normalized to FLAG signals in the immunoprecipitation. Control extracts were derived from 293T cells lacking any exogenous FLAG-tagged protein.
(B) Western blot analysis of nuclear extracts (left) and immunoprecipitations from either 293T control nuclear extract, or nuclear extracts derived from stable cell lines expressing either FLAG-hINTS8-WT or FLAG-hINTS8-WFEF/A mutation.
(C) Western blot analysis of cell lysates derived from 293T cells transfected with either control siRNA or INTS8-targeting siRNA. Lysates were probed with a series of antibodies labeled on the right side of the blots. Note that hSpt5-Ser666 is the equivalent residue of Drosophila Spt5-Ser707, and hSp55-Thr806 is the equivalent of fly Spt5-Thr847.
(D) Results of malachite green assay using a CTD-Ser7P peptide substrate represented as pmoles released above control. Integrator-PP2A was purified from 293T nuclear extracts containing either FLAG-hINTS8-WT or FLAG-hINTS8-WFEF/A.
To test whether human PP2A association with Integrator requires the same N-terminal motif within INTS8 identified in Drosophila, we created 293T cell lines stably expressing either FLAG-tagged hINTS8-WT or hINTS8-WFEF/A mutant. Using anti-FLAG affinity resin, we purified Integrator from nuclear extracts derived from both of these cell lines as well as control 293T extracts from cells lacking a FLAG-tagged protein. Similar to our findings in Drosophila, we observed comparable levels of other Integrator subunits associated with the FLAG-tagged hINTS8-WT and hINTS8-WFEF/A mutant but found a significant reduction in both PR65 and PP2Ac associated with hINTS8-WFEF/A (Figure 6B). We thus conclude that the hINTS8-WFEF/A mutant disrupts a conserved motif required for PP2A association with Integrator in human cells.
To determine if human Integrator-PP2A has similar substrate specificity to the Drosophila complex, we depleted INTS8 from 293T cells using siRNA. The INTS8 siRNA effectively depleted endogenous INTS8 but did not affect levels of PR65 or PP2Ac (Figure 6C). Using antibodies specific to phosphorylated residues within the Pol II CTD and Spt5, we observed significant and specific increases in the level of Ser5P and Ser7P in INTS8-depleted cells, as well as Spt5 at Ser666 (Ser707 in Drosophila). We also incubated Integrator-PP2A purified using either FLAG-hINTS8-WT or FLAG-hINTS8-WFEF/A with the CTD Ser7P peptide and measured activity using the malachite green assay. Consistently, activity measured from Integrator-PP2A containing INTS8-WFEF/A was >70% lower than that observed with INTS8-WT (Figure 6D). Overall, these data reveal a striking conservation in phosphorylation targets of Integrator-PP2A between human and Drosophila cells.
Depletion of Human INTS8 Causes Upregulation of Genes in Signal-Responsive Pathways
To determine if Integrator-PP2A functions similarly in humans as it does in Drosophila, we identified Integrator target genes in human 293T cells treated with either control siRNA or INTS8-targeting siRNA for 48 h. Effective INTS8 depletion was observed in each of the three independent biological replicates evaluated by total RNA-seq (Figure 7A). This analysis identified 420 upregulated and 53 downregulated genes after INTS8 depletion (Figure 7B, Table S3). Example upregulated immediate early genes ARC and GADD45B (Figure 7C) were independently validated using RT-qPCR (Figure S7A). Gene ontology analysis highlighted immediate early response genes within several signaling pathways (e.g. receptor and MAPK signaling, Figure S7B) as enriched among INTS8 targets, consistent with data from depletion of other Drosophila and human Integrator subunits (Elrod et al., 2019; Gardini et al., 2014; Yue et al., 2017).
Figure 7. Function of INTS8 is conserved in human cells.

(A) Western blot analysis of cell lysates derived from 293T cells transfected with control or INTS8 targeting siRNA. Lysates were harvested after 48 h and probed with antibodies against INTS8 or GAPDH as a loading control.
(B) 293T cells were transfected with control or INTS8 targeting siRNA. After 48 h, RNA was harvested for total RNA-seq (n=3). Normalized counts are shown for mRNA genes (N=12059). Affected genes are those with a fold-change > 2 and adjusted p-value <0.001.
(C) RNA-seq tracks are shown for ARC and GADD45B, genes upregulated upon INTS8 depletion.
(D) Schematic of the ARC and GADD45B reporter systems, wherein promoter and 5’ UTRs were cloned upstream of GFP. Top: In wild type cells, Integrator-PP2A drives Pol II termination and prevents GFP expression. Bottom: Upon loss of Integrator-PP2A association through INTS8 depletion or expression of INTS8-WFEF/A, Pol II productively elongates through the GFP ORF, yielding GFP expression.
(E) Western blot analysis of 293T cells transfected with the GFP reporters, along with control or IntS8 targeting siRNA, or IntS8 targeting siRNA plus RNAi-resistant INTS8-WT or INTS8-WFEF/A mutant cDNA.
To determine if PP2A association with Integrator is necessary to maintain repression of ARC and GADD45B in human cells, we cloned the promoters and 5’UTR sequences from these genes upstream of GFP to create reporter plasmids. If PP2A normally functions with Integrator to drive termination at ARC and GADD45B genes, then wild type cells should exhibit low basal GFP expression. However, if PP2A association with Integrator is lost, either by depletion of INTS8 or expression of the hINTS8-WFEF/A mutant, then we predict that paused Pol II would transition to productive elongation and transcribe GFP mRNA, increasing GFP levels (Figure 7D). As expected, depletion of INTS8 from cells transfected with either the ARC-GFP or GADD45B-GFP reporter resulted in increased GFP expression compared to that observed in control cells (Figure 7E). Importantly, expression of RNAi-resistant FLAG-hINTS8-WT cDNA reduced GFP expression to levels observed in control cells, whereas RNAi-resistant FLAG-hINTS8-WFEF/A expression restored GFP expression (Figure 7E). Finally, to determine if direct PP2A inhibition is sufficient to mediate similar upregulation of Integrator target genes, we treated 293T cells with two different PP2A inhibitors and found significant upregulation of both ARC and GADD45B (Figure S7C). Altogether, these results demonstrate that association of PP2A with Integrator is critical for gene repression and that this function is conserved from fly to man.
DISCUSSION
In this report, we identified a 4 amino acid, conserved motif within IntS8 that is critical for the association of PP2A with the Integrator complex. Disruption of this motif abolishes promoter-proximal termination mediated by Integrator. We conclude that Integrator utilizes PP2A to dephosphorylate components of the paused Pol II complex, thereby antagonizing the activity of transcriptional kinases and preventing the transition of paused Pol II to productive elongation.
The results presented here and previously (Elrod et al., 2019; Tatomer et al., 2019), establish that Integrator-mediated transcription termination requires two distinct activities: cleavage of nascent RNA through the β-CASP domain of IntS11, and dephosphorylation of the early elongation complex through the PP2A protein phosphatase associated through IntS8. Both activities are essential to inhibit gene expression as evidenced by broad upregulation of common Integrator-target transcripts in Drosophila cells expressing a catalytically inactive IntS11 (E203Q) (Elrod et al., 2019) and an IntS8 mutant (IntS8-WFEF/A) that blocks association with PP2A (Figures S1 and 3). PRO-seq analysis from IntS8-depleted cells or from cells expressing the IntS8-WFEF/A mutant (Figure 4) reveal that loss of Integrator-PP2A interactions causes a significant decrease in the levels of promoter-proximally paused Pol II (located 25–50nt downstream of the TSS) and a concomitant increase in the levels of Pol II downstream within gene bodies (Figure 4D). This change in Pol II profile relative to cells expressing IntS8-WT is consistent with increased pause release in the absence of PP2A. Consistent with this, we observe increased phosphorylation of the Pol II CTD and the central elongation factor Spt5 when Integrator-PP2A interactions are disrupted (Figure 5).
Our findings highlight an antagonistic relationship between phosphorylation events associated with pause release and the phosphatase function of Integrator-PP2A. We posit that PP2A removes stimulatory phosphorylation on the CTD and Spt5 to counteract Cdk7 and P-TEFb activity. In particular, we envision that removal of phosphorylation on Spt5 reduces elongation potential of promoter-proximal Pol II, much as it does at gene 3’ ends. Indeed, to facilitate termination at the 3’ end of genes the phosphatase PP1 dephosphorylates Spt5 at a distinct position (Spt5-Thr806P/Thr847). The removal of phosphate(s) from Spt5 functions to slow down Pol II elongation, facilitating CPSF73-dependent termination. Likewise, we propose that rapid removal of Spt5-Ser666P/Ser707P by PP2A would reduce the likelihood of pause release, trapping Pol II near the promoter to provide an increased window of opportunity for the nascent RNA to be targeted by IntS11 for cleavage.
Our data also shed light on Pol II turnover and reinitiation at enhancers, which we previously reported to harbor less stably paused Pol II than mRNA promoters, and to exhibit faster termination and recycling of polymerases (Henriques et al., 2018). The rapid initiation of Pol II at enhancers required for such recycling is supported by our data here, wherein increased pause release at enhancers upon IntS8-depletion is not accompanied by a clear reduction of Pol II in the promoter-proximal window. This finding indicates that Pol II released from pausing at enhancers is quickly replaced by a new polymerase, in agreement with rapid transcription initiation and a highly dynamic behavior of Pol II at enhancers.
A large body of structural and biochemical work support a model where the heterodimeric enzymatic core of PP2A, comprised of PR65 and PP2Ac, differentially associates with many distinct B regulatory subunits (Shi, 2009). This mechanism is thought to specifically target PP2A to diverse cellular substrates. Indeed, recent proteomics approaches have shown that the B56 regulatory subunit can identify >300 substrates that contain a consensus short linear motif (SLiM) and recruit those proteins to PP2A for dephosphorylation (Hertz et al., 2016; Wang et al., 2016). Our results, however, indicate that the PP2A core enzyme associates with Integrator without any of the known regulatory B subunits (Figures 2 and 6). This observation raises the question as to why Integrator evolved this atypical mechanism to associate with PP2A. One possible answer is that Integrator has evolved to target PP2A to a specific set of proteins, including Spt5-Ser666P/Ser707 and residues within the CTD, particularly Ser7P and Ser5P (Figure 5 and 6). It is also possible that Integrator utilizes PP2A to dephosphorylate its own subunits but our data suggests that at least in the case of INTS12, which is subject to extensive phosphorylation(Chen et al., 2013), there are no overt changes in phosphorylation status (Figure 2F). Moreover, we envision that Integrator-PP2A could target additional transcription regulators for dephosphorylation under particular conditions, a possibility that will be valuable to probe in future.
In summary, this work has revealed an essential role for IntS8 in recruitment of PP2A to Pol II early elongation complexes. While mutation of IntS8 significantly reduces PP2A association with Integrator, it remains to be seen if IntS8 is the only Integrator subunit involved in mediating PP2A interaction. Indeed, we observed that depletion of IntS8 in 293T cells causes significant upregulation of IntS6 (Table S3) suggesting a potential compensatory pathway. These findings suggest that other Integrator subunits could also have important functions in the regulation of coding and non-coding RNA production. Notably, the structure of the IntS13/IntS14 heterodimer has recently been solved revealing a previously unsuspected nucleic acid binding motif (Sabath et al., 2020). It will be interesting in future to define the means by which Integrator-mediated gene repression is alleviated to enable gene activation, and which subunits govern this behavior.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric J. Wagner (ejwagner@utmb.edu).
Materials Availability
Unique and stable reagents generated in this study are available upon request.
Data and Code Availability
GEO Accession numbers: all datasets generated in this study are available for download from GEO: GSE150844. Original image data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/bt76mktdfx.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Drosophila DL1 cells were cultured at 27°C in Schneider’s Drosophila medium (Gibco, #21720–024), supplemented with 10% (v/v) fetal bovine serum (HyClone, #SH30910.03), 1% (v/v) Antibiotic-Antimycotic (Gibco, #15240–062). Drosophila S2 cells were grown at 27°C in Sf-900 II SFM (Gibco, #10902–088), supplemented with 1% (v/v) Antibiotic-Antimycotic. HEK 293T cells were grown at 37°C with 5% CO2 in DMEM (Gibco, #11965–092), supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin (Gibco, #15070–063).
METHOD DETAILS
Plasmid construction and stable cell lines generation
For the alanine-scanning of Drosophila IntS8, every four amino acids from the conserved N-terminus were mutated to alanine using site-directed PCR mutagenesis. Wild type and the WFEF/A mutant dIntS8 were subsequently either cloned into pUB-3xFLAG vector (Chen et al., 2013) to stably express transgenes in S2 cells or pMT-3xFLAG-puro vector (Elrod et al., 2019) to inducibly express in DL1 cells. The PCR primers are provided in Table_S1. All plasmids were sequenced to confirm identity. To generate cells stably expressing the FLAG-IntS8-WT, FLAG-IntS8-WFEF/A, and eGFP control transgenes, 2×106 cells were first plated in regular maintenance media in a 6-well dish overnight. 2 μg of expressing plasmids were transfected using Fugene HD (Promega, #E2311). After 24 hours, 2.5 μg/mL puromycin was added to the media to select and maintain the cell population.
Nuclear extract preparation
Cells were collected and washed in cold PBS. Cells were then resuspended in five times the cell pellet volume of Buffer A (10mM Tris pH8, 1.5mM MgCl2, 10mM KCl, 0.5mM DTT, and 0.2mM PMSF). Resuspended cells were allowed to swell during a 15-minute rotation at 4°C. After pelleting down at 1,000g for 10 minutes, two volumes of the original cell pellet of Buffer A was added and cells were homogenized with a dounce pestle B for 20 strokes on ice. Nuclear and cytosolic fractions were then separated by centrifugation at 2,000g for 10 minutes. To attain a nuclear fraction, the pellet was washed once with Buffer A before resuspending in an equal amount of the original cell pellet volume of Buffer C (20mM Tris pH8, 420mM NaCl, 1.5mM MgCl2, 25% glycerol, 0,2mM EDTA, 0.5mM PMSF, and 0.5mM DTT). The sample was then homogenized with a dounce pestle B for 20 strokes on ice and rotated for 30 minutes at 4°C before centrifuging at 15,000g for 30 minutes at 4°C. Finally, supernatants were collected and subjected to dialysis in Buffer D (20mM HEPES, 100mM KCl, 0.2mM EDTA, 0.5mM DTT, and 20% glycerol) overnight at 4°C. Prior to any downstream applications, nuclear extracts were centrifuged again at 15,000g for 3 minutes at 4°C to remove any precipitate.
Western blotting and anti-FLAG affinity purification
Protein was extracted directly by adding 2X SDS sample buffer (120mM Tris pH6.8, 4% SDS, 200mM DTT, 20% Glycerol, and 0.02% Bromophenol blue) to cells while on the plate. Samples were then incubated at room temperature while on the plate with periodic swirling prior to a 10-minute boiling at 95°C and a short sonication. Denatured protein samples were then resolved on a 4–15% gradient gel (Bio-Rad, #456–1086) and transferred to a PVDF membrane (Bio-Rad, #1620177). For both commercial and custom-designed antibodies, blots were probed as previously described (Elrod et al., 2019). For phospho-specific antibodies, TBS-0.1% Tween supplemented with 5% BSA was used instead of PBS-0.1% Tween supplemented with 5% nonfat milk, which was used for all other blots.
To purify FLAG-tagged Integrator complexes, 2mg of nuclear extract was mixed with 50μl anti-Flag M2 affinity agarose slurry (Sigma, #A2220) equilibrated in binding buffer (20mM HEPES pH7.4, 150mM KCl, 10% Glycerol, 0.1% NP-40) and rotated for 2 hours at 4°C. Following the two-hour incubation/rotation, five sequential washes were carried out in binding buffer with a 10-minute rotation at 4°C followed by a 1,000g centrifugation at 4°C. After the final wash, the binding buffer supernatant was removed using a pipette and the protein complexes were eluted from the anti-FLAG resin by adding 50μl of 2X sample buffer and boiled at 95°C for five minutes. For Western blots, input samples were generated by adding equal volume of 2X loading buffer to nuclear extract and 1/10 of the immunoprecipitation was loaded as estimated by protein mass.
Antibody generation and purification
Commercial antibodies that were used to detect proteins are listed in the Key Resources Table. The remainder of the antibodies were custom-made and were raised against recombinant proteins expressed in E. coli. Specifically, these proteins were: full-length Drosophila PR65 or the first 100 amino acids of the N-termini of either Drosophila IntS4, Drosophila IntS7, or Drosophila IntS8. These recombinant proteins were then shipped to a commercial vendor (Cocalico Biologicals, PA) and used to inoculate guinea pigs. Sera was isolated and tested for specific reactivity to target proteins using Western blot analysis initially with nuclear extract to confirm that protein bands matched predicted size. Sera passing this filter was subsequently tested by probing lysates from DL1 cells treated with dsRNA targeting the protein of recognition to confirm loss of specific bands.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Flag M2-HRP | Sigma #A8592 | AB_439702 |
| Drosophila IntS1 | Ezzeddine et al., 2011 | |
| Drosophila IntS4 | This paper | |
| Drosophila IntS7 | This paper | |
| Drosophila IntS8 | This paper | |
| Drosophila IntS9 | Ezzeddine et al., 2011 | |
| Drosophila IntS11 | Ezzeddine et al., 2011 | |
| Drosophila IntS12 | Chen et al., 2012 | |
| Drosophila PR65 | This paper | |
| PP2Ac | Cell Signaling #2038 | AB_2169495 |
| alpha-tubulin | abcam #ab15246 | AB_301787 |
| GAPDH (GA1R) | ThermoFisher #MA5-15738 | AB_10977387 |
| RNA Pol II CTD Tyr1-p (3D12) | Active motif #61383 | AB_2793613 |
| RNA Pol II CTD Ser2-p (3E10) | Active motif #61083 | AB_2687450 |
| RNA Pol II CTD Thr4-p (6D7) | Active motif #61361 | AB_2750848 |
| RNA Pol II CTD Ser5-p (3E8) | Active motif #61085 | AB_2687451 |
| RNA Pol II CTD Ser7-p (4E12) | Active motif #61087 | AB_2687452 |
| RNA Pol II CTD Ser7-p (3D4A12) | Active motif #61703 | AB_2793742 |
| Spt5-Ser707P | This paper | |
| Spt5-Thr806P | This paper | |
| Spt5 | BD #611107 | AB_398420 |
| hINTS1 | Bethyl #A300–361A | AB_2127258 |
| hINTS3 | Proteintech #16620–1 | AB_2127274 |
| hINTS4 | Bethyl #A301–296A | AB_937909 |
| hINTS7 | Bethyl #A300–271A | AB_2127399 |
| hINTS10 | Proteintech #15271–1 | AB_2127260 |
| hINTS11 | Bethyl #A301–274 | AB_937779 |
| hPR65 (81G5) | Cell Signaling #2041 | AB_2168121 |
| siRNA | ||
| Universal Negative Control #2 | Sigma | Cat. #SIC002 |
| Human IntS8 (NM_017864) | Sigma | SASI_Hs01_00121954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AzNTPs | TriLink Technologies | Cat. #K-1005 |
| Biotin-11-NTPs | Perkin Elmer | Cat. #NEL54(2/3/4/5)001 |
| Okadaic Acid | Cell Signaling | Cat. #5934S |
| Calyculin A | Cell Signaling | Cat. #9902S |
| Phendione | Sigma | Cat. #496383 |
| Critical Commercial Assays | ||
| NEB Next Ultra II DNA library kit | NEB | Cat. #E7103S |
| PP2A Immunoprecipitation Phosphatase Assay Kit | Sigma | Cat. #17–313 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE150844 |
| Raw image files | Mendeley | http://dx.doi.org/10.17632/bt76mktdfx.1 |
| RNA-seq from IntS9 and IntS11-depleted DL1 cells, IntS1 and IntS12 ChIP-seq | Elrod et al., 2019 | GEO: GSE114467 |
| Experimental Models: Cell Lines | ||
| DL1 | Dr. Sara Cherry, UPenn | |
| DL1 FLAG-eGFP | This paper | |
| DL1 FLAG-IntS8-WT | This paper | |
| DL1 FLAG-IntS8-WFEF/A | This paper | |
| S2-DGRC clone 6 | DGRC | |
| S2 Flag-PR65 | This paper | |
| S2 Flag-IntS1 | This paper | |
| S2 Flag-IntS5 | This paper | |
| S2 Flag-IntS8 | This paper | |
| S2 Flag-IntS8-WFEF/A | This paper | |
| S2 Flag-IntS11 | This paper | |
| 293T | ATCC | CRL-3216 |
| 293T CRISPR-en-Flag-PR65 | This paper | |
| 293T CRISPR-en-Flag-INTS1 | This paper | |
| 293T CRISPR-en-Flag-INTS5 | This paper | |
| 293T CRISPR-en-Flag-INTS8 | This paper | |
| 293T CRISPR-en-Flag-INTS11 | This paper | |
| 293T Flag-INTS8-WT | This paper | |
| 293T Flag-INTS8-WFEF/A | This paper | |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Table_S1 | This paper | |
| Recombinant DNA | ||
| pUB-3xFLAG vector | Chen et al., 2012 | |
| pUB-3xFLAG-dIntS8-WT | This paper | |
| pUB-3xFLAG-dIntS8-WFEF/A | This paper | |
| pMT-3xFLAG-puro vector | Elrod et al., 2019 | |
| pMT-3xFLAG-dIntS8-WT-RESC-puro | This paper | |
| pMT-3xFLAG-dIntS8-WFEF/A-RESC-puro | This paper | |
| pMT-3xFLAG-eGFP-puro | This paper | |
| pcDNA6–3xFlag-hINTS8-WT | Oegema et al., 2017 | |
| pcDNA6–3xFlag-hINTS8-WFEF/A | This paper | |
| Software and Algorithms | ||
| bowtie 1.2.2 | Langmead et al., 2009 | |
| R v3.3.1 | www.r-project.org | |
| Rstudio v1.0.136 | www.rstudio.com | |
| featureCounts | Liao et al., 2014 | |
| DESeq2 | Love et al., 2014 | |
| MISO | Katz et al., 2010 | |
| Prism v8.1.2 | GraphPad | |
| Partek Genomics Suite v6.15.0127 | www.partek.com | |
To raise antibodies that recognize specific phosphorylated epitopes in Spt5, synthetic peptides were created (21st Century Biochemicals) bearing the following sequences: Spt5-Ser666 in humans, CVGGFTPM[pS]PRISSP, Spt5-Thr806 in humans, CPHYGSQ[pT]PLHDGS. These peptides were used to raise antibodies in rabbits using a commercial vendor (Covance, Princeton NJ). Exsanguinated samples showed specificity as compared to pre-immune sera. In order to detect the phosphorylated serine 707 in Drosophila Spt5 (Homologous of serine 666 in human Spt5), further purification against fly sequence was performed to ensure epitope specificity. Spt5-Ser707-P in fly: VGGLGFM[pS]PRIQSP (synthesized by United Biosystems). To purify antibody from crude serum, AminoLink Coupling Resin (Thermo, #20381) column was built per manufacturer’s instructions. Briefly, 4ml of resin slurry was packed into a 10ml capacity chromatography column (Bio-Rad). One full column volume of a 1xPBS wash was performed to equilibrate resin before adding 8mls of diluted phospho-peptide [5mg peptide, which were resuspended in H2O at 2mg/ml, were mixed in PBS solution to the final concentration of 1XPBS and 50mM NaCNBH3 (Thermo, #44892)]. The mixture was incubated overnight at 4°C before washing resin with 1xPBS using gravity flow. Nonspecific binding sites on resin were blocked using a quenching buffer (1M Tris-HCl, pH7.4 and 50mM NaCNBH3) that was added to the column for 60 minutes on a rotator at room temperature. Resin was then washed twice with one full column volume of 1M NaCl and then 1xPBS, sequentially. Serum was then loaded onto the column and incubated overnight at 4°C with rotation. Following the overnight incubation, the column was washed three times with PBS and the antibody was eluted with 15ml of 0.1M Glycine, pH2.7. Every 1ml eluted antibody solution was immediately neutralized by collecting it into a tube containing 60μl of 1M Tris pH8.8. Eluates having positive signal for protein using a Bradford Assay (Bio-Rad) were subsequently pooled and sodium chloride (150mM final) was added into the pooled solution before concentrating through 10KDa cut-off Amicon Ultra-15 spin tube (Millipore). The concentration was adjusted to ~1mg/ml with 50mM Tris [pH7.5] before adding equal amount of Glycerol (50% final) and BSA (100μg/ml final). Antibody specificity was then confirmed by immunoblotting as described above.
In vitro phosphatase assay
Phospho-peptides were synthesized (United BioSystems) and dissolved in H2O to a concentration of 1mg/ml. INT-PP2A was purified as described above using FLAG-tagged PR65 nuclear extract with some additional modifications. Specifically, ~1mg of nuclear extract was used and incubated with 25μl of anti-FLAG agarose slurry and incubated as described. Beads were then washed four times in HEPES buffer (20mM HEPES pH7.4, 150mM KCl, 10% Glycerol, 0.1% NP-40) and resuspended in 20μl of pNPP Ser/Thr reaction buffer (50mM Tris-HCl, pH7 and 100μM CaCl2). Phospho-peptides (60μl at 1μg/μl) were individually mixed with purified INT-PP2A while still associated with the agarose (no elution) and further incubated at 30°C for 30 minutes. Following that incubation, 10μl of supernatant was removed and added to 40μl Malachite Green working solution (Sigma, #17–313) in a 384-well plate and incubated for 3 minutes at room temperature. The absorbance was measured at OD620 in Cytation5 reader (BioTek). Each experimental condition was done in triplicate. Phosphate standards were used to calculate pmoles produced. In experiments where PP2A inhibitors were used, the inhibitors were added while resuspending agarose in pNPP Ser/Thr reaction buffer.
Mass spectrometry sample digestion
The samples were prepared similar to as described (Anderson et al., 2020). Briefly, the agarose bead-bound proteins were washed several times with 50mM Triethylammonium bicarbonate (TEAB) pH 7.1, before being solubilized with 40μL of 5% SDS, 50mM TEAB, pH 7.55 followed by a room temperature incubation for 30 minutes. The supernatant containing the proteins of interest was then transferred to a new tube, reduced by making the solution 10mM Tris(2-carboxyethyl)phosphine (TCEP) (Thermo, #77720), and further incubated at 65°C for 10 minutes. The sample was then cooled to room temperature and 3.75 μL of 1M iodoacetamide acid was added and allowed to react for 20 minutes in the dark after which 0.5μL of 2M DTT was added to quench the reaction. Then, 5 μl of 12% phosphoric acid was then added to the 50μL protein solution followed by 350μL of binding buffer (90% Methanol, 100mM TEAB final; pH 7.1). The resulting solution was administered to an S-Trap spin column (Protifi, Farmingdale NY) and passed through the column using a bench top centrifuge (30 second spin at 4,000g). The spin column was then washed three times with 400μL of binding buffer and centrifuged (1200rpm, 1min). Trypsin (Promega, #V5280, Madison, WI) was then added to the protein mixture in a ratio of 1:25 in 50mM TEAB, pH=8, and incubated at 37°C for 4 hours. Peptides were eluted with 80uL of 50mM TEAB, followed by 80μL of 0.2% formic acid, and finally 80 μL of 50% acetonitrile, 0.2% formic acid. The combined peptide solution was then dried in a speed vacuum (room temperature, 1.5 hours) and resuspended in 2% acetonitrile, 0.1% formic acid, 97.9% water and aliquoted into an autosampler vial.
NanoLC MS/MS Analysis
Peptide mixtures were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS) using a nano-LC chromatography system (UltiMate 3000 RSLCnano, Dionex, Thermo Fisher Scientific, San Jose, CA). The nanoLC-MS/MS system was coupled on-line to a Thermo Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA) through a nanospray ion source (Thermo Scientific). A trap and elute method was used to desalt and concentrate the sample, while preserving the analytical column. The trap column (Thermo Scientific) was a C18 PepMap100 (300um X 5mm, 5um particle size) while the analytical column was an Acclaim PepMap 100 (75μm X 25 cm) (Thermo Scientific). After equilibrating the column in 98% solvent A (0.1% formic acid in water) and 2% solvent B (0.1% formic acid in acetonitrile (ACN)), the samples (2 μL in solvent A) were injected onto the trap column and subsequently eluted (400 nL/min) by gradient elution onto the C18 column as follows: isocratic at 2% B, 0–5 min; 2% to 32% B, 5–39 min; 32% to 70% B, 39–49 min; 70% to 90% B, 49–50 min; isocratic at 90% B, 50–54 min; 90% to 2%, 54–55 min; and isocratic at 2% B, until the 65 minute mark.
All LC-MS/MS data were acquired using XCalibur, version 2.1.0 (Thermo Fisher Scientific) in positive ion mode using a top speed data-dependent acquisition (DDA) method with a 3 second cycle time. The survey scans (m/z 350–1500) were acquired in the Orbitrap at 120,000 resolution (at m/z = 400) in profile mode, with a maximum injection time of 100 msec and an AGC target of 400,000 ions. The S-lens RF level was set to 60. Isolation was performed in the quadrupole with a 1.6 Da isolation window, and CID MS/MS acquisition was performed in profile mode using rapid scan rate with detection in the ion-trap using the following settings: parent threshold = 5,000; collision energy = 32%; maximum injection time 56 msec; AGC target 500,000 ions. Monoisotopic precursor selection (MIPS) and charge state filtering were on, with charge states 2–6 included. Dynamic exclusion was used to remove selected precursor ions, with a +/− 10 ppm mass tolerance, for 15 seconds after acquisition of one MS/MS spectrum.
Database Searching
Tandem mass spectra were extracted and charge state deconvoluted using Proteome Discoverer (Thermo Fisher, version 2.2.0388). Deisotoping was not performed. All MS/MS spectra were searched against a Uniprot Drosophila database (version 04–04-2018) using Sequest. Searches were performed with a parent ion tolerance of 5 ppm and a fragment ion tolerance of 0.60 Da. Trypsin was specified as the enzyme, allowing for two missed cleavages. Fixed modification of carbamidomethyl (C) and variable modifications of oxidation (M) and deamidation were specified in Sequest.
Yeast two-hybrid analysis
Yeast two-hybrid assays was carried out in PJ69–4α and PJ49–4a strains as described (Albrecht et al., 2018). In brief, Drosophila IntS1 through IntS14 was well as PR65 were cloned into pGAD or pOBD vectors using conventional cloning. Deletion or point mutants of dIntS8 and dPR65 were created from full length constructs using oligonucleotides described in Table_S1. pOBD plasmids were then transformed into PJ69–4α yeast and were selected on tryptophan-dropout medium (Clontech, #630413); pGAD plasmids were transformed into PJ49–4a yeast and were selected on leucine-dropout medium (Clontech, #630414). Yeast strains were then mated and subsequently selected on medium lacking both tryptophan and leucine. Interactions were tested through serial dilution (one to five) of diploid yeast followed by plating on medium lacking tryptophan and leucine or on medium lacking tryptophan, leucine, and histidine that also was supplemented with 5mM 3-amino-1,2,4-triazole.
RNA Interference (Drosophila cells)
Double-stranded RNAs targeting the C-terminal region of Drosophila IntS8 were generated by in vitro transcription of PCR templates containing the T7 promoter sequence on both ends using MEGAscript kit (Thermo, #AMB13345). Primer sequences are provided in Table_S1. For RNA interference experiments, 1.5 × 106/ml of DL1 cells were washed into serum free media and seeded into a 6-well plate along with 10 μg of dsRNA. After a 1-hour incubation, 2mls of complete growth medium was added followed by 60 hours of incubation before harvest. To perform rescue experiments while knocking down, cells were also treated with 100 μM CuSO4 throughout the 60 hours incubation period to induce expression of the RNAi-resistant FLAG-IntS8-WT or FLAG-IntS8-WFEF/A transgenes.
RNA Interference (Human cells)
INTS8 targeting and control siRNAs (7.5 μL each of 20 μM stock) were incubated in 250 μL of pre-warmed (room temperature) Opti-MEM I reduced serum medium (Gibco) for 5 minutes at room temperature. Similarly, RNAiMax (7.5 μL per well) was incubated in 250 μL pre-warmed Opti-MEM I reduced serum medium for 5 minutes. The siRNA and RNAiMAX dilutions were mixed and incubated for 30 minutes at room temperature. 1 × 106 293T cells were seeded into a 6-well plate and the prepared transfection mixes of INTS8 or control nontargeting siRNAs were added to the cells at a final concentration of 60nM. The cells were expanded into 10 cm plates after 24 hours of incubation under standard mammalian cell culture conditions. The cells were further incubated for 24 hours before harvest.
PP2A inhibition
DL1 (2X106/well) and 293T (6×105/well) were seeded in 12-well plates in standard growth medium. Calyculin A and Phendione were dissolved in DMSO to make stock solutions of 20 μM and 20 mM, respectively. Four hours after initial cell seeding, an equal volume of medium containing 40 nM Calyculin A or 40 μM Phendione were added resulting in a further two-fold dilution of each drug. Cells were incubated for an additional 24hr before harvesting for RNA extraction. The same volume of DMSO was added in control wells.
RT-qPCR
Total RNA was isolated using Trizol and cDNA was reverse transcribed using Superscript III Reverse Transcriptase (Thermo, #18080085) according to the manufacturer’s instructions. Random hexamers were used for cDNA synthesis and RT-qPCR was then carried out in triplicate using Bio-Rad iTaq Universal SYBR Green Supermix (Bio-Rad, #1725120) and measured in CFX Connect Real-Time System (Bio-Rad). All RT-qPCR primers are provided in Table_S1.
CRISPR genomic editing
To make 293T cells harboring a homozygous, genomically-encoded N-terminal FLAG tag within an Integrator subunit or PR65 genes individually, CRISPR/Cas9 was used to precisely incorporate FLAG sequence (GAT TAC AAG GAT GAC GAC GAT AAG) as previously described (Baillat et al., 2016). In brief, a 100bp genomic sequence flanking the translation start site was input into the CRISPOR program for gRNAs prediction (http://crispor.tefor.net/crispor.py) (Haeussler et al., 2016). gRNAs were selected based upon 1) a preferred minimal distance between the Cas9 cutting site (NGG is PAM motif) and the FLAG insertion site. 2) the specificity score judging from the number of off targets. Selected sgRNA (Table_S1) were then cloned into pGL3-U6-sgRNA-PGK-puromycin using an annealed oligonucleotide strategy. For single stranded DNA donor template design, 200bp of genomic sequence (including 24 bases of the FLAG sequence in the middle) surrounding the translation start site were synthesized (Integrated DNA Technologies, IA). The PAM motif within the donor template was silently mutated to avoid iterative cleavage by the transfected sgRNA and Cas9. Equal amounts of sgRNA and Cas9 (addgene, #44758) plasmids (720ng in total) were mixed with 10pmole (~660ng) of donor template and transfected into 293T cell with Lipofectamine 2000 according to manufacturer’s instructions (Thermo, #11668019). Selection (10μg/ml Blasticidin and 1μg/ml Puromycin) was started 24 hours after transfection and occurred for a total of 48 hours. Cells were then expanded in normal growth medium without antibiotics. Western Blotting was performed to verify the FLAG signal from pools of transfected cells and clonal lines were selected followed by additional Western Blotting using anti-FLAG antibodies. Genomic DNA was extracted from selected clonal cell lines that displayed a positive FLAG signal using Western Blot analysis. PCR was then performed to amplify the genomic region containing FLAG sequence (Table_S1), and the product was resolved on agarose gel to check homogeneity of FLAG insertion on all alleles of the target gene. Ultimately, all PCR products were cloned and sequenced to confirm identity.
QUANTIFICATION AND STATISTICAL ANALYSIS
RT-qPCR quantification and analysis
Data were analyzed using the ΔΔCt method with Rps17 as the reference gene and LacZ dsRNA-treated cells as the control, as described previously (Ezzeddine et al., 2011). Results presented in Figure S1 and S3 are shown from biologically independent replicates, depicting averages and standard deviations (mean +/− SD, N=3).
Generation of Transcript Annotations
Transcript annotations used here were the same as in (Elrod et al., 2019). Briefly, all transcript annotations for D. melanogaster r5.57 were downloaded from flybase.org in GTF format and filtered such that only “exon” entries for the feature types considered for re-annotation remained. Annotations from chrY, chrM, and random chromosomes were also excluded. Unique “gene_id” values were assigned to each transcript, such that those grouped and represented by a single member in TSS-based analyses were identical. Precise TSS locations employed were based on high-resolution Start-seq data as described previously (Henriques et al., 2013; Henriques et al., 2018; Nechaev et al., 2010). The start location of each transcript was adjusted to the observed TSS from Start-seq when this resulted in truncation, rather than extension of the model. If the observed TSS fell within an intron, all preceding exons were removed, and the transcript start was set to the beginning of the following downstream exon.
Metagene analysis
Metagene plots were generated by summing reads at each indicated position with respect to the TSS and dividing by the number of TSSs within each group. Values were graphed across a range of distances as indicated in figure legends. Heatmaps were generated using Partek Genomics Suite version 6.16.0812.
RNA-seq library generation and mapping (Drosophila DL1 cells)
DL1 cells were treated for 60 hours with a control dsRNA (targeting β-galactosidase) or a dsRNA targeting IntS8 (see RNAi details above). For the samples expressing exogenous FLAG-IntS8-WT, FLAG-IntS8-WFEF/A, or an empty vector control; CuSO4 was added to 200 uM final concentration 24 hours prior to harvest. Total RNA was isolated with Trizol (Thermo, #15596026) following manufacturer’s instructions. RNA quality was confirmed with a BioAnalyzer (Agilent). Using Oligo d(T)25 Magnetic Beads (NEB, #S1419S), polyA+ RNA from 2.5 μg of total RNA was enriched and then RNA-seq libraries (3 independent biological replicates per condition) prepared with the Click-seq library preparation method using a 1:35 azido-nucleotide ratio (Jaworski and Routh, 2018). Libraries were sequenced using a single-end 75 bp cycle run on an Illumina NextSeq 500.
Reads were filtered to require a mean quality score ≥ 20, trimmed to 50 nt, and mapped to the dm3 Drosophila genome assembly using STAR. Default parameters were used, except that multimappers were randomly assigned (outMultimapperOrder Random), spurious junctions were filtered (outFilterType BySJout), minimum overhang for non-annotated junctions was set to 8 nt (alignSJoverhangMin 8), and non-canonical alignments were removed (outFilterIntronMotifs RemoveNoncanonicalUnannotated). The total number of RNA-seq reads aligned in the control (beta-galactosidase), IntS8-dep., or rescue samples is described below (upper table for Figure 1, lower table for Figure 3):
| Sample | Total Reads | Mappable Fragments (Percent of total) | Agreement between replicates (Spearman’s rho) | Depth Normalization Factor |
|---|---|---|---|---|
| Control No Cu | 65,094,896 | 71.61% | >0.97 | 1.187186 |
| IntS8-dep. No Cu | 61,078,878 | 72.46% | >0.97 | 0.9825324 |
| Sample | Total Reads | Mappable Fragments (Percent of total) | Agreement between replicates (Spearman’s rho) | Normalization Factor |
|---|---|---|---|---|
| Control | 69,412,709 | 90.08% | >0.97 | 0.993314358 |
| IntS8-dep. | 62,360,768 | 89.36% | >0.97 | 1.105641298 |
| IntS8-dep.+FLAG-IntS8-WT | 60,765,646 | 90.02% | >0.98 | 1.134664815 |
| IntS8-dep.+FLAG-IntS8-WFEF/A | 83,255,439 | 91.57% | >0.96 | 0.828157792 |
Differentially expressed genes in RNA-seq
Read counts were calculated on a per-gene basis in a strand-specific manner using featureCounts (Liao et al., 2014) in R version 3.6.1. Differentially expressed genes were identified using DESeq2 (Love et al., 2014). For Control versus IntS8-dep. comparisons, an adjusted p-value threshold of < 0.0001 and fold change of > 1.5 was used. This revealed 1099 (out of 9303) genes to be upregulated and 182 to be downregulated.
MISO Analysis
Mixture of Isoform analysis (MISO) (Katz et al., 2010) was performed following the directions for an exon-centric analysis on the documents section of the program’s website (https://miso.readthedocs.io/en/fastmiso/). Differential expression was compared between control (β-galactosidase dsRNA-treated) and IntS8-depleted RNA-seq BAM files for retained introns, skipped exons, alternative 5’ splice sites, alternative 3’ splice sites, and mutually exclusive exons using the Drosophila annotations described above. Results were filtered using the developer’s suggested default settings to retain only events with: (a) ≥ 10 inclusion reads, (b) 10 exclusion reads such that (c) the sum of inclusion and exclusion reads is ≥ 30, and (d) the ΔΨ ≥ 0.25 with a (e) Bayes factor ≥ 20, and (a)-(e) true in one of the samples. Using this filter, locations of alternative splicing events were compared to Flybase annotated chromosomal regions using the UCSC Genome Browser table browser to identify the FBgnIDs of affected genes. The number of changes in splicing events are described below.
| Splicing Event Type | Events Compared | Events Passing Filter | Percent Events Passing Filter |
|---|---|---|---|
| Retained Intron | 20213 | 518 | 2.56% |
| Alternative 5’SS | 3191 | 109 | 3.42% |
| Alternative 3’SS | 1566 | 66 | 4.21% |
| Skipped Exon | 1386 | 44 | 3.17% |
All Flybase genes that included any splicing event that passed filter in MISO were removed from the list of active genes, such that a total of 7472 active genes were investigated for the effects of IntS8 depletion.
RNA-seq library generation and mapping (Human 293T cells)
293T cells were treated for 48 hours with either control or INTS8 targeting siRNAs as described above. Cells were harvested and washed with PBS. Cells were counted, and 1 × 106 cells were resuspended in 1mL of Trizol and spiked with 1 μL of 1:10 diluted ERCC Spike-in Mix (Invitrogen) and RNA was purified. 1 μg of RNA from each sample was diluted in 10uL of water. The input RNA was subjected to RNA-seq library generation using the TruSeq Stranded Total RNA sequencing kit with RiboZero rRNA depletion (Illumina). The final libraries were amplified to 10 cycles and purified using AMPure XP beads (Beckman Coulter). Pooled libraries (10nM) were sequenced (PE150) on the HiSeq platform at Novogene.
Reads were filtered to require a mean quality score ≥ 20, trimmed to 100 nt, and mapped to the hg38 human genome assembly using STAR. Default parameters were used, except that multimappers were randomly assigned (outMultimapperOrder Random), spurious junctions were filtered (outFilterType BySJout), minimum overhang for non-annotated junctions was set to 8 nt (alignSJoverhangMin 8), and non-canonical alignments were removed (outFilterIntronMotifs RemoveNoncanonicalUnannotated).
The total number of RNA-seq reads aligned in the control or INTS8-dep. samples is described below:
| Sample | Total Reads | Mappable Fragments (Percent of total) | Agreement between replicates (Spearman’s rho) |
|---|---|---|---|
| Control | 271,095,957 | 83.58% | >0.99 |
| INTS8-dep. | 242,045,837 | 85.27% | >0.99 |
Differentially expressed genes in 293T RNA-seq
To account for variation in ERCC spike recovery and read depth, the following normalization factors were applied in DEseq2:
| Raw Reads | Reads Uniquely Mapping to hg38 | Reads Mapping to ERCC | Spike Normalization Ratio | |
|---|---|---|---|---|
| Control Rep 1 | 109,669,541 | 92,512,073 | 71,836 | 0.53755777 |
| Control Rep 2 | 89,600,985 | 74,150,258 | 51,433 | 0.750802014 |
| Control Rep 3 | 71,825,431 | 60,050,506 | 41,881 | 0.922041021 |
| INTS8-dep. Rep 1 | 79,839,087 | 69,074,139 | 42,029 | 0.918794166 |
| INTS8-dep. Rep 2 | 85,448,446 | 73,303,454 | 42,240 | 0.914204545 |
| INTS8-dep. Rep 3 | 76,758,304 | 64,109,089 | 38,616 | 1 |
For control versus INTS8-dep. comparisons, an adjusted p-value threshold of < 0.001 and fold change of > 2 was used. This revealed 420 (out of 12059) protein-coding genes to be upregulated and 53 to be downregulated.
UCSC Genome Browser tracks representing read coverage were generated from the combined replicates per condition. Here, the following condition-level normalization factors were applied: Control: 0.74; INTS8-dep.: 0.94.
PRO-seq library preparation and data analysis
DL1 cells that were treated with control (β-galactosidase-targeting dsRNA) or IntS8 targeting dsRNA for 60 hours and rescued with either RNAi-resistant FLAG-IntS8-WT or FLAG-IntS8-WFEF/A (200 μM CuSO4 treatment for 24hours) were permeabilized as described below. All procedures were performed on ice unless otherwise noted. Cells were washed once in ice-cold 1x PBS and resuspended in Buffer W (10 mM Tris-HCl pH 8.0, 10% glycerol, 250mM sucrose, 10 mM KCl, 5 mM MgCl2, 0.5 mM DTT, protease inhibitors cocktail (Roche), and 4 u/mL RNase inhibitor [SUPERaseIN, Ambion]) at a cell density of 2 × 107 cells/mL. 9x volume of Buffer P (10 mM Tris-HCl pH 8.0, 10% glycerol, 250 mM sucrose, 10 mM KCl, 5 mM MgCl2, 0.5 mM DTT, 0.1% Igepal, protease inhibitors cocktail (Roche), 4 u/mL RNase inhibitor [SUPERaseIN, Ambion]) was then immediately added. Cells were gently resuspended and incubated for up to 2 minutes on ice. Cells were then recovered by centrifugation (800g for 4 min) and washed in Buffer F (50 mM Tris-HCl pH 8.0, 40% glycerol, 5 mM MgCl2, 0.5 mM DTT, 4 u/mL RNase inhibitor [SUPERaseIN, Ambion]). Washed permeabilized cells were finally resuspended in Buffer F at a density of 1 × 106 cells/30 μL and immediately frozen in liquid nitrogen. Permeabilized cells were stored in −80°C until usage.
PRO-seq run-on reactions were performed by adding 1 × 106 permeabilized DL1 cells (spiked with 5 × 104 permeabilized human HEK293T cells) to the same volume of 2x Nuclear Run-On reaction mixture (10 mM Tris-HCl pH 8.0, 300 mM KCl, 1% Sarkosyl, 5 mM MgCl2, 1 mM DTT, 200 μM biotin-11-A/C/G/UTP (Perkin-Elmer), 0.8 u/μL SUPERaseIN inhibitor [Ambion]) and incubating for 5 min at 30°C. Nascent RNA was extracted using a Total RNA Purification Kit following the manufacturer’s instructions (Norgen Biotek Corp.), and was followed by fragmentation by base hydrolysis in 0.25 N NaOH on ice for 9 min and neutralized by adding 1x volume of 1 M Tris-HCl pH 6.8. Fragmented nascent RNA was bound to 30 μL of Streptavidin M-280 magnetic beads (Thermo Fisher Scientific) in Binding Buffer (300 mM NaCl, 10 mM Tris-HCl pH 7.4, 0.1% Triton X-100). The beads were washed twice with high salt buffer (2 M NaCl, 50 mM Tris-HCl pH 7.4, 0.5% Triton X-100), twice with Binding buffer, and twice with Low salt buffer (5 mM Tris-HCl pH 7.4, 0.1% Triton X-100). Bound RNA was extracted from the beads using Trizol (Invitrogen) followed by ethanol precipitation.
3’ adaptor ligation was performed by dissolving the fragmented nascent RNA in water and incubating with 10 pmol of reverse 3’ RNA adaptor (/5Phos/rGrArUrCrGrUrCrGrGrArCrUrGrUrArGrArArCrUrCrUrGrArArC/3InvdT/) and T4 RNA ligase I (NEB) for 2 hours at room temperature under conditions recommended by manufacturer. Another round of Streptavidin bead binding was performed to enrich for biotin-labeled product. The beads were washed twice each with High, Binding, and Low salt buffer and once with 1x Thermo Pol Buffer (NEB). The 5’ ends were decapped by treating the RNA products with RNA 5’ Pyrophosphohydrolase (RppH, NEB) at 37°C for 1 hour followed by one wash of High, Low, and T4 PNK Buffer. To repair 5’ ends, the RNA products were treated with Polynucleotide Kinase (PNK, NEB) at 37°C for 1 hr.
The 5’ repaired RNA was then ligated to reverse 5’ RNA adaptor (5’-rCrCrUrUrGrGrCrArCrCrCrGrArGrArArUrUrCrCrA-3’) with T4 RNA ligase I (NEB) at room temperature for 2 hours under conditions suggested by manufacturer. Ligation was followed by two washes each of High, Binding, and Low salt buffers and one wash in 0.25X FSS Buffer 9Thermo Fisher Scientific). The products were reverse transcribed using 25 pmol RT primer (5’-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3’) for TRU-seq barcodes (RP1 primer, Illumina). RT products were eluted from beads by two incubations at 95°C for 30 seconds. The optimal number of PCR cycles was determined by using a portion of the RT product in a round of test amplification. For the final amplification, 12.5 pmol of RPI-index primer (for TRU-seq barcodes, Illumina) and RT primer was added to the RT product with Q5 polymerase (NEB) under standard PCR conditions. The product was amplified for 11–12 cycles and size selected using ProNex Size-Selective Purification System (Promega). The final products were sequenced on NextSeq 500 in a high output 150 bp cycle run.
Paired-end reads were trimmed to 40nt, for adaptor sequence and low quality 3’ ends using cutadapt 1.14, discarding those containing reads shorter than 20nt (-m 20 –q 10), to allow successful alignment with Bowtie 1.2.2 (Langmead et al., 2009). Remaining pairs were paired-end aligned to the hg38 genome index to determine spike-normalization ratios based on uniquely mapped reads. Mappable pairs were omitted from subsequent analysis, and unmapped pairs were aligned to the dm3 genome assembly. Identical parameters were used in each alignment described above: up to 2 mismatches, and unmappable pairs routed to separate output files (-v2, -un, --best). Pairs mapping to dm3, representing biotin-labeled RNA 3’ ends, were separated, and strand-specific counts of the 3’ mapping positions determined at single nucleotide resolution, genome-wide, and expressed in bedGraph format with “plus” and “minus” strand labels swapped for each 3’ bedGraph, to correct for the “forward/reverse” nature of Illumina paired-end sequencing (Mahat et al., 2016). Based on similar recovery of spike-in reads, depth normalization was performed and bedGraphs from replicates of each condition were combined by summing counts per nucleotide.
| Sample | Total Raw Reads | Mapped Reads | Agreement between replicates (Spearman’s rho) | Depth normalization factor |
|---|---|---|---|---|
| Control | 81,944,375 | 42,043,220 | > 0.97 | 1 |
| IntS8-dep. | 98,159,133 | 66,830,046 | > 0.99 | 0.629 |
| IntS8-dep+FLAG-IntS8-WT | 98,229,493 | 70,678,471 | > 0.99 | 0.595 |
| IntS8-dep.+FLAG-IntS8-WFEF/A | 97,356,999 | 59,136,534 | > 0.99 | 0.711 |
Normalization was performed after combining the two replicates, adjusting for library depth. Bigwigs and bedgraphs used for generating UCSC Genome Browser tracks and metagene analyses represent the 3’ end of each mapped read.
Statistical tests
For RNA-seq and PRO-seq, statistical significance was determined using Mann-Whitney pairwise tests unless otherwise noted. The details of violin plots, statistical tests and error bars are explained in their respective figure legends.
Supplementary Material
The oligonucleotide sequences used for RT-qPCR, ChIP-qPCR, plasmid cloning, dsRNA synthesis and siRNA targeting are provided.
List of genes that were identified as upregulated (N=1099) or downregulated (N=182) in RNA-seq experiments performed on Control and IntS8-depleted Drosophila DL1 cells. The threshold used to define INTS8-affected genes was p<0.001 and fold change >2. Mean normalized counts in cells treated with control or siRNA targeting IntS8 is shown for each gene, along with the corresponding adjusted p value. An additional column indicates the 441 upregulated genes that are within the top decile of pausing indices. For this analysis, the Pausing Index was calculated as the PRO-seq signal density from the TSS to +150 nt, divided by the signal density from +250 to +2250 downstream of the TSS, using data from control cells.
List of genes that were identified as upregulated (N=420) or downregulated (N=53) in RNA-seq experiments performed on Control and INTS8-depleted 293T cells. The threshold used to define INTS8-affected genes was p<0.001 and fold change >2. Mean normalized counts in cells treated with control or siRNA targeting INTS8 is shown for each gene, along with the corresponding adjusted p value.
Highlights:
A short motif in IntS8 mediates association with Protein Phosphatase 2A (PP2A)
Recruitment of PP2A is necessary for Integrator-mediated gene repression
Integrator-bound PP2A dephosphorylates residues within the Pol II CTD and Spt5
PP2A antagonizes transcriptional kinases to prevent productive elongation
ACKNOWLEDGEMENTS
We thank the BPF Next-Gen Sequencing Core Facility at Harvard Medical School and members of the Wagner and Adelman labs for helpful discussions. This work was supported by The Cancer Prevention Research Institute of Texas (CPRIT) to K.L.H., National Institutes of Health grant R01-GM134539 (K.A. and E.J.W.), the Intramural Research Program of the NIH, National Institutes of Environmental Health Sciences to K.A. (Z01ES101987), the Welch Foundation grant H-1889–20180324 to The University of Texas Medical Branch at Galveston (E.J.W.). The UTMB Mass Spectrometry Facility is supported in part by CPRIT grant number RP190682 (W.K.R).
Footnotes
DECLARATION OF INTERESTS
K.A. and E.J.W. consult for Syros Pharmaceuticals and K.A. is on scientific advisory board of CAMP4 Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, and Ansari AZ (2009). TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell 34, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht TR, Shevtsov SP, Wu Y, Mascibroda LG, Peart NJ, Huang KL, Sawyer IA, Tong L, Dundr M, and Wagner EJ (2018). Integrator subunit 4 is a ‘Symplekin-like’ scaffold that associates with INTS9/11 to form the Integrator cleavage module. Nucleic Acids Res 46, 4241–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht TR, and Wagner EJ (2012). snRNA 3’ end formation requires heterodimeric association of integrator subunits. Mol Cell Biol 32, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AP, Luo X, Russell W, and Yin YW (2020). Oxidative damage diminishes mitochondrial DNA polymerase replication fidelity. Nucleic Acids Res 48, 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, and Shiekhattar R. (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276. [DOI] [PubMed] [Google Scholar]
- Baillat D, Russell WK, and Wagner EJ (2016). CRISPR-Cas9 mediated genetic engineering for the purification of the endogenous integrator complex from mammalian cells. Protein Expr Purif 128, 101–108. [DOI] [PubMed] [Google Scholar]
- Baillat D, and Wagner EJ (2015). Integrator: surprisingly diverse functions in gene expression. Trends Biochem Sci 40, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. (2012). mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell 46, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ezzeddine N, Waltenspiel B, Albrecht TR, Warren WD, Marzluff WF, and Wagner EJ (2012). An RNAi screen identifies additional members of the Drosophila Integrator complex and a requirement for cyclin C/Cdk8 in snRNA 3’-end formation. RNA 18, 2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Waltenspiel B, Warren WD, and Wagner EJ (2013). Functional analysis of the integrator subunit 12 identifies a microdomain that mediates activation of the Drosophila integrator complex. J Biol Chem 288, 4867–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, and Price DH (2007). Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem 282, 21901–21912. [DOI] [PubMed] [Google Scholar]
- Cheng L, Zhang Q, Yang S, Yang Y, Zhang W, Gao H, Deng X, and Zhang Q. (2013). A 4-gene panel as a marker at chromosome 8q in Asian gastric cancer patients. Genomics 102, 323–330. [DOI] [PubMed] [Google Scholar]
- Cho US, and Xu W. (2007). Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57. [DOI] [PubMed] [Google Scholar]
- Cohen P, Klumpp S, and Schelling DL (1989). An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett 250, 596–600. [DOI] [PubMed] [Google Scholar]
- Core L, and Adelman K. (2019). Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev 33, 960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar MA, Sheridan RM, Erickson B, Fong N, Glover-Cutter K, Brannan K, and Bentley DL (2019). Control of RNA Pol II Speed by PNUTS-PP1 and Spt5 Dephosphorylation Facilitates Termination by a “Sitting Duck Torpedo” Mechanism. Mol Cell 76, 896–908 e894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czudnochowski N, Bosken CA, and Geyer M. (2012). Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition. Nat Commun 3, 842. [DOI] [PubMed] [Google Scholar]
- Eaton JD, Davidson L, Bauer DLV, Natsume T, Kanemaki MT, and West S. (2018). Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev 32, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Francis L, Davidson L, and West S. (2020). A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev 34, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara H, Yokoyama T, Shigematsu H, Yokoyama S, Shirouzu M, and Sekine SI (2017). Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357, 921–924. [DOI] [PubMed] [Google Scholar]
- Ehrensberger AH, Kelly GP, and Svejstrup JQ (2013). Mechanistic interpretation of promoter-proximal peaks and RNAPII density maps. Cell 154, 713–715. [DOI] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K. (2019). The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol Cell 76, 738–752 e737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzeddine N, Chen J, Waltenspiel B, Burch B, Albrecht T, Zhuo M, Warren WD, Marzluff WF, and Wagner EJ (2011). A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3’-end formation. Mol Cell Biol 31, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrega C, Hausmann S, Shen V, Shuman S, and Lima CD (2004). Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell 13, 77–89. [DOI] [PubMed] [Google Scholar]
- Federico A, Rienzo M, Abbondanza C, Costa V, Ciccodicola A, and Casamassimi A. (2017). Pan-Cancer Mutational and Transcriptional Analysis of the Integrator Complex. Int J Mol Sci 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R. (2014). Integrator Regulates Transcriptional Initiation and Pause Release following Activation. Mol Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geladopoulos TP, Sotiroudis TG, and Evangelopoulos AE (1991). A malachite green colorimetric assay for protein phosphatase activity. Anal Biochem 192, 112–116. [DOI] [PubMed] [Google Scholar]
- Haberle V, and Stark A. (2018). Eukaryotic core promoters and the functional basis of transcription initiation. Nat Rev Mol Cell Biol 19, 621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K. (2013). Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K. (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz EPT, Kruse T, Davey NE, Lopez-Mendez B, Sigurethsson JO, Montoya G, Olsen JV, and Nilsson J. (2016). A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol Cell 63, 686–695. [DOI] [PubMed] [Google Scholar]
- Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, et al. (2012). Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. (1989). Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun 159, 871–877. [DOI] [PubMed] [Google Scholar]
- Jaworski E, and Routh A. (2018). ClickSeq: Replacing Fragmentation and Enzymatic Ligation with Click-Chemistry to Prevent Sequence Chimeras. Methods Mol Biol 1712, 71–85. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kecman T, Kus K, Heo DH, Duckett K, Birot A, Liberatori S, Mohammed S, Geis-Asteggiante L, Robinson CV, and Vasiljeva L. (2018). Elongation/Termination Factor Exchange Mediated by PP1 Phosphatase Orchestrates Transcription Termination. Cell Rep 25, 259–269 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ahn SH, Krogan NJ, Greenblatt JF, and Buratowski S. (2004). Transitions in RNA polymerase II elongation complexes at the 3’ ends of genes. EMBO J 23, 354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, and Schubeler D. (2017). Genome-wide Single-Molecule Footprinting Reveals High RNA Polymerase II Turnover at Paused Promoters. Mol Cell 67, 411–422 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R. (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lis JT, Mason P, Peng J, Price DH, and Werner J. (2000). P-TEFb kinase recruitment and function at heat shock loci. Genes Dev 14, 792–803. [PMC free article] [PubMed] [Google Scholar]
- Logan J, Falck-Pedersen E, Darnell JE Jr., and Shenk T. (1987). A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouse beta maj-globin gene. Proc Natl Acad Sci U S A 84, 8306–8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, Ding C, Shi Y, Yucer N, Krenciute G, et al. (2011). Analysis of the human endogenous coregulator complexome. Cell 145, 787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malovannaya A, Li Y, Bulynko Y, Jung SY, Wang Y, Lanz RB, O’Malley BW, and Qin J. (2010). Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc Natl Acad Sci U S A 107, 2431–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, and Tong L. (2006). Polyadenylation factor CPSF-73 is the pre-mRNA 3’-end-processing endonuclease. Nature 444, 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, and Adelman K. (2010). Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327, 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema R, Baillat D, Schot R, van Unen LM, Brooks A, Kia SK, Hoogeboom AJM, Xia Z, Li W, Cesaroni M, et al. (2017). Human mutations in integrator complex subunits link transcriptome integrity to brain development. PLoS Genet 13, e1006809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Booth GT, Sanso M, Benjamin B, Tanny JC, Lis JT, and Fisher RP (2018). A Cdk9-PP1 switch regulates the elongation-termination transition of RNA polymerase II. Nature 558, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, and Price DH (2006). Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23, 297–305. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ (2016). Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 352, aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath K, Staubli ML, Marti S, Leitner A, Moes M, and Jonas S. (2020). INTS10-INTS13-INTS14 form a functional module of Integrator that binds nucleic acids and the cleavage module. Nat Commun 11, 3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury S, Bernecky C, and Cramer P. (2015). Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol 16, 129–143. [DOI] [PubMed] [Google Scholar]
- Sanso M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. (2016). P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev 30, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreieck A, Easter AD, Etzold S, Wiederhold K, Lidschreiber M, Cramer P, and Passmore LA (2014). RNA polymerase II termination involves C-terminal-domain tyrosine dephosphorylation by CPF subunit Glc7. Nat Struct Mol Biol 21, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshacharyulu P, Pandey P, Datta K, and Batra SK (2013). Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 335, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. [DOI] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3’ processing complex. Mol Cell 33, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson HM, Khan RZ, Song C, Sharma D, Sadashivaiah K, Furusawa A, Liu X, Nagaraj S, Sengamalay N, Sadzewicz L, et al. (2015). Concurrent Mutations in ATM and Genes Associated with Common gamma Chain Signaling in Peripheral T Cell Lymphoma. PLoS One 10, e0141906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Ferris AL, Wu X, Saraf A, Khanna KK, Florens L, Washburn MP, Hughes SH, and Pagano M. (2015). The Integrator complex controls the termination of transcription at diverse classes of gene targets. Cell Res 25, 288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmayer B, Micas G, Gamot A, Martin P, Malirat N, Koval S, Raffel R, Sobhian B, Severac D, Rialle S, et al. (2014). Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at coding genes. Nat Commun 5, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev 33, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P. (2018a).Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560, 607–612. [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Urlaub H, and Cramer P. (2018b). Structure of paused transcription complex Pol II-DSIF-NELF. Nature 560, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, Winston F, et al. (1998a). DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev 12, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Watanabe D, and Handa H. (1998b). Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 17, 7395–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bajaj R, Bollen M, Peti W, and Page R. (2016). Expanding the PP2A Interactome by Defining a B56- Specific SLiM. Structure 24, 2174–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, Gromak N, and Proudfoot NJ (2004). Human 5’ --> 3’ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432, 522–525. [DOI] [PubMed] [Google Scholar]
- Whitelaw E, and Proudfoot N. (1986). Alpha-thalassaemia caused by a poly(A) site mutation reveals that transcriptional termination is linked to 3’ end processing in the human alpha 2 globin gene. EMBO J 5, 2915–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Chen Y, Zhang P, Jeffrey PD, and Shi Y. (2008). Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell 31, 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav L, Tamene F, Goos H, van Drogen A, Katainen R, Aebersold R, Gstaiger M, and Varjosalo M. (2017). Systematic Analysis of Human Protein Phosphatase Interactions and Dynamics. Cell Syst 4, 430–444 e435. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, and Handa H. (2006). P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21, 227–237. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, and Handa H. (1999). NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97, 41–51. [DOI] [PubMed] [Google Scholar]
- Yue J, Lai F, Beckedorff F, Zhang A, Pastori C, and Shiekhattar R. (2017). Integrator orchestrates RAS/ERK1/2 signaling transcriptional programs. Genes Dev 31, 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Vendramin R, Liu F, Lopez O, Valencia MG, Gomes Dos Santos H, Gaidosh G, Beckedorff F, Blumenthal E, Speroni L, et al. (2020). Targeted chemotherapy overcomes drug resistance in melanoma. Genes Dev 34, 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li T, and Price DH (2012). RNA polymerase II elongation control. Annu Rev Biochem 81, 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The oligonucleotide sequences used for RT-qPCR, ChIP-qPCR, plasmid cloning, dsRNA synthesis and siRNA targeting are provided.
List of genes that were identified as upregulated (N=1099) or downregulated (N=182) in RNA-seq experiments performed on Control and IntS8-depleted Drosophila DL1 cells. The threshold used to define INTS8-affected genes was p<0.001 and fold change >2. Mean normalized counts in cells treated with control or siRNA targeting IntS8 is shown for each gene, along with the corresponding adjusted p value. An additional column indicates the 441 upregulated genes that are within the top decile of pausing indices. For this analysis, the Pausing Index was calculated as the PRO-seq signal density from the TSS to +150 nt, divided by the signal density from +250 to +2250 downstream of the TSS, using data from control cells.
List of genes that were identified as upregulated (N=420) or downregulated (N=53) in RNA-seq experiments performed on Control and INTS8-depleted 293T cells. The threshold used to define INTS8-affected genes was p<0.001 and fold change >2. Mean normalized counts in cells treated with control or siRNA targeting INTS8 is shown for each gene, along with the corresponding adjusted p value.
Data Availability Statement
GEO Accession numbers: all datasets generated in this study are available for download from GEO: GSE150844. Original image data have been deposited to Mendeley Data: http://dx.doi.org/10.17632/bt76mktdfx.1