

Abstract
Among targets selected for studies aimed at identifying potential inhibitors against COVID-19, SARS-CoV2 main proteinase (Mpro) is highlighted. Mpro is indispensable for virus replication and is a promising target of potential inhibitors of COVID-19. Recently, monomeric SARS-CoV2 Mpro, drug repurposing, and docking methods have facilitated the identification of several potential inhibitors. Results were refined through the assessment of dimeric SARS-CoV2 Mpro, which represents the functional state of enzyme. Docking and molecular dynamics (MD) simulations combined with molecular mechanics/generalized Born surface area (MM/GBSA) studies indicated that dimeric Mpro most significantly impacts binding affinity tendency compared with the monomeric state, which suggests that dimeric state is most useful when performing studies aimed at identifying drugs targeting Mpro. In this study, we extend previous research by performing docking and MD simulation studies coupled with an MM/GBSA approach to assess binding of dimeric SARS-CoV2 Mpro to 12 FDA-approved drugs (darunavir, indinavir, saquinavir, tipranavir, diosmin, hesperidin, rutin, raltegravir, velpatasvir, ledipasvir, rosuvastatin, and bortezomib), which were identified as the best candidates for the treatment of COVID-19 in some previous dockings studies involving monomeric SARS-CoV2 Mpro. This analysis identified saquinavir as a potent inhibitor of dimeric SARS-CoV2 Mpro; therefore, the compound may have clinical utility against COVID-19.
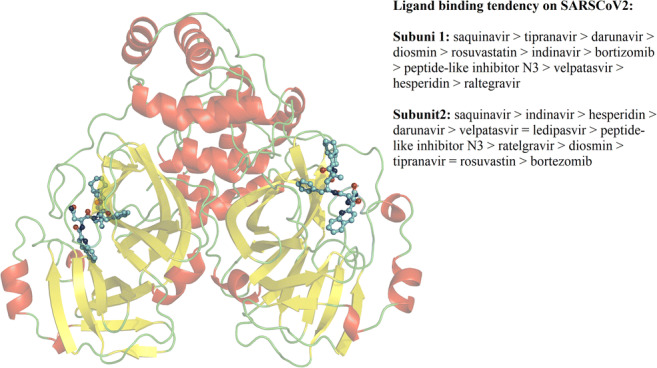
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00894-020-04600-4.
Keywords: Protease, SARS-CoV2, Docking, MD simulations
Introduction
The outbreak of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) was first reported on December 30, 2019, in Wuhan China [1]. SARS-CoV2 belongs to the beta coronavirus group and is similar to SARS-coronaviruses. Despite sequence diversity, its spike protein binds strongly to the human ACE2 receptor [1]. The disease caused by SARS-CoV2 was named coronavirus disease 2019 (COVID-19) by World Health Organization and represents a grave menace to global public health and local economies. As of May 2, 2020, over 3,362,778 cases of COVID-19 have been reported in 187 countries and have caused 239,227 total deaths (https://coronavirus.jhu.edu/map.html). Therefore, we must urgently identify effective, available, and affordable drugs to treat COVID-19 to reduce the toll of the epidemic.
A potential treatment for COVID-19 involves combining two HIV-1 protease inhibitors (lopinavir and ritonavir), which have been useful against severe acute respiratory syndrome coronavirus (SARS-CoV) [2] via targeting SARS-CoV main proteinase (SARS-CoV Mpro) [3]. SARS-CoV Mpro mediates replicase polyprotein proteolytic activity, which is essential for viral replication, and represents an important target for reducing the impact of COVID-19. Alignment of SARS-CoV Mpro and SARS-CoV2 main proteinase showed that both proteins share up to 95% sequence identity, suggesting that SARS-CoV Mpro inhibitors may function similarly against SARS-CoV2 Mpro. Theoretical methods repurposed from studies of SARS-CoV Mpro have been used to identify FDA-approved small ligands that are potential inhibitors of monomeric SARS-CoV2 Mpro [4–8]. Additional theoretical studies compare binding to dimeric forms of SARS-CoV Mpro and SARS-CoV2 Mpro, which is the active form of the enzyme [9]. This highlighted the differences between the binding affinities of monomeric versus dimeric forms of SARS-CoV Mpro or SARS-CoV2 Mpro, and revealed that praziquantel (first option) and perampanel (second option) may be useful for treating COVID-19 via inhibition of SARS-CoV-2 Mpro [10]. In addition, previous work demonstrated that docking and MD simulations should be performed using homodimeric, rather than monomeric, SARS-CoV2 Mpro.
Here, we elaborate upon previous research by performing docking and MD simulation studies coupled with the molecular mechanics/generalized Born surface area (MM/GBSA) to assess binding of dimeric SARS-CoV2 Mpro to 12 FDA-approved drugs, which were identified as being the most attractive candidates for treatment of COVID-19 in previous studies that virtually screened candidates targeting monomeric SARS-CoV2 Mpro [11–14]. Of the 12 FDA-approved drugs identified, darunavir, indinavir, saquinavir, and tipranavir are HIV protease inhibitors, and raltegravir is a HIV integrase inhibitor used to treat HIV infections. Three other drugs identified, diosmin, hesperidin, and ruitn, are flavonoids used in the treatment of vascular disease, velpatasvir and ledipasvir are used to treat chronic hepatitis C, rosuvastatin is a statin, and bortezomib, a single boron atom compound [15], is an inhibitor of proteasomal functioning used in the treatment of relapsed multiple myeloma [16]. First, we generated complexes between the 12 FDA-approved drugs and dimeric SARS-CoV2 Mpro using a docking method. Subsequently, complexes were submitted to MD simulations coupled with MM/GBSA to elucidate the molecular mechanism by which the molecules were able to inhibit dimeric SARS-CoV2 Mpro. The analysis identified saquinavir based on its therapeutic potential against COVID-19.
Methods
Preparation of systems
Twelve FDA-approved small drugs, darunavir (DB01264), indinavir (DB00224), saquinavir (DB01232), tipranavir (DB00932), diosmin (DB08995), hesperidin (DB04703), rutin (DB01698), raltegravir (DB06817), velpatasvir (DB11613), ledipasvir (DB09027), rosuvastatin (DB01098), and bortezomib (DB00188), were obtained from the DrugBank version 5.0 [17] and optimized at the AM1 level using the Gaussian 09W software [18]. The X-ray structure of SARS-CoV2 Mpro (PDB code 6LU7) was used to build protein-ligand complexes.
Molecular docking
The fourteen FDA-approved small compounds were coupled with dimeric SARS-CoV2 Mpro using AutoDock Tools 1.5.6 and AutoDock 4.2 programs [19]. Hydrogen atoms were added to ligand and protein atoms, and partial charges were assigned to receptor (Kollman) and ligand Gasteiger. The grid box was focused on the receptor with the following grid points: 70 × 70 × 70 Å in the x, y, and z positions, respectively, and a grid space of 0.375 Å. Ligand placement was performed using a Lamarckian-genetic algorithm. The protein-ligand complex with the lowest binding energy was used to initiate MD simulations, and validation of the method was performed by replicating the experimental binding mode of co-crystallized ligand present on SARS-CoV2 Mpro (PDB code 6LU7).
MD simulations, binding free energy, and per-residue decomposition calculations
MD simulations were performed using the AMBER16 software package [20] and ff14SB force field [21]. Ligand force fields were constructed employing AM1-BCC atomic charges and GAFF force field [22]. Each system was solvated using the TIP3P water model [23] in a 12.0-Å dodecahedric box and neutralized with 0.10 M of NaCl. Once systems were equilibrated, triplicate 100-ns-long MD simulations were performed using an NPT ensemble at 310 K. The SHAKE algorithm [24] was employed to constrain bond at their equilibrium lengths, and temperature and pressure were preserved using the weak-coupling algorithm [25]. Electrostatic forces were defined using the PME method [26], and a 10-Å cut-off was designated for van der Waals interactions. Simulations were analyzed using the AmberTools16 software. Images were built using PyMOL [27].
The MM/GBSA [28, 29] approach was used to determine binding free energies (ΔGbind) of complexes and per-residue decomposition contributions, which were determined as described previously [30]. Specifically, we used the single-trajectory MM/GBSA approach, and under this method, the receptor and ligand conformations are extracted from MD simulations; as a consequence, there is no contribution of the internal energy to the net molecular mechanics energy. Prior to calculations, counterions and water molecules were removed over 500 snapshots during the last 50 ns of MD simulations, simulation time where the RMSD, RG, and average energies converged (Fig. 1, supplementary material). A salt concentration of 0.10 M and the Born implicit solvent model of 2 (igb = 2) was selected to perform the calculations.
Results and discussion
Docking between ligands and dimeric SARS-CoV2 Mpro
Docking studies between compounds and SARS-CoV2 Mpro showed that all ligands, darunavir, indinavir, saquinavir, tipranavir, diosmin, hesperidin, raltegravir, rosuvastatin, and bortezomib, reached the catalytic binding site of SARS-CoV-2 Mpro (Fig. 2A, supplementary material), with the exception of rutin, which was bounded at the entrance of the catalytic binding site, whereas for ledipasvir, velpatasvir were coupled both at the entrance and the catalytic site (Fig. 2B, supplementary material). Analysis of the residues stabilizing the ligands was typically dispersed between domains 1 (residues 8–101) and 2 (residues 102–184), with similar interactions than those found in the co-crystallized complex between the SARS-CoV-2 Mpro and the inhibitor N3 (PDB ID: 6LU7).
Convergence of MD simulations
Evaluation of root means square deviation (RMSD) and radius of gyration (RG) revealed that systems reached the equilibrium in 10–20 ns with average RMSG and RG values that fluctuated between 1.5 ± 0.2 and 2.4 ± 0.2 Å, and 25.7 ± 0.3 and 26.1 ± 0.2 Å, respectively (Table 1 and Fig. 1A and B, supplementary material). Values determined here were similar to those previously observed for the bound dimeric system [10]. Therefore, the first 30 ns of the 100 ns simulation was excluded from further analyses.
MD simulations of ligand-dimeric SARS-CoV2 Mpro binding
MD simulations showed that ligands remained bound to both subunits of the dimer, with the exception of systems assessing rutin and ledipasvir with dimeric SARS-CoV2 Mpro. In this case, rutin diffused from both subunits of SARS-CoV2 Mpro, and ledipasvir remained bound to one dimeric SARS-CoV2 Mpro catalytic site. Peptide-like inhibitor N3 was bound to subunit 1 via hydrophobic contacts involving T25, L27, H41, C44, T45, S46, M49, and Q189. N3 also established polar interactions with the side chain of Q189 (Fig. 1a). At subunit 2, inhibitor N3 was stabilized by contact with L27, H41, M49, N142, G143, S144, C145, M165, P168, D187, and Q189, and hydrogen bonds with backbone C145, and side chain S144 (Fig. 1b). Of these residues, T25, H41, M49, N142, G143, S144, C145, M165, P168, D187, and Q189 were observed in the co-crystallized complex (PDB code 6LU7).
Fig. 1.

Interactions between complexes comprised of ligands and dimeric SARS-CoV2 Mpro. Peptide-like inhibitor N3 bound to subunits 1 (a) and 2 (b), darunavir bound to subunits 1 (c) and 2 (d), and indinavir bound to subunits 1 (e) and 2 (f) of dimeric SARS-CoV2 Mpro are shown. Each complex resembles the most populated complex generated via a molecular docking simulation. The receptor is shown in the green cartoon representation, interacting residues are shown with green sticks, and the ligand is shown using a ball and stick representation. The figure was constructed with PyMOL 0.99rc6 [27]
Darunavir in subunit 1 was bound by L27, H41, M49, F140, N142, G143, S144, C145, H163, M165, D187, and Q189 via non-polar interactions and formed hydrogen bonds with backbone G143 and D187 residues (Fig. 1c). On subunit 2, coordination of darunavir occurred through non-polar interactions with T25, H41, C44, S46, M49, N142, C145, M165, D187, R88, and Q189, and polar interactions with T25 and N142 side chains and backbone atoms of H41 and C44 (Fig. 1d).
Indinavir was stabilized at subunit 1 through non-polar interactions with H41, M49, M165, E166, P168, R188, Q189, A191, and Q192, and established polar interactions with the side chain of Q189 (Fig. 1e). On subunit 2, the ligand was coordinated by H41, M49, C145, M165, E166, L167, P168, A191, and Q192 through hydrophobic contacts, and by polar interactions with backbone and side chain atoms of E166 (Fig. 1f).
Saquinavir bound subunit 1 through non-polar interactions with T25, H41, S46, M49, F140, L141, N142, S144, C145, M165, E166, P168, D187, and polar interactions with the side chain of Q189 and backbone atoms of D187 (Fig. 2a). Saquinavir bound subunit 2 via non-polar interactions involving H41, V42, M49, F140, L141, N142, S144, C145, M165, E166, P168, D187, and Q189, and through polar interactions with side chain groups of N142 and Q189, and with backbone atoms of E166 (Fig. 2b).
Fig. 2.

Interaction map of ligand and dimeric SARS-CoV2 Mpro complex formation. Saquinavir bound to subunits 1 (a) and 2 (b), tipranavir bound to subunits 1 (c) and 2 (d), and diosmin bound to subunits 1 (e) and 2 (f) of dimeric SARS-CoV2 Mpro are shown
Tipranavir bound subunit 1 through hydrophobic contacts with T25, T26, L27, H41, V42, C44, M49, N142, G143, S144, C145, and M165, and polar interactions with backbone atoms of G143 and T26 (Fig. 2c). On subunit 2, tipranavir was bound via polar interactions with T24, T25, L27, C44, T45, S46, M49, N142, and G143, and polar interactions with the side chain of S46 (Fig. 2d).
Interactions between diosmin and subunit 1 were stabilized through hydrophobic interactions with T25, L27, H41, M49, P52, F140, N142, G143, S144, C145, H163, M165, E166, R188, and Q189, polar interactions with backbone atoms of M49 and S144, and side chain atoms of H41 and E166 (Fig. 2e). On the subunit 2, diosmin bound H41, M49, M165, L167, P168, D187, R188, Q189, and Q192 via non-polar interactions, and backbone atoms of R188 through polar interactions (Fig. 2f).
Hesperidin bound subunit 1 through hydrophobic contacts with L27, H41, T45, C145, H164, M165, E166, L167, P168, D187, Q189, T190, and A191, and hydrogen bonds with the side chains of H41 and E166 (Fig. 3a). On subunit 2, hesperidin bound via hydrophobic interactions with L27, H41, M49, N142, C145, M165, and Q189, and formed polar interaction with the side chain of the latter residue (Fig. 3b).
Fig. 3.

Interaction map of ligand and dimeric SARS-CoV3 Mpro complex formation. Hesperidin coupled to subunits 1 (a) and 2 (b), raltegravir bound to subunits 1 (c) and 2 (d), and velpatasvir bound subunits 1 (e) and 2 (f) of dimeric SARS-CoV2 MPRO
Raltegravir bound subunit 1 through non-polar interactions with T25, L27, H41, T45, S46, M49, and N142 (Fig. 3c). On subunit 2, binding to raltegravir occurred through polar contacts with H41, M49, M165, D187, R188, and Q189, and hydrogen bonding with R188 (Fig. 3d). Velpatasvir was stabilized at subunit 1 through hydrophobic contacts with T25, L27, S46, M49, L50, G143, C145, Q189, T190, and A191, and by one hydrogen bond with the side chain of S46 (Fig. 3e). In contrast, at subunit 2, the ligand bound via non-polar interactions with T25, T26, L27, S46, M49, L50, N142, G143, P168, Q189, T190, A191, and polar interaction with backbone atoms of T26, Q189, and A191 (Fig. 3f).
Ledipasvir bound subunit 1 via hydrophobic contacts with L141, N142, Q189, T190, A191, and polar interactions with both the backbone and side chain of N142 (Fig. 4a).
Fig. 4.

Interaction map of ligand and dimeric SARS-CoV2 Mpro. Ledipasvir bound subunit 1 (a), rosuvastatin bound to subunits 1 (c) and 2 (d), bortezomib bound subunits 1 (e) and 2 (f) of dimeric SARS-CoV2 Mpro
Rosuvastatin was stabilized through hydrophobic interactions with T25, L27, H41, M49, N142, C145, H163, M165, E166, Q189, and Q192, and formed polar interaction with the side chain of the latter residue (Fig. 4b). At subunit 2, rosuvastatin was stabilized via hydrophobic interactions with T25, L27, H41, T45, N142, G143, C145, M165, and Q189 (Fig. 4c).
Bortezomib bound subunit 1 through hydrophobic contacts with L27, H41, M49, L141, N142, G143, S144, F140, C145, H163, and M165, and established polar interactions with the side chain of N142, and backbone atoms of G143 and L27 (Fig. 4d). At subunit 2, bortezomib bound L27, H41, C44, T45, A46, M49, M165, D187, R188, and Q189 via hydrophobic interactions, and formed hydrogen bonds with backbone atoms of R188, and the side chain of Q189 (Fig. 4e).
Binding was primarily stabilized via non-polar interactions involving L27, H41, M49, and M165 residues. T25, T26, H41, C44, S46, M49, N142, G143, S144, C145, E166, D187, R188, Q189, A191, and Q192 established polar interactions through their backbones or side chains with some of the compounds assessed (Figs. 2, 3 and 4). H41 and C145 are part of the catalytic site, and H41, M49, G143, S144, M165, E166, D187, R188, Q189, A191, and Q192 form part of the substrate binding region [31, 32], indicating that all ligands fit into their respective active sites. In addition, we observed that complexes involving each subunit were generally stabilized by an uneven number of residues. This suggested that the conformations of the two catalytic sites of the dimer were variable, which was a result not observed via crystallographic methods.
Free energy of binding
Affinity for complex formation was calculated using the MM/GBSA approach. We observed that all binding was thermodynamically favorable and occurred through the formation of non-polar interactions, van der Waals energy (ΔEvdw), and non-polar free energy of desolvation (ΔGnpol,sol). ΔGbind values for compounds bound to the first subunit of dimeric SARS-CoV2 Mpro demonstrated the following binding tendencies: saquinavir > tipranavir > darunavir > diosmin > rosuvastatin > indinavir > bortezomib > peptide-like inhibitor N3 > velpatasvir > hesperidin > raltegravir. ΔGbind values associated with binding to the second subunit were as follows: saquinavir > indinavir > hesperidin > darunavir > velpatasvir = ledipasvir > peptide-like inhibitor N3 > raltegravir > diosmin > tipranavir = rosuvastatin > bortezomib (Table 1). Differing affinity for each subunit is consistent with observed differences in the number of interactions involved in binding to each subunit, which were determined through structural analyses (Figs. 1, 2, 3, and 4). This highlights the importance of evaluating affinity to both subunits using end-point free energy methods. Binding tendency determined here is in agreement with a previous study in which enhanced inhibitory properties were associated with saquinavir relative to darunavir [33], but contrasting with the enhanced inhibitory activity experimentally determined for nelfinavir compared to saquinavir. This may suggest nelfinavir inhibits the activity of more than one target [34]. Saquinavir and darunavir had increased the affinity for dimeric SARS-CoV Mpro than that which was previously determined for praziquantel, perampanel, ritonavir, and nelfinavir using similar methodology, whereas affinity of darunavir was similar to that which had previously been reported for lopinavir [10].
Table 1.
Binding free energies of components of ligand and dimeric SARS-CoV2 and SARS-CoV2 Mpro complexes (in units of kcal/mol)
| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | DGmmgbsa |
|---|---|---|---|---|---|
| Dimmeric SARS-CoV2 Mpro | |||||
| SARS-CoV2sub1-inhibitor N3 | − 42.2 ± 7.0 | − 14.7 ± 6.0 | 36.7 ± 7.0 | − 5.4 ± 0.90 | −25.5 ± 6.0 |
| SARS-CoV2sub2-inhibitor N3 | − 49.6 ± 7.0 | − 30.9 ± 10.0 | 55.9 ± 10.0 | − 6.3 ± 0.70 | − 30.9 ± 5.0 |
| SARS-CoV2sub1-darunavir | − 54.0 ± 5.0 | − 19.4 ± 6.0 | 40.3 ± 6.0 | − 6.8 ± 0.60 | − 40.0 ± 6.0 |
| SARS-CoV2sub2-darunavir | − 45.7 ± 9.0 | − 18.8 ± 6.0 | 36.4 ± 7.0 | − 5.8 ± 1.0 | − 33.9 ± 9.0 |
| SARS-CoV2sub1-indinavir | − 39.3 ± 5.0 | − 197.2 ± 22.0 | 213.2 ± 22.0 | − 4.9 ± 0.5 | − 28.2 ± 5.0 |
| SARS-CoV2sub2-indinavir | − 46.6 ± 5.0 | − 259.9 ± 23.0 | 273.8 ± 23.0 | − 6.0 ± 0.6 | − 38.8 ± 4.0 |
| SARS-CoV2sub1-saquinavir | − 62.4 ± 4.0 | − 116.6 ± 7.7 | 142.5 ± 7.0 | − 7.3 ± 0.3 | − 43.9 ± 4.0 |
| SARS-CoV2sub2-saquinavir | − 60.9 ± 4.0 | − 125.0 ± 9.0 | 151.2 ± 10.0 | − 7.6 ± 0.5 | − 42.4 ± 4.0 |
| SARS-CoV2sub1-tipranavir | − 54.9 ± 3.0 | 44.5 ± 7.4 | − 25.1 ± 7.0 | − 6.6 ± 0.3 | − 42.0 ± 3.0 |
| SARS-CoV2sub2-tipranavir | − 33.7 ± 5.0 | 65.0 ± 12.0 | − 46.6 ± 11.0 | − 4.6 ± 0.7 | − 20.0 ± 6.0 |
| SARS-CoV2sub1-diosmin | − 54.1 ± 4.0 | − 26.7 ± 8.0 | 54.0 ± 8.0 | − 6.7 ± 0.3 | − 33.5 ± 4.0 |
| SARS-CoV2sub2-diosmin | − 40.2 ± 6.0 | − 40.3 ± 16.0 | 62.0 ± 14.0 | − 5.2 ± 0.4 | − 23.0 ± 6.0 |
| SARS-CoV2sub1-hesperidin | − 45.8 ± 4.0 | − 21.8 ± 10.0 | 50.2 ± 9.0 | − 5.8 ± 0.5 | − 23.2 ± 5.0 |
| SARS-CoV2sub2-hesperidin | − 57.4 ± 3.0 | − 26.0 ± 10.0 | 53.6 ± 8.0 | − 6.6 ± 0.2 | − 36.4 ± 4.0 |
| SARS-CoV2sub1-rutin | ND | ND | ND | ND | ND |
| SARS-CoV2sub2-rutin | ND | ND | ND | ND | ND |
| SARS-CoV2sub1-raltegravir | − 28.0 ± 6.0 | − 13.6 ± 7.0 | 28.8 ± 7.0 | − 3.7 ± 0.7 | − 16.5 ± 5.0 |
| SARS-CoV2sub2-ratelgravir | − 40.3 ± 3.0 | − 11.6 ± 5.0 | 30.9 ± 4.0 | − 4.5 ± 0.3 | − 25.6 ± 4.0 |
| SARS-CoV2sub1-velpatasvir | − 38.9 ± 8.0 | − 14.2 ± 7.0 | 33.5 ± 8.0 | − 4.8 ± 0.9 | − 24.5 ± 7.0 |
| SARS-CoV2sub2-velpatasvir | − 47.8 ± 5.0 | − 25.6 ± 8.0 | 47.5 ± 7.0 | − 6.0 ± 0.5 | − 32.0 ± 5.0 |
| SARS-CoV2sub1-ledipasvir | ND | ND | ND | ND | ND |
| SARS-CoV2sub2-ledipasvir | − 47.8 ± 5.0 | − 25.6 ± 8.0 | 47.5 ± 7.0 | − 6.0 ± 0.5 | − 32.0 ± 5.0 |
| SARS-CoV2sub1-rosuvastatin | − 41.3 ± 4.0 | 58.0 ± 11.0 | − 41.4 ± 10.0 | − 5.4 ± 0.5 | − 30.1 ± 5.0 |
| SARS-CoV2sub2-rosuvastatin | − 36.0 ± 6.0 | 60.8 ± 12.0 | − 40.2 ± 11.0 | − 4.9 ± 0.7 | − 20.2 ± 6.0 |
| SARS-CoV2sub1-bortezomib | − 38.1 ± 3.3 | 65.4 ± 14.0 | − 48.4 ± 11.0 | − 4.7 ± 0.40 | − 25.9 ± 4.0 |
| SARS-CoV2sub2-bortezomib | − 32.9 ± 4.3 | 73.1 ± 11.0 | − 53.4 ± 11.0 | − 4.3 ± 0.60 | − 17.4 ± 5.0 |
Based on this analysis, it is clear that both saquinavir and darunavir, both known to possess potent antiviral protease inhibitory activity [35, 36], are attractive anti-COVID-19 clinical drug candidates. The two drugs had stronger affinity for both subunits of Mpro than that which was experimentally determined for peptide-like inhibitor N3, which displayed strong antiviral effects in the micromolar concentration range in SARS-CoV-2 virus-infected Vero cells [37]. Assessment of ΔGbind values for indinavir and tipranavir indicated that they may also possess moderate activities against COVID-19. A comparison of the 12 FDA-approved compounds evaluated, after excluding ledipasvir and rutin that exhibited ligand diffusion in one or both subunits, revealed that the worst candidates to inhibit activity of dimeric SARS-CoV Mpro were raltegravir and bortezomib. These compounds had the greatest affinities of all inhibitors assessed via virtual screening, which employed docking studies involving monomeric SARS-CoV Mpro [13, 14].
Comparative analysis of the affinity tendency observed for three of the 12 evaluated compounds previously reported by Farag et al. (darunavir > rosuvastatin > saquinavir) [11], Chen et al. (ledipasvir > velpatasvir) [12], Adem et al. (hesperidin > rutin > diosmin) [38], and Kumar et al. (tipranavir > raltegravir) [13] revealed discrepancies when compared with our findings. These observations highlight the degree to which the computational strategy employed to identify potential inhibitors of SARS-CoV Mpro impacted affinity tendency and underscored the utility of combining docking, MD simulations, and end-point binding free energy methods.
Per-residue free energy decomposition
Investigation of the residues contributing to ΔGbind in ligands-dimeric SARS-CoV2 complex revealed that complex stabilization could generally be attributed to 5 to 14 residues (Tables 2, 3, and 4). Complex formation between dimeric SARS-CoV2 Mpro and peptide-like inhibitor N3 involved T25, C44, T45, and S46 of subunit 1. The energetic contribution of N142, G143, S144, C145, M165, P168, and D187 residues was only observed when ligands bound subunit 2 (Table 2), while L27, H41, M49, and Q189 contributed to the stabilization of binding to both subunits. For darunavir, L27, F140, G143, S144, and H163 stabilized binding subunit 1, and T25, C44, S46, and R188 stabilized binding to subunit 2. H41, M49, N142, C145, M165, D187, and Q189 residues were involved in binding to both subunits.
Table 2.
Per-residue free energies of ligand-dimeric SARS-CoV2 Mpro complexes (values kcal/mol)
| Residue | Lig1sub2 | Lig1Sub2 | Lig2Sub1 | Lig2Sub2 | Lig3Sub1 | Lig3Sub2 | Lig4Sub1 | Lig4Sub2 | Lig5Sub1 | Lig5Sub2 |
|---|---|---|---|---|---|---|---|---|---|---|
| T24 | − 0.809 | |||||||||
| T25 | − 0.604 | − 0.740 | − 0.567 | − 2.816 | − 2.750 | |||||
| T26 | − 3.063 | |||||||||
| L27 | − 0.550 | − 0.569 | − 0.669 | − 1.807 | − 0.874 | |||||
| H41 | − 1.806 | − 0.925 | − 1.217 | − 1.955 | − 0.595 | − 0.785 | − 0.865 | − 1.337 | − 2.202 | |
| V42 | − 0.549 | − 1.561 | ||||||||
| C44 | − 1.030 | − 0.646 | − 0.756 | − 0.558 | ||||||
| T45 | − 0.818 | − 1.562 | ||||||||
| S46 | − 0.603 | − 0.658 | − 0.659 | − 1.445 | ||||||
| M49 | − 2.051 | − 2.381 | − 1.779 | − 2.172 | − 0.888 | − 0.589 | − 2.127 | − 1.954 | − 0.630 | − 1.429 |
| F140 | − 1.353 | − 0.577 | − 0.671 | |||||||
| L141 | − 0.578 | − 0.525 | ||||||||
| N142 | − 1.571 | − 2.836 | − 0.786 | − 1.080 | − 1.421 | − 3.386 | − 1.418 | |||
| G143 | − 0.986 | − 2.027 | − 3.138 | − 0.713 | ||||||
| S144 | − 0.651 | − 0.624 | − 0.794 | − 0.767 | − 0.684 | |||||
| C145 | − 1.297 | − 0.717 | − 0.506 | − 0.633 | − 0.601 | − 0.731 | − 1.229 | |||
| H163 | − 0.686 | |||||||||
| M165 | − 1.214 | − 1.549 | − 2.264 | − 3.263 | − 4.215 | − 3.455 | − 3.395 | − 1.935 | ||
| D166 | − 0.683 | − 2.464 | − 0.810 | − 0.955 | ||||||
| L167 | − 0.545 | |||||||||
| P168 | − 0.719 | − 0.547 | − 0.652 | − 1.085 | − 1.072 | |||||
| D187 | − 0.650 | − 0.556 | − 1.468 | − 0.971 | − 0.803 | |||||
| R188 | − 1.194 | − 0.513 | ||||||||
| Q189 | − 1.214 | − 1.930 | − 2.431 | − 1.037 | − 3.544 | − 3.109 | − 3.197 | |||
| A191 | − 0.586 | − 1.057 | ||||||||
| Q192 | − 0.523 | − 1.074 |
Inhibitor N3, lig1; darunavir, lig2; indinavir, lig3; saquinavir, lig4; tipranavir, 5
Table 3.
Per-residue free energies of ligand-dimeric SARS-CoV2 Mpro complexes (values kcal/mol)
| Residue | Lig6Sub1 | Lig6Sub2 | Lig7Sub1 | Lig7Sub2 | Lig8Sub1 | Lig8Sub2 | Lig9Sub1 | Lig9Sub2 |
|---|---|---|---|---|---|---|---|---|
| T25 | − 0.511 | − 0.712 | − 0.741 | − 2.032 | ||||
| T26 | − 1.202 | |||||||
| L27 | − 0.651 | − 0.840 | − 0.782 | − 0.782 | − 0.606 | − 0.640 | ||
| H41 | − 2.058 | − 0.927 | − 1.461 | − 1.856 | − 0.631 | − 1.276 | ||
| T45 | − 0.593 | − 0.899 | ||||||
| S46 | − 1.504 | − 0.747 | − 0.505 | |||||
| D48 | ||||||||
| M49 | − 2.339 | − 1.860 | − 1.189 | − 1.567 | − 1.032 | − 2.156 | − 2.725 | |
| L50 | − 1.922 | − 2.552 | ||||||
| P52 | − 0.509 | |||||||
| F140 | − 0.550 | |||||||
| N142 | − 0.686 | − 0.932 | − 0.608 | − 1.733 | ||||
| G143 | − 0.724 | − 0.550 | − 1.069 | |||||
| S144 | − 1.506 | |||||||
| C145 | − 1.713 | − 0.989 | − 0.967 | − 0.792 | ||||
| H163 | − 2.198 | |||||||
| H164 | − 0.517 | |||||||
| M165 | − 3.400 | − 1.907 | − 2.454 | − 1.658 | − 1.931 | |||
| E166 | − 0.858 | − 0.836 | ||||||
| L167 | − 0.851 | − 0.677 | ||||||
| P168 | − 0.983 | − 0.743 | − 0.507 | |||||
| D187 | − 1.274 | − 0.678 | − 1.636 | |||||
| R188 | − 0.945 | − 0.920 | − 1.169 | |||||
| Q189 | − 1.648 | − 2.271 | − 2.137 | − 1.799 | − 2.550 | − 1.528 | − 1.319 | |
| T190 | − 0.734 | − 0.535 | − 1.130 | |||||
| A191 | − 0.878 | − 0.742 | − 0.682 | |||||
| Q192 | − 0.656 | |||||||
Diosmin, lig6; hesperidin, lig7; raltegravir, lig8; and velpatasvir, lig9
Table 4.
Per-residue free energies of ligand-dimeric SARS-CoV2 Mpro complexes (values kcal/mol)
| Residue | Lig10Sub2 | Lig11Sub1 | Lig11Sub2 | Lig12Sub1 | Lig12Sub2 | |||
|---|---|---|---|---|---|---|---|---|
| T25 | − 0.614 | − 0.522 | ||||||
| L27 | − 0.512 | − 1.001 | − 1.365 | − 0.573 | ||||
| H41 | − 1.226 | − 1.746 | − 0.579 | − 1.622 | ||||
| C44 | − 0.608 | |||||||
| T45 | − 0.767 | − 0.508 | ||||||
| S46 | − 0.701 | |||||||
| M49 | − 0.707 | − 0.558 | − 3.131 | |||||
| F140 | − 0.980 | |||||||
| L141 | − 1.688 | − 0.534 | ||||||
| N142 | − 3.207 | − 0.830 | − 1.403 | − 2.251 | ||||
| G143 | − 1.142 | − 3.204 | ||||||
| S144 | − 0.866 | |||||||
| C145 | − 0.918 | − 1.131 | − 3.803 | |||||
| H163 | − 0.539 | − 1.775 | ||||||
| M165 | − 4.104 | − 1.833 | − 0.912 | − 0.942 | ||||
| E166 | − 0.587 | |||||||
| D187 | − 1.333 | |||||||
| R188 | − 0.589 | |||||||
| Q189 | − 1.656 | − 2.145 | − 1.485 | − 1.798 | ||||
| T190 | − 1.785 | |||||||
| A191 | − 2.481 | |||||||
| Q192 | − 2.258 |
Ledipasvir, lig10; rosuvastatin, lig11; bortezomib, lig12
For indinavir, R188 and Q189 contribute to the stabilization of binding to subunit 1, whereas C145 and L167 facilitated subunit 2 binding. H41, M49, M165, D166, P168, A191, and Q192 were involved in binding to both subunits. For saquinavir, T25 and S46 of subunit 1 stabilized binding, and V42 stabilized interactions involving subunit 2. H41, M49, F140, L141, N142, S144, C145, M165, D166, P168, D187, and Q189 contributed energetically to binding to both subunits. T26, H41, V42, S144, C145, and M165 residues of subunit 1, and T24, T45, and S46 of subunit 2, were involved in binding to tipranavir. T25, L27, C44, M49, N142, and G143 facilitated binding to both subunits of Mpro. T25, L27, P52, F140, N142, G143, S144, C145, H163, and E166 stabilized diosmin binding to subunit 1, whereas L167 and P168 promoted binding to subunit 2. H41, M49, M165, R188, and Q189 contribute at both subunits of dimeric SARS-CoV2 Mpro (Table 3).
T45, H164, E166, L167, P168, D187, T190, and A191 facilitated hesperidin-subunit 1 binding, and M49 and N142 exclusively facilitated binding to subunit 2. L27, H41, C145, M165, and Q189 of both subunits of dimeric SARS-CoV2 were involved in binding hesperidin (Table 3). T25, L27, T45, S46, and N142 of subunit 1 were determined to be the principal stabilizers of raltegravir binding, whereas M165, D187, R188, and Q189 of subunit 2 promoted binding to the inhibitor. H41 and M49 were involved in binding both Mpro subunits (Table 3). C145 was the only residue of subunit 1 involved in binding velpatasvir, while T26, N142, and P168 of subunit 2 stabilized binding, while T25, L27, S46, M49, L50, G143, Q189, T190, and A191 of both subunits were involved in binding (Table 3). Regarding ledipasvir, ligand only remained bound to subunit 2 via the energetic contributions of L141, N142, Q189, and T190 (Table 4). With regard to rosuvastatin, M49, H163, E166, and Q192 of subunit 1, and T45 and G143 of subunit 2, stabilized interactions with ligands. L27, H41, N142, N145, M165, and Q189 of both subunits facilitated binding. For bortezomib, it was observed that F140, L141, N142, G143, S144, C145, and H163 stabilized the ligand binding to subunit 1. C44, T45, S46, D187, R188, and Q189 stabilized the ligand binding to subunit 2. However, L27, H41, M49, and M165 stabilized the ligand binding to both subunits.
An analysis of residues that enhanced the ligand binding affinity revealed that, in general, L27, T25, T26, H41, M49, V42, T45, L50, S46, F140, N142, G143, S144, C145, H163, M165, D166, P168, D187, R188, Q189, T190, A191, and Q192 significantly contributed to ΔGbind values of some complexes, but only L27, H41, M49, and M165 were involved in protein-ligand complex formation consistently. This highlights the importance of residues that comprise the catalytic (H41) and substrate binding (M49 and M165) sites [31, 32], and reveals the importance of the participation of other residues involved in ligand stabilization, which were also identified previously [10].
Principal component analysis
PCA allowed researchers to quantitatively approximate the degree of mobility change that occurred upon ligand complexation. Therefore, the trace of the diagonalized covariance matrix of backbone atoms was calculated for bound SARS-CoV2 Mpro systems. This analysis showed that darunavir, hesperidin, and raltegravir binding was linked to a reduced degree of conformational change of dimeric SARS-CoV2 Mpro relative to that which occurred when the apo state [10] of the protein was bound. Peptide-like inhibitor N3, indinavir, saquinavir, tipranavir, velpatasvir, and bortezomib binding to dimeric SARS-CoV2 Mpro produced a small increase in conformational mobility. However, diosmin and ledipasvir binding did not produce conformational change upon ligand binding. Importantly, binding of rosuvastatin increased the conformational mobility of dimeric SARS-CoV2 Mpro, which suggested binding of most compounds was not likely to significantly impact affinity for rosuvastatin (Table 1), while decreases in the predicted affinity for rosuvastatin could be expected as a result of favorable entropic components that affect the degree of conformational mobility upon complex formation.
Conclusion
Through a combination of structural data, docking, and MD simulations using an MM/GBSA approach, previous research identified new inhibitors (praziquantel and perampanel) of SARS-CoV2 Mpro. MD simulations used in combination with end-point free energy methods revealed that ligand binding was enhanced when dimeric SARS-CoV2 Mpro, rather than its monomeric form, was used. In this research, we elaborated upon previous work by employing docking and MD simulation studies coupled with the MM/GBSA approach to assess interactions between SARS-CoV2 Mpro and 12 FDA-approved drugs identified previously for their potential activity against COVID-19 via virtual screening of monomeric SARS-CoV2 Mpro and the peptide-like inhibitor, N3, which was demonstrated to have strong antiviral activity against COVID19 in vitro. Our results indicated that saquinavir is predicted to have a greater affinity for dimeric SARS-CoV Mpro than that which is predicted for praziquantel, perampanel, ritonavir, lopinavir, or nelfinavir, using comparable methodology. Therefore, saquinavir shows great potential as a strong anti-COVID-19 therapeutic candidate, particularly because it displayed higher affinity for both subunits than peptide-like inhibitor N3. Although these data are insufficient for confirming antiviral activity, they provide a basis for future studies focused on in vitro and in vivo testing of viral activity.
Supplementary Information
(DOCX 1405 kb)
Funding information
The work was supported by grants from CONACYT (CB-A1-S-21278) and SIP/IPN (20201015).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020;63(3):457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vastag B. Old drugs for a new bug. JAMA. 2003;290(13):1695–1696. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- 3.Nukoolkarn V, Lee VS, Malaisree M, Aruksakulwong O, Hannongbua S. Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J Theor Biol. 2008;254(4):861–867. doi: 10.1016/j.jtbi.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Z, Peng C, Shi Y, Zhu Z, Mu K, Wang X, Zhu W (2020) Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. 10.1101/2020.01.27.921627
- 5.Sang P, Tian S, Meng Z & Yang L (2020) Insight derived from molecular docking and molecular dynamics simulations into the binding interactions between HIV-1 protease inhibitor s and SARS-CoV-2 3CLpro. ChemRxiv. Preprint. 10.26434/chemrxiv.11932995.v1
- 6.Andrianov AM, Kornoushenko YV, Karpenko AD, Bosko IP, Tuzikov AV (2020) Computational discovery of small drug-like compounds as potential inhibitors of SARS-CoV-2 main protease. J Biomol Struct Dyn:1–13 [DOI] [PMC free article] [PubMed]
- 7.Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019- nCoV) Nat Rev Drug Discov. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- 8.Jiménez-Alberto A, Ribas-Aparicio RM, Aparicio-Ozores G, Castelán-Vega JA (2020) Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput Biol Chem 107325 [DOI] [PMC free article] [PubMed]
- 9.Graziano V, McGrath WJ, Yang L, Mangel WF. SARS CoV main proteinase: the monomer− dimer equilibrium dissociation constant. Biochemistry. 2006;45(49):14632–14641. doi: 10.1021/bi061746y. [DOI] [PubMed] [Google Scholar]
- 10.Bello M (2020) Prediction of potential inhibitors of the dimeric SARS-CoV-2 main proteinase through the MM/GBSA approach. Res Square. 10.21203/rs.3.rs-21205/v1 [DOI] [PMC free article] [PubMed]
- 11.Farag A, Wang P, Ahmed M & Sadek H (2020) Identification of FDA approved drugs targeting COVID-19 virus by structure-based drug repositioning. ChemRxiv. Preprint. 10.26434/chemrxiv.12003930.v3
- 12.Chen YW, Yiu CB, Wong KY. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res. 2020;9:129. doi: 10.12688/f1000research.22457.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar Y & Singh H (2020) In silico identification and docking-based drug repurposing against the main protease of SARS-CoV-2, causative agent of COVID-19. ChemRxiv. Preprint. 10.26434/chemrxiv.12049590.v1
- 14.Vega Valdez IR, Santiago-Quintana JM, ROSALEZ M, et al (2020) Theoretical evaluation of bortezomib and other boron-containing compounds as inhibitors of SARS-CoV-2 main protease. ChemRxiv. Preprint. 10.26434/chemrxiv.12047346.v1
- 15.Bello M. Advances in theoretical studies on the design of single boron atom compounds. Curr Pharm Des. 2018;29:3466–3475. doi: 10.2174/1381612824666181102142432. [DOI] [PubMed] [Google Scholar]
- 16.Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control. 2003;10(5):361–369. doi: 10.1177/107327480301000502. [DOI] [PubMed] [Google Scholar]
- 17.Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frisch MJT, Schlegel HBGW, Scuseria GE et al (2009) Gaussian Gaussian Inc.
- 19.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor fexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Case DA, Darden TA, Cheatham TE, III, et al. AMBER 12. San Francisco: University of California; 2010. [Google Scholar]
- 21.Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 24.van Gunsteren WF, Berendsen HJC. Algorithms for macromolecular dynamics and constraint dynamics. Mol Phys. 1977;34:1311–1327. doi: 10.1080/00268977700102571. [DOI] [Google Scholar]
- 25.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 26.Darden T, York D, Pedersen L. Particle mesh Ewald-an N.Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 27.DeLano WL. The PyMOL molecular graphics system. San Carlos: Delano Scientific; 2002. [Google Scholar]
- 28.Miller BR, McGee TD, Swails JM, et al. MMPBSA.py: an efcient program for end-state free energy calculations. J Chem Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 29.Gohlke H, Kiel C, Case DAJ. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. Mol Biol. 2003;330:891–913. doi: 10.1016/S0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 30.Bello M, Mendieta-Wejebe JE, Correa-Basurto J. Structural and energetic analysis to provide insight residues of CYP2C9, 2C11 and 2E1 involved in valproic acid dehydrogenation selectivity. Biochem Pharmacol. 2014;90(2):145–158. doi: 10.1016/j.bcp.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C, Wei P, Fan K, et al. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto N, Matsuyama S, Hoshino T, Yamamoto N (2020) Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. BioRxiv. 10.1101/2020.04.06.026476
- 34.Xu Z, Yao H, Shen J et al (2020) Nelfinavir is active against SARS-CoV-2 in Vero E6 cells. ChemRxiv. Preprint. 10.26434/chemrxiv.12039888.v1
- 35.Schapiro JM, et al. The effect of high-dose saquinavir on viral load and CD4+ T-cell counts in HIV-infected patients. Ann Intern Med. 1996;124(12):1039–1050. doi: 10.7326/0003-4819-124-12-199606150-00003. [DOI] [PubMed] [Google Scholar]
- 36.Dierynck I, De Wit M, Gustin E et al (2007) Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol 81(24):13845–13851 [DOI] [PMC free article] [PubMed]
- 37.Jin Z, Du X, Xu Y, et al (2020) Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 1–5 [DOI] [PubMed]
- 38.Adem S, Eyupoglu V, Sarfraz I et al (2020) Identification of potent COVID–19 main protease (Mpro) inhibitors from natural polyphenols: an in silico strategy unveils a hope against CORONA. Preprints. 10.20944/preprints202003.0333.v1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 1405 kb)