

Abstract
Sphingosine 1-phosphate (S1P), a metabolic product of cell membrane sphingolipids, is bound to extracellular chaperones, enriched in circulatory fluids and binds to G protein-coupled S1P receptors (S1PRs) to regulate embryonic development, postnatal organ function and disease. Indeed, S1PRs regulate essential processes such as adaptive immune cell trafficking, vascular development and homeostasis. Moreover, S1PR signaling is a driver of multiples diseases. The past decade has witnessed an exponential growth in this field, in part due to multidisciplinary research focused on this lipid mediator and the application of S1PR-targeted drugs in clinical medicine. This has revealed fundamental principles of lysophospholipid mediator signaling that not only clarify the complex and wide ranging actions of S1P but also guide the development of therapeutics and translational directions in immunological, cardiovascular, neurological, inflammatory and fibrotic diseases.
Introduction
Lipids are best known for their roles in energy storage and formation of cellular membranes. The diversity of membrane lipid structures ensures structural flexibility and integrity of cellular membranes needed for adaptation to varying environments. As vertebrate evolution ensued, metabolites of membrane lipids were repurposed as extracellular ligands for G protein-coupled receptors (GPCRs), which in turn activate numerous intracellular signaling pathways, thus coupling membrane phospholipid breakdown with intercellular communication (1). This coupling was first illustrated by platelet activating factor, a bioactive lipid synthesized from membrane phospholipids of hematopoietic and vascular cells that activates GPCRs in an autocrine and paracrine manner to evoke allergic reactions and anaphylaxis (2). Similarly, sphingosine 1-phosphate (S1P), which is produced and secreted by red blood cells, endothelial cells and platelets into the extracellular environment, acts on specific GPCRs expressed by most, if not all, cell types (3).
S1P was discovered in the 1960s as a terminal product of sphingolipid metabolism (4). Studies in cultured cells suggested its bioactive nature even though the prevailing notion at the time considered it more akin to classical second messengers such as diacylglycerol and Ca2+ (5, 6). We now know that vertebrates possess five S1P receptors (S1PR1–5) that respond to extracellular S1P to regulate embryonic development, physiological homeostasis and pathogenic processes in multiple organ systems (3). These high affinity S1PRs couple to key intracellular signaling pathways controlled by heterotrimeric G proteins, Rho family small guanosine triphosphatases (GTPases) and the protein kinase AKT, resulting in changes in cytoskeleton, cell adhesion and survival (7). Although the extracellular actions of S1P are well-established, distinct intracellular functions of S1P have also been suggested. For example, S1P was purported to bind to and regulate the activities of cytosolic signal transduction mediator tumor necrosis factor (TNF)-α-receptor-associated factor 2 (TRAF-2) (8), the chromatin modifying enzyme histone deacetylase-1 (HDAC-1) (9), the mitochondrial regulator prohibitin-2 (10), atypical protein kinase-C (11) and the catalytic subunit of telomere reverse transcriptase (12). Whether intracellular S1P signaling is physiologically relevant is not clear because genetic studies do not support the essential and/or functional roles of intracellular S1P targets (13–16).
The unique physicochemical properties of S1P endow its multifunctional nature in developmental, physiological and pathological contexts (17). For example, resistance to oxidative modification makes S1P suitable as a modulator of inflammatory processes, which are characterized by the influx of cells with high oxidative capacity. Poor aqueous solubility of S1P necessitates its association with protein molecules for transport in the blood and lymph circulation and access to S1PRs (18). Such interactions also facilitate the formation of S1P gradients, which are spatial concentration differences across biological compartments, thus allowing selective receptor activation at various locations (18–21). These properties enable S1P to be involved in multiple physiological processes. The essential nature of S1P functions has been illustrated in humans with congenital conditions such as Sjögren-Larsson syndrome, nephrosis, adrenal insufficiency, hearing impairment, and in mouse genetic studies in which major embryologic and post-natal defects in various organs were observed when S1PRs, transporters and S1P metabolic enzymes were mutated (3, 22, 23).
S1P-regulated processes contribute to several cardiovascular, autoimmune, inflammatory, neurological, oncologic and fibrotic diseases (3). Moreover, the successful US Food and Drug Administration (FDA) approval of an S1PR-targeted drug, fingolimod, to treat multiple sclerosis (MS) in 2010 (24) has led to considerable growth of research and drug development efforts. However, fingolimod binds and activates four S1PRs (S1PR1,3,4 and 5) while also functionally antagonizing S1PR1. Recently, a more specific S1PR1 and S1PR5-targeted drug – sipondimod was approved for the treatment of MS. In this Review, we discuss how conceptual advances in understanding S1P signaling have reached a critical threshold to allow rational design of new therapeutic strategies.
S1P spatial gradients
Extracellular gradients of growth factors, cytokines and developmental morphogens are critical for achieving fidelity and specificity of biological responses (25). Lipid mediators, small molecules with limited aqueous solubility and affinity for hydrophobic molecules adopted distinct signaling mechanisms to overcome these biophysical challenges. For example, tissue- and cell-specific secretion, chaperone association and extracellular metabolism of S1P result in the formation of S1P spatial gradients that are important for its numerous biological activities.
The enzymes that generate S1P, namely ceramidase and sphingosine kinase (SPHK), do double duty in intracellular sphingolipid metabolism as well as extracellular signaling (26). In many tissues, S1P is rapidly degraded by the endoplasmic reticulum-resident S1P lyase, resulting in very low intracellular S1P concentrations. By contrast, S1P can also be coupled to export processes via specific transporters. These flip the S1P molecule from the inner to the extracellular leaflet of the plasma membrane, thereby allowing extracellular action of S1P (3). Once exported, S1P is capable of activating S1PRs expressed in the same cell in an autocrine manner. However, extraction by chaperone proteins, which bind to S1P and enable aqueous solubility, lead to diffusible S1P in the extracellular environment, thus creating spatial gradients that activate S1PRs in a paracrine and/or endocrine manner (Figure 1).
Figure 1. Establishing and maintaining S1P gradients in vessels.

S1P is transported by chaperones- ApoM+HDL and albumin in the circulation and presented to S1PRs. S1P export by vascular endothelial cells (VECs) and lymphatic endothelial cells (LECs) (by SPNS2) and by RBCs (by MFS2B), as well as degradation of S1P by the phosphatase LPP3 modify the extracellular S1P gradient. Low interstitial tissue S1P is achieved by the S1P lyase. Tissue concentrations of S1P are indicated in parentheses.
S1PR: sphingosine 1-phosphare receptor, SPNS2: Sphingolipid Transporter 2, MFSD2B: Major Facilitator Superfamily Domain Containing 2B, LPP3: Lipid phosphate phosphatase 3
Tissue-specific transporters that belong to the “major facilitator” superfamily export S1P to establish extracellular gradients. For example, spinster homolog 2 (SPNS2), first identified in a zebrafish screen for cardia bifida (a developmental condition that leads to the formation of two beating hearts), functions in the extraembryonic yolk syncytial layer to export S1P, which binds to S1PR2 on the cells of the endoderm, which in turn allows directional cardiac progenitor cell migration to the midline and proper cardiac development (27). This is the first example of S1P extracellular gradients created by a lipid transporter that is required for a developmental event (28, 29). In mice, SPNS2 exports S1P from vascular and lymphatic endothelial cells (LECs) to activate S1PRs on lymphocytes, thus allowing their egress from lymph nodes into the circulatory system (30, 31). High circulating S1P concentrations together with low amounts of S1P in interstitial fluid in secondary lymphoid organs, such as lymph nodes, are critical for lymphocyte egress into lymphatic circulation (19).
Mouse models that use reporters to sense extracellular S1P revealed S1P gradients at higher resolution. In the spleen, extracellular S1P gradients formed by the phosphatase enzyme lipid phosphate phosphohydrolase 3 (LPP3) are essential for marginal zone B cell trafficking (32), whereas LEC-expressed SPNS2 in lymph nodes regulated medullary cord natural killer (NK) cell location and function (33). The transporter, major facilitator superfamily domain-containing protein 2B (MFSD2B), which exports S1P from red blood cells (RBCs) into plasma, is also important for their normal function and turnover (34). Loss of function of S1P transporters leads not only to developmental defects, but also to early onset hearing loss (35), and retinal defects (36). These observations suggest that spatial S1P signaling, which is critical in embryogenesis and post-natal physiological processes, leads to pathological conditions when dysregulated. Cellular mechanisms by which S1P transporters are controlled are poorly understood. Strategies to restore normal transporter function could be useful in the treatment of various diseases in which extracellular S1P gradients are perturbed.
Chaperones, biased signaling and S1PR regulators
S1P chaperone proteins, which stably bind and transport S1P in circulatory and interstitial fluids, facilitate S1PR activation on recipient cells. The prototypical S1P chaperone is apolipoprotein M (ApoM), a component of high density lipoprotein (HDL), a circulating lipoprotein associated with vascular health (20). In addition, serum albumin binds to S1P at lower affinity (37). HDL-bound ApoM binds S1P stably in blood plasma and activates S1PRs on endothelial cells to suppress inflammation, promote barrier function, allow liver regeneration after hepatectomy and protect from bacterial endotoxin-induced inflammatory responses (38–41). In hematopoietic progenitor cells, ApoM-bound S1P suppresses lymphopoiesis (42). HDL-bound S1P activates S1PR1 as a biased agonist that selectively induces certain biological responses. Other HDL-bound factors such as apolipoprotein A1, cellular HDL receptor scavenger receptor-B1, and the ability of HDL to modulate membrane raft-based organization of receptor-effector complexes may all be involved in biased signaling (43). Additionally, the relative stability of HDL-bound S1P compared to albumin-bound S1P may also play a role in differential kinetics of receptor activation and signaling (38) (Figure 2). By contrast, albumin-bound S1P is short-lived in plasma (44) and activates S1PRs in a manner distinct from that of HDL-S1P.
Figure 2. S1PR1 Signaling Regulation.

Schematic representation of regulators of S1P receptor-1 (S1PR1) cell surface expression, signaling and turnover. Albumin-S1P, HDL-S1P and FTY720-P activate S1PR1. Internalization of S1PR1 eventually leads to proteasomal degradation by the ubiquitin ligase WW Domain Containing E3 Ubiquitin Protein Ligase 2 (WWP2). CD44 and activated protein C (APC)/ endothelial protein C receptor (EPCR) transactivate S1PR1, while CD69 binds to and downregulates S1PR1 at the cell surface. Endocytosis of S1PR1 is achieved via GPCR kinase 2 (GRK2)-dependent phosphorylation, and endocytic regulators (dynamin, moesin, β-arrestin and AP2/Clathrin). The intracellular signaling of S1PR1 is transmitted via the hetero-trimeric Gαi family proteins, the inhibition of adenylyl cyclase and activation of PI3K-AKT and ERK pathways. Palmitoylation by DHHC5 affects the coupling of S1PR1 to Gαi, while α-synuclein modulates the coupling of S1PR1 with Gαi by removing S1PR1 from lipid rafts. Regulators of other S1PRs are not well characterized.
Once activated by small molecule agonists or albumin-bound S1P, S1PR expression on the cell surface is down-regulated, which makes vascular and hematopoietic cells exposed to circulatory S1P refractory to further stimulation. Indeed, regulation of S1PR1 endocytosis is precisely controlled by G protein-coupled receptor kinase 2, dynamin and moesin (45–48). Loss of such S1PR1 regulatory events leads to changes in biological outcomes such as altered immune cell trafficking (46) and vascular endothelial barrier function (49). Sustained endocytosis of lymphocyte S1PR1 explains the therapeutic efficacy of S1PR1 functional antagonists in the treatment of autoimmune diseases, as discussed below.
The recent development of S1PR reporter mice has revealed plasma S1P-dependent β-arrestin activation in vascular endothelium upon systemic endotoxemia (50, 51) and in the aortic endothelium at areas of disturbed flow (38). This suggests that S1PR1 is downregulated in injured and inflamed endothelium. The ability to monitor S1PR function in vivo in real time and at single cell resolution is anticipated to lead to new insights into how S1P signaling is regulated physiologically and in disease.
In addition to the natural ligand S1P, other molecules as well as co-receptors modulate S1PR activity. For example, conjugated bile acids (CBAs) activate S1PR2, which may be important in hepatobiliary and intestinal systems during cholestatic injury (52). In endothelial cells, activated protein C receptor and the hyaluronic acid receptor CD44 transactivate S1PR1 to increase barrier function and thereby prevent plasma leakage, thus limiting inflammatory responses (53, 54). In lymphocytes, activation-induced CD69 binds to S1PR1 and downregulates its expression, an event that is critical for regulation of lymphocyte egress and tissue residence (55). In the central nervous system (CNS), the binding of the extracellular domain of NOGOA, a neuronal repulsive molecule that regulates neural network formation, to S1PR2 induces signals via the GPCR Gα13-RhoA signaling pathway to repress neurite outgrowth, thus achieving synaptic plasticity in the hippocampus and motor cortex (56). An unbiased, genome-wide CRISPR/Cas9 screen for S1PR1 modulators identified lysophosphatidic acid receptor-1 as a negative regulator. This axis is important in lymphatic endothelial cells of the lymph node sinuses (57). In γδ T cells, S1PR2 inhibition of S1PR1 function restrains them in the skin, thus promoting immunity in the local environment by gaining tissue residency (58).
These examples illustrate that S1PRs evolved to interact with many other signaling pathways involved in embryonic development, inflammation, host defense and homeostasis. Mechanistic details of how S1P transfer takes place from the chaperones to S1PRs, the action of various receptor modulatory molecules, and the functions of regulated endocytosis and plasma membrane retention of S1PRs need to be further elucidated. Interestingly, the crystal structure of agonist-bound S1PR1 suggested that ligand (i.e. S1P) entry takes place via the transmembrane domains of S1PR1 (59). Therefore, it is likely that HDL docking on the plasma membrane enables efficient S1P transfer to S1PRs via a transmembrane route. New structural analysis methods, such as cryo-electron microscopy, may reveal dynamic receptor conformations that are anticipated to lead to insights for the development of highly specific receptor modulatory agents. This may also lead to general understanding of how lipid mediators function in regulating cellular and organ function.
Cardiovascular development and pathophysiology
The vasculature, one of the first organ systems to develop in vertebrate embryogenesis, is regulated by circulatory S1P signaling (60, 61) (Figure 3). Initial phases of angiogenesis encompass hypoxia-induction of vascular endothelial growth factor (VEGF) that induces endothelial sprouts, in which S1P signaling is minimal. When vascular sprouts fuse to form primary vascular networks, circulatory S1P accesses endothelial cells. These fusion events coincide with the induction of S1PR1 expression in the endothelium, and consequently the formation of adherens junctions and cell-extracellular matrix adhesions that enable the vascular barrier to form (62, 63). Activation of endothelial nitric oxide synthase (eNOS) dilates vessels and allows proper blood flow (61). Multiple redundant sources of circulating S1P from RBCs, endothelial cells and platelets and the existence of sensing mechanisms to monitor and maintain circulating S1P levels during vascular development reinforces the essential nature of spatial S1P gradients (64, 65).
Figure 3. S1P function in vascular development, homeostasis and pathology.

Vascular network development, maturation and stabilization is regulated by endothelial S1PR1. Activation of S1PR1 inhibits sprouting angiogenesis and vascular endothelial growth factor receptor 2 (VEGRF2), and promotes vascular stability via adherens junctions and cell-extracellular matrix adhesion (integrins). Loss of cell surface S1PR1 and destabilization of the endothelium causes the leakage of plasma and immune cells into the tissue, leading to inflammation. Vascular damage also leads to thrombosis (platelet aggregation) and fibrin deposition in the interstitial space, which activates fibroblasts and macrophages. Loss of S1PR1 signaling on endothelial cells and activated S1PR2 and S1PR3 signaling on macrophages and fibroblasts ultimately leads to tissue fibrosis, with extracellular matrix deposition, and organ dysfunction.
Postnatally, S1P signaling regulates normal vascular homeostasis. For instance, S1P-S1PR1 signaling maintains endothelial cell barrier functions to limit plasma and leukocyte extravasation (66, 67). Deficiency in this homeostatic signaling system, such as reduced HDL-S1P, leads to compromised barrier function and vascular tone, which is needed to control blood flow and systemic blood pressure (39, 68). In addition, S1P signaling in the endothelium protects the glycocalyx, a protective, glycoprotein-rich layer that lines the apical surface of blood vessels (69). Recent identification of the endoplasmic reticulum-resident protein, NOGO-B, as a negative regulator of de novo sphingolipid synthesis and autocrine S1P signaling in the endothelium suggests the presence of local, as well as systemic, factors in the regulation of vascular tone and blood pressure (70, 71). The control of local S1P signaling may be vascular bed specific, that is, in arteries versus veins and possibly even organ-specific to enable precise control of the vascular system in response to metabolic and physiological cues of a specific organ system (72).
Diabetes and hypertension as well as premature birth frequently lead to vascular diseases of the eye (retina), such as retinopathy, characterized by abnormal blood flow, excessive vascular leak and blood vessel overgrowth. Neutralizing S1P antibodies reduced retinal neovascularization and S1pr2-deficient mice showed attenuated angiogenesis in a mouse model of retinopathy of prematurity, a condition that occurs in premature neonates exposed to high levels of oxygen (73, 74). In diabetes as well as retinopathy of prematurity, the motility of Müller glial cells, which monitor retinal homeostasis, support neurons and contribute to retinal structure and function, is regulated by S1P activation of S1PR3, suggesting a role for pathologic S1P signaling in retinal astrogliosis (scar formation) (75). S1P signaling may be a useful therapeutic target for proliferative retina diseases, such as diabetic retinopathy and age-related macular degeneration (wet form).
Cardiac development is also regulated by S1PR signaling. In zebrafish, the SPHK2-SPNS2-S1PR2-hippo signaling pathway is essential for endoderm development and myocardial progenitor cell migration (14, 28, 76). In mice, S1PR1 function in the endocardium, cardiomyocytes and endothelial cells is essential for embryonic heart development (77). In human induced pluripotent stem cells, S1P induces cardiomyocyte differentiation (78), suggesting a critical role of S1P in development of the myocardium (heart muscle). Although S1P pathway alterations have not been causally linked with congenital heart defects in humans, these observations suggest S1P is involved in post-natal cardiac remodeling events, including heart failure, a complex disease in which cardiac tissue is remodelled and the pumping efficiency of the heart diminishes.
Postnatally, cardiac S1PRs regulate several pathophysiological processes such as cardiac rhythm, vascular perfusion and myocardial protection. S1P induces bradycardia (slow heart rate) by activating the S1PR1-regulated G protein-coupled inwardly-rectifying potassium (GIRK) channels, which hyperpolarize myocardial cells and reduce the rate of action potential firing (79). HDL-induced S1P signaling via endothelial S1PRs allow efficient coronary circulation via eNOS-dependent vasodilatation and protection from myocardial ischemia-reperfusion injury, an important clinical problem following heart attacks (80). Importantly, the S1P pathway is cardioprotective. During ischemic preconditioning, a phenomenon in which small and transient ischemia protects from massive ischemic insults, SPHK is induced in the myocardium resulting in enhanced synthesis of S1P, which protects the heart by promoting circulation and reducing the heart rate (81). Further, in a porcine model of ischemia-reperfusion injury, activation of S1PRs by fingolimod, reduced infarct size and improved cardiac function (82). Moreover, in a mouse model of acute myocardial infarction, treatment with an engineered S1P chaperone (ApoM-Fc) suppressed cardiac injury and enhanced the recovery of cardiac function (67).
An endogenous regulator of S1P metabolism, NOGO-B was shown to modulate plasma and inflammatory cell extravasation from blood vessels in pressure-overloaded hearts and pathological cardiac hypertrophy via endothelial S1PR1 autocrine signaling (83). The complex developmental and pathophysiological roles of S1P in the cardiovascular system provides therapeutic opportunities. For example, endothelial cell-targeted S1PR1 agonism may be useful in limiting tissue damage during ischemia-reperfusion injury. In addition, downregulating S1PRs in autoimmune disease leads to adverse cardiovascular effects, such as bradycardia and vascular leakage, which were predicted by preclinical and mechanistic studies (84). Therefore, S1PR-targeted drugs that are more selective for either the vascular or immune S1PRs may provide better safety profiles in the control of cardiovascular and autoimmune disorders, respectively.
Complex actions in immunity
In addition to its established roles in immune cell trafficking, S1P signaling regulates diverse immunological processes, including cell fate switching, cell survival, innate immunity and anti-tumor immune responses (Figure 4).
Figure 4. S1P function in immunity.

Immune cell function, egress and survival are highly dependent on the regulation of S1P and S1PR signaling. Innate immune cell localization to inflammatory sites, egress of B and T cells from bone marrow and thymus, towards and out of secondary lymphoid organ marginal zones rely on S1PR cell surface expression and S1P gradient. The metabolism of T cell ATP, as well as the regulation of fate switching between TH17 and regulatory T cells are also S1PR1-dependent. The confinement of activated CD4+ T cells in germinal centers within secondary lymphoid organs and spleen is regulated by S1PR2, while SPNS2 and S1PR1 are shown to be implicated in the recruitment of effector T cells to the tumor microenvironment and suppression of metastasis.
Recent studies have illustrated the role of S1P gradients and S1PR signaling in the migration and retention of adaptive immune cells in resident tissues, thus influencing the specificity and magnitude of immune responses. Endothelial cell SPNS2, via the export of S1P, mediates the egress of immature B cells from the bone marrow and naïve T cells from the thymus (30). In the spleen, mature B cell localization and shuttling at the specific immunological region of this organ, called the marginal zone (MZ), is critical for the adpative immune response against blood borne antigens that get trapped in the spleen (85). B cell S1PR1 ensures MZ localization whereas S1PR3 inhibits MZ shuttling, both of which are important for optimal immune responses (86).
B cell confinement and growth regulation in the germinal centers of lymphoid follicles, which are sites for B cell maturation and clonal selection, is controlled by S1PR2 signaling via the Gα13 GPCR signaling pathway (87). In the absence of this spatial restriction, B cell mislocalization is observed, and may contribute to the dissemination of lymphoma (a B cell tumor). A recent study showed that in glioblastoma brain tumor patients, naïve T cell S1PR1 is downregulated, leading to sequestration of such cells in bone marrow, resulting in lymphopenia and decreased anti-tumor immune responses (88). Prevention of this tumor-induced S1PR1 loss of function in T cells could enhance anti-tumor immunotherapy treatments. These illustrative examples demonstrate how spatial S1P gradients and cell-specific S1PR isotypes shape adaptive immune responses.
CD8+ resident memory T cells (CD8+TRM), long-lived, non-circulating cells that are critical for immunological memory and host defense, show reduced expression of the transcription factor Kruppel-like factor 2 (KLF2) and its downstream target gene S1pr1 in mice, thus effectively minimizing egress and achieving tissue retention (89). In addition, the cell-surface lectin CD69 down-regulates S1PR1 in T cells, which also blocks tissue egress and reinforces TRM cell differentiation. CD69 expression is also critical for tissue retention of not only TRM cells, but also early effector T cells, thus impacting rapid immunological memory responses and the magnitude of the adaptive immune response (90). The migration of human effector memory T cells (which are circulating TRM cells), TRM cells and recently activated T cells is inhibited by S1P due to the resulting downregulation of S1PR1 and enhanced expression of S1PR2 on these cells (91). Thus, S1PR-dependent signaling in various memory T cells shape long term immunological memory, a key defense mechanism against repeated infections.
In addition, innate immune cell localization and function, which are important for host defense and inflammatory processes are regulated by S1PR signaling. The positioning of NK cells in the medulla of lymph nodes, which is important for rapid production of the inflammatory cytokine interferon-γ (IFN-γ) after infection, is regulated by S1PR5 (33). Indeed, S1PR5 expression is induced during NK cell maturation and regulates the egress of NK cells from the bone marrow and secondary lymphoid organs (92). In plasmacytoid dendritic cells, S1PR1 blunts the amplification of inflammatory IFN-α-dependent downstream immune responses (93). Intestinal type-2 innate lymphoid cells undergo interorgan migration, a property that is essential to their protective role in parasitic infections, in an S1P-dependent manner (94). S1PR2 promotes the retention of macrophages at sites of inflammation by inhibiting chemokine-induced cellular motility (95). The generality of S1PR-dependent immune cell trafficking influencing pathological processes was recently illustrated by the discovery that the S1P transporter SPNS2 is essential for the recruitment of anti-tumor CD8+ T cells and NK cells to the tumor microenvironment, resulting in the inhibition of metastasis (96).
Moreover, S1PRs also modulate immune cell energy metabolism, which modulates cell fate during immune responses. Naïve T cells constantly survey lymphoid organs for foreign antigens, which necessitates high metabolic flux to generate intracellular adenosine trisphosphate (ATP). In LECs, SPNS2 exports S1P, which activates lymphocyte S1PR1 and helps maintain the mitochondrial content of naïve T cells, thus ensuring high ATP generation by mitochondria (97). S1P is also necessary for the persistent activation of CD4+ T cells in inflamed tissues (98).
S1P signaling in specific contexts appears to regulate T cell subtype fate switching, which determines divergent immune responses. S1PR1 phosphorylation and subsequent internalization in T cells regulates T helper 17 (Th17) cell polarization, which induces pro-inflammatory immune reactions. Specifically, S1PR1 on the plasma membrane of immature T cells promotes differentiation into Th17 cells via interleukin-6 (IL-6)-dependent signal transducer and activator of transcription 3 (STAT3) signaling and enhances autoimmune neuroinflammation (99). S1pr1-deficient mice and MS patients treated with fingolimod (which downregulates S1PR1), exhibit increased levels of regulatory T cells (Treg), which suppress inflammatory responses (100). Thus, S1PR1 regulates proper Th17 and Treg cell distribution across peripheral organs and thereby influence the magnitude of adaptive immune responses. Permanent deletion of S1pr1 in Treg cells leads to spontaneous autoimmunity whereas acute deletion of S1pr1 induces a higher inflammatory response in the experimental autoimmune encephalomyelitis (EAE) model, an animal model for MS in which adaptive immune cells enter the CNS to induce myelin destruction (101). A key unexplored question is how altered signaling of S1PR1 translates to T cell fate switching. It is possible that altered T cell localization brought about by S1PR1-dependent trafficking and retention mechanisms might change the magnitude of cytokine responses that induce fate switching. Alternatively, S1PR signaling may influence T cell intrinsic mechanisms that induce fate switching, i.e., forkhead box P3 (FOXP3) for Treg and retinoid-related orphan receptor-γ (RORγ) and STAT3 for T helper 17 (TH17) cells. However, modulation of the transcription factor STAT3 expression may be involved in S1PR1-dependent pathological immune reactions (102, 103). Although many mechanistic steps have been uncovered, much needs to be learned about receptor specificity, redundant signaling, and involvement in normal and pathological immune responses. This could allow rational design of immunological and anti-inflammatory therapeutics that target S1P signaling.
The central nervous system
The neurovascular unit of the CNS forms a selective barrier called the blood–brain barrier (BBB), composed of endothelial cells, pericytes, astrocytes and microglia, which protects the brain parenchyma from circulatory elements. Dysregulated S1P signaling disrupts the BBB, which is an early event that contributes to many CNS diseases including MS, Alzheimer’s disease (AD) and ischemic stroke (104) (Figure 5). Brain endothelial cell S1PR1 regulates tight junction protein functions, thus leading to the establishment of the BBB. Studies in mice revealed that loss of this mechanism resulted in a size-selective (< 3 Kd) breach of the BBB (105), presumably due to defective tight junction function and increased leakage via a paracellular pathway (that is, between cells). Because transient pharmacological inhibition of brain endothelial cell S1PR1 leads to increased CNS penetration of small molecules, this approach may be potentially useful in enhancing drug delivery in neurological diseases (105). In addition, P glycoprotein, which exports small molecules out of the CNS into the circulation, is inhibited by S1PR inhibitors, which thereby allows enhanced penetration of small molecule drugs into the CNS (106). Such preclinical studies have sparked interest in inhibiting S1PR1 to enhance CNS-targeted therapeutics, a major bottleneck in modern medicine.
Figure 5. S1P in the central nervous system.

S1PRs are important for normal CNS development, neural stem cell self-renewal and differentiation, as well as neural tube closure and the formation of the neurovascular unit (NVU). Dysregulation of S1PRs is also implicated in stroke, subarachnoid hemorrhage (SAH), multiple sclerosis and neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD).
In acute ischemic stroke, SPHK2-dependent S1P production in the brain tissue is neuroprotective, presumably due to extracellular signaling by S1PRs (107). By contrast, S1PR2, which is induced in vascular endothelial cells after ischemia, was associated with increased infarct size, edema, cerebrovascular permeability, hemorrhage and neurovascular injury (108). Fingolimod was effective in rat models of intracerebral hemorrhage and stroke via immunological and non-immunological mechanisms (109). These preclinical studies have prompted the initiation of clinical trials to use fingolimod in the treatment of stroke (110). These studies also highlight the S1P axis in protecting the vasculature during ischemic stress and hence the underlying neuronal tissue following stroke.
The expression of S1PR2 was higher in vascular endothelium and astrocytes within neuroinflammatory disease-susceptible CNS regions of an inbred strain (SJL) of female mice, and in the white matter of female MS patients than in males. In mouse EAE models, S1PR2 function is responsible for the breakdown of adherens junctions, BBB leakage and chemokine-dependent inflammatory responses, thus contributing to disease severity (111). Given that autoimmune neuroinflammatory diseases, such as MS, occur more commonly in females, the identification of S1PR2 as a sex-dimorphic regulator of the BBB permeability raises a number of interesting questions. For example, what are the mechanisms by which blood vessels in the brain of females express higher levels of S1PR2? Would targeting this receptor provide a therapeutic opportunity for the treatment of MS, especially in treatment resistant female MS patients?
In MS, autoreactive immune cells enter the CNS and induce inflammation, leading to demyelination. In addition to S1PR1 activation in autoreactive lymphocytes, which is an important target of S1P therapeutics, S1PR1 and S1PR3 expressed by astrocytes may influence neuroinflammatory processes (112, 113). Indeed, recent studies suggest that SPHK1 and S1PR3 are associated with inflammatory phenotypes of glia (114, 115). More defined mouse genetic studies are needed to precisely elucidate S1PR-mediated inflammatory processes in both immune cells and resident microglial cells of the CNS in various forms of MS. This is an important issue to resolve for the development of more effective therapeutics.
Recent studies also suggest the involvement of S1PR signaling in the pathogenesis of Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD). Associations between decreased S1P levels with AD (116, 117), PD (118) and HD (119) were observed, especially in early stages of these diseases. In addition, defective S1PR signaling was seen in cell culture (120) and preclinical models of neurodegenerative disease (121). Given the high unmet medical need for these conditions, the prominent signaling of S1PRs in the CNS suggests that better understanding of the exact roles of S1P signaling in AD, PD and HD could lead to new therapeutic approaches.
The pathogenesis of fibrosis
Chronic tissue damage coupled with reduced resolution of inflammatory and wound healing responses lead to fibrosis (scarring), thus impairing normal organ function. Abnormal S1P signaling plays an important role in fibrosis of multiple organ systems (Figure 3). When the microvasculature is injured or dysfunctional, tissue parenchyma becomes flooded with plasma S1P, which leads to the activation of S1PRs in interstitial cells. In addition, platelet activation and thrombosis also cause increased local secretion of S1P, which leads to abnormal S1PR activation. Attenuated vascular S1PR1 and increased parenchymal and immune cell S1PR2 and S1PR3 leads to activation of Rho GTPase and Hippo-Yes-associated protein (YAP) signaling pathways that drive fibrosis (76). Thus, pathological S1P signaling in parenchymal and infiltrating immune cells promotes fibrotic diseases of the kidney, liver and the lung.
Acute kidney injury caused by perioperative renal ischemia and reperfusion leads to damage and dysfunction of the tissue, resulting in upregulation of S1PR2 expression in renal proximal tubule cells, which promotes kidney fibrosis. By contrast, SPHK1 and S1PR1 signaling attenuated renal injury, presumably by salvaging the vasculature (122). Treatment of mice with fingolimod decreased the infiltration of immune cells and fibrosis in the kidney after unilateral urethral obstruction (UUO), a model of kidney injury, likely due to transient activation of vascular S1PR1 and inhibition of immune cell trafficking to the kidney (123). In tubulointerstitial renal fibrosis induced in mice, SPHK2 is upregulated. Sphk2-deficient mice show less severe fibrosis in the UUO model; and this correlates with suppression of transforming growth factor-β (TGF-β) signaling in the kidney parenchymal cells and increased macrophage polarization towards pro-healing and anti-inflammatory (M2) macrophages (124, 125). Further, genetic deletion of Sphk2 in bone marrow in mice is sufficient to protect from folic acid-induced renal fibrosis via the signaling of IFN-γ, CXC ligand 9 (CXCL9) and CXCL10 (126) on infiltrated hematopoietic cells as well as kidney parenchymal cells. Because kidney injury and fibrosis occur in many clinical conditions, including diabetes, hypertension, autoimmune diseases and infections, the identification of S1PR signaling activation as a renal protective mechanism has revealed numerous therapeutic opportunities such as S1PR1-selective agonists and SPHK inhibitors.
S1P pathway dysregulation is also involved in liver fibrosis, a serious chronic disease that is associated with fatty liver disease, hepatitis and alcoholism. In human tissues, SPHK1, SPNS2 and S1PR2 expression correlate with the severity of liver fibrosis (127). Increased liver S1P levels activate hepatic stellate cells, which are located between sinusoidal vessels and hepatocytes, to undergo fibrotic changes (127, 128), and inhibition of S1PR2 with a pharmacological antagonist attenuated liver fibrosis in mice (129). S1P signaling in bone marrow-derived macrophages recruited to the injured liver in mice may be involved in the amplification of fibrotic responses (130, 131). Liver regeneration in mice following partial hepatectomy or bile duct ligation is dependent on S1PR1 expressed by liver sinusoidal endothelial cell (LSEC) activation by ApoM+HDL-bound S1P and suppression of Rho signaling. Hepatic sinusoidal vascular remodeling and the restoration of functional liver tissue is impaired in Apom-deficient mice, which showed characteristics of perivascular fibrosis and thrombosis. Conversely, ApoM transgenic mice showed minimal fibrosis via endothelial S1PR1 function in liver sinusoids (40).
Idiopathic pulmonary fibrosis (IPF), a progressive lung disease that occurs in ~0.1% of the population worldwide, leads to disability and death. The S1P pathway is implicated in normal function of the pulmonary vasculature and appears to be impaired in acute lung dysfunction while it is ectopically induced in chronic fibrosis. Patients with IPF have increased S1P levels in serum and bronchoalveolar lavage (BAL) and increased SPHK1 expression in lung alveolar macrophages, which correlated with markers of epithelial mesenchymal transition (EMT) and fibrosis such as α-smooth muscle actin (α-SMA), vimentin and collagen type 1. In alveolar type II cells in the lung, S1P activates S1PR2 and S1PR3, and induces EMT through the activation of phosphorylated-SMAD3, RhoA-GTP, oxidative stress and TGF-β1 release (132). The use of receptor-selective agonists revealed that although endothelial cell S1PR1 protects from fibrosis and promotes normal lung function, S1PR2 and S1PR3 expressed in fibroblasts and infiltrating myeloid cells, promotes fibrosis, such as EMT marker expression, increased proliferation of myofibroblasts and extracellular matrix deposition (133). The fibrosis-inducing potential of the DNA damaging agent bleomycin in mouse models was attenuated in S1pr2- and S1pr3-deficient mice (134, 135). In these preclinical models, collaboration of inflammatory cells and lung parenchymal cells occurs in the complex S1P signaling network to drive fibrotic responses. These observations suggest agonism of vascular endothelial S1PR1 and antagonism of parenchymal S1PR2 and/or S1PR3 as potential therapeutic approaches in fibrotic diseases of multiple organ systems. Whether the results from mouse models will be relevant to human fibrotic disease is not clear and requires large animal pre-clinical models and ultimately clinical trials with appropriate S1PR-selective agents.
Therapeutic opportunities
Fingolimod, the first FDA approved S1PR-targeted drug, is an effective first line therapy in relapsing remitting multiple sclerosis (RRMS). The primary mechanism of action is as a functional antagonist of S1PR1 expressed on lymphocytes (24), thereby preventing the egress of autoreactive lymphocytes from secondary lymphoid organs and reducing their migration into the CNS, thus slowing disease progression. Substantial adverse events such as first-dose bradycardia (reduction of heart rate) and macular edema (vascular leak into the center of the retina) limits the usage of fingolimod (84). The adverse events can be explained by on-target actions on S1PRs expressed by the heart and retinal vasculature.
Currently, several clinical trials are underway to test the effectiveness of the next generation S1PR-targeted agents (Table 1) (136). Such agents also induce functional antagonism of S1PR1 in lymphocytes and are more selective in their interaction with other S1PRs. One such compound, siponimod, is a selective antagonist of S1PR1 and S1PR5, was shown to have efficacy in both RRMS and progressive forms of MS in human patients, but had similar adverse event profiles as fingolimod (137), which confirms that interference with S1PR1 in the heart and vasculature is the cause of these effects. Siponimod achieved FDA approval in 2019. Siponimod antagonism on S1PR5 is thought to enhance survival and/or differentiation of mouse oligodendrocytes, which are defective in MS and neurodegenerative diseases (138). Subsequent studies of fingolimod, as well as the recent phase III studies with siponimod (137) found that S1PR functional antagonist therapy of MS patients protects from accelerated brain atrophy. This finding warrants further studies to examine if this therapeutic axis is useful in neurodegenerative diseases in general. Furthermore, because S1PR1-dependent autoreactive immune cell trafficking to target organs is common to many autoimmune diseases, this therapeutic approach is likely applicable to other non-CNS autoimmune diseases. As such, clinical trials are underway to treat patients with psoriasis, inflammatory bowel disease and systemic lupus erythromatosus (Table 1).
Table 1. S1P modulators under development or in clinical testing.
RRMS: Relapsing Remitting Multiple Sclerosis, PPMS: Primary Progressive Multiple Sclerosis, SPMS: Secondary Progressive Multiple Sclerosis, RMS: Relapsing Multiple Sclerosis, UC: Ulcerative Colitis, CIPD: Chronic Inflammatory Demyelinating Polyradiculoneuropathy, ALS: Amyotrophic Lateral Sclerosis, SLE: Systemic Lupus Erythematosus, SCLE: Subacute Cuteaneous Lupus Erythematosus RA: Rheumatoid Arthritis, AMD: Age-related Macular Degeneration.
| MOLECULE NAME | DRUG NAME | TARGETED PROTEIN | SPONSOR | PROPOSED OR APPROVED USE | CURRENT TRIAL PHASE | CT ID |
|---|---|---|---|---|---|---|
| Fingolimod (Gilenya) | S1PR1, 3, 4, 5 | Novartis | RRMS | Marketed | ||
| Transplant | III | NCT00099801 | ||||
| CIPD | III | NCT01625182 | ||||
| PPMS | III | NCT00731692 (INFORMS) | ||||
| ALS | II | NCT01786174 | ||||
| Asthma | II | NCT00785083 | ||||
| BAF312 | Siponimod (Mayzent) | S1PR1, 5 | Novartis | SPMS | FDA approved | NCT01665144 (EXPAND) |
| RRMS | II | NCT01185821 | ||||
| Active dermatomyositis | II | NCT02029274 | ||||
| Hemorrhagic stroke, ICH | II | NCT03338998 | ||||
| Hepatic impairment | I | NCT01565902 | ||||
| KRP-203 | S1PR1, 5 | Novartis | UC | II | NCT01375179 | |
| SCLE | II | NCT01294774 | ||||
| Hematological malignancies | I | NCT01830010 | ||||
| ACT-128800 | Ponesimod | S1PR1, 3, 5 | Actelion | MS | III | NCT03232073 (OPTIMUM-LT) |
| Psoriasis | II | NCT01208090 | ||||
| ACT-334441 | Cerenimod | S1PR1, 5 | Actelion | SLE | II | NCT02472795 |
| APD334 | Etrasimod | S1PR1 | Arena Pharmaceuticals | UC | III | NCT03945188 |
| RPC1063 | Ozanimod | S1PR1, 5 | Receptos (Celgene) | Crohn | III | NCT03440372 |
| NCT03440385 | ||||||
| UC | III | NCT02435992 | ||||
| NCT02531126 | ||||||
| RMS | III | NCT02576717 | ||||
| ONO-4641 | Ceralifimod | S1PR1, 5 | Ono | RRMS | II | NCT01081782 (DreaMS) |
| MT-1303 | Amiselimod | S1PR1, 4, 5 | Mitsubishi Tanabe | RRMS | II | NCT01742052 |
| Crohn | II | NCT02389790 | ||||
| Psoriasis | II | NCT01987843 | ||||
| SLE | I | NCT02307643 | ||||
| AKP11 | S1PR1 | Akaal Pharma | Atopic dermatitis | II | ACTRN12617000763347 | |
| RA | II | ACTRN12617001223325 | ||||
| GSK-2018682 | S1PR1 | GlaxoSmithKline | RRMS | I | NCT01431937 (P1A114347) | |
| BMS-986104 | S1PR1 | Bristol-Myers Squibb | RA | I | NCT02211469 | |
| CS-0777 | S1PR1 | Daiichi Sankyo, Inc | MS | I | NCT00616733 | |
| LT1009 (Sonepcizumab) | ASOPNEP | S1P antibody | Lpath Inc / Pfizer | Renal cell carcinoma | II | NCT01762033 |
| iSONEP | AMD | II | NCT01414153 (Nexus) | |||
| ABC294640 | Opaganib (Yeliva®) | SphK2 | RedHill Biopharma Limited | Cholangiocarcinoma | II | NCT03377179 |
| Hepatocellular carcinoma | II | NCT02939807 | ||||
| Solid Tumor | I | NCT01488513 (ABC-101) | ||||
| DHS (L-threo-Dihydrosphingosine) | Safingol | SphK1 | MSKKC/NCI | Locally advanced or metastatic solid tumors | 1 | NCT00084812 |
| LX3305 | SPGL1 | Lexicon Pharmaceuticals | RA | II | NCT00903383 |
Biased agonists of S1PR1 may achieve vascular protective effects in diabetes, metabolic and cardiovascular diseases (38). Activation by S1P of S1PR1 in the endothelium leads to receptor downregulation (endocytosis), which limits the response to agonists. However, biased agonists of endothelial S1PR1 that do not induce S1PR1 endocytosis and thus receptor degradation may induce sustained endothelial protective effects such as NO synthesis, increased barrier function, endothelial cell survival, among others, thus protecting the vasculature. However, it is possible that such agents could have distinct adverse effect profiles, for example, hypotension. Biased agonists for other GPCRs such as μ-opioid receptor, dopamine D2 receptor and angiotensin receptor-1 are being actively developed to achieve clinical efficacy and reduced adverse effect profiles (139). In some cases, as in the case of the μ-opioid receptor, a biased agonist potently achieved analgesia with limited adverse effects (140).
Small molecule pharmacological antagonists have been used to target other S1PRs in preclinical models of inflammation and fibrosis (129, 131, 135, 141). This approach may potentially lead to adverse events such as hearing defects (35), lymphoid neoplasms (87), and epileptic seizures (142), which have been observed in S1pr2 genetic loss of function mouse models. S1PR redundancy may also lead to limited efficacy in targeting a single receptor isoform and therefore, targeting multiple receptor isoforms that carry out similar biological effects may prove to be more effective.
S1P chaperones allow an additional opportunity for S1PR modulation. For example, therapeutic delivery of engineered ApoM, namely, ApoM-Fc, suppresses inflammation (39, 41) and promotes vascular homeostasis in mouse models of myocardial ischemia-reperfusion injury, chronic hypertension and experimental stroke (67). In sharp contrast to small molecule S1PR1-targeted drugs, ApoM-S1P treatment did not lead to lymphopenia, suggesting the inability of chaperone-bound S1P to downregulate S1PR1 in secondary lymphoid organs. This ApoM-S1P therapeutic approach may selectively activate endothelial cell S1PRs without inducing functional antagonism of immune cell S1PRs. However, ApoM-S1P can activate multiple receptors and therefore could potentially lead to undesirable side effects by activating pro-inflammatory S1PR2 and S1PR3. Selective activation of S1PRs by modified chaperone-based approaches may need to be developed to provide therapeutic utility.
Selective activation of S1P transporters may have utility in cancer metastasis and anemia (34, 96). In metastasis, SPNS2 activation, which allows S1P export, signaling by immune cell S1PRs to regulate trafficking, may enhance CD8+ T cell and NK cell infiltration in tumors and potentially enhance anti-tumor immunity. In anemia, activation of the MFSD2B transporter in RBCs may help reduce the concentration of sphingosine and S1P in the cell membranes of RBCs, which was shown to reduce recovery from anemia in mice.
The inhibition of the metabolic enzymes involved in S1P biosynthesis may also have therapeutic utility. An S1P lyase inhibitor induced lymphopenia and suppressed autoimmune inflammation by enhancing S1P levels (143) (Table 1). However, broad spectrum sphingolipid alterations induced by inhibition of S1P lyase could lead to side effects. Similarly, even though SPHK inhibitors have been tested in preclinical models to inhibit the over-active SPHK1 enzyme in cancer (144), recent work with highly specific and potent inhibitors suggest they have limited utility, indicating that off-target effects of early SPHK-targeted agents may be involved (145). SPHK2 inhibitors led to increased plasma S1P and attenuation of inflammatory and fibrotic responses (146). Because modulation of S1P metabolic enzymes led to broad changes in the metabolite concentrations and flux of not only sphingolipids but also other phospholipids, this approach will need to carefully consider lipid metabolism prior to clinical application.
Conclusions and future perspectives
The use of mouse models and modern pharmacological tools have led to our current understanding of the biology of the S1P-S1PR signaling system. With the application of S1PR-targeted drugs in humans, much has been learned about the role of this pathway in the immune system and its potential future application in immunological, neurological and perhaps cardiovascular and fibrotic diseases. S1PR1 functional antagonists that induce irreversible S1PR internalization and degradation are the most advanced class of compounds, whereas competitive antagonists have not yet been tested in clinical trials. Perhaps functional antagonism of the receptors may be preferable to block S1PRs in ligand-rich compartments and may improve efficacy in the clinic. However, the emerging biological understanding and clinical research suggest additional opportunities for therapeutic application, including transporter modulators, biased agonists of S1PRs, chaperone-based strategies and ligand neutralization approaches. Such approaches, which attenuate abnormal S1PR signaling and normalize spatial S1P gradients, may be useful to combat diseases in the context of the complex S1P signaling mechanisms that are prevalent in many organ systems.
PRINT SUMMARY.
Background:
Sphingosine 1-phosphate (S1P), a product of membrane sphingolipid metabolism, is secreted and acts via G protein-coupled S1P receptors (S1PRs) in vertebrates. S1PR isoforms mediate complex cellular actions either alone or in combination in most organ systems. This stable lysolipid circulates as a complex with protein chaperones that not only enables aqueous solubility, but also helps facilitate specific modes of receptor signaling. However, differential concentration gradients of S1P are normally present in various compartments and are perturbed under disease conditions. The abundance of circulatory S1P and the high expression of S1PRs in exposed cells, that is, vascular and hematopoietic cells, poses a key question of how this signaling axis is regulated. Indeed, this question is of clinical relevance because the first S1PR-targeted drug, fingolimod, has been approved for the treatment of multiple sclerosis since 2010. Recent findings from basic research as well as insights gleaned from clinical and translational studies have enriched our understanding of how this simple lysolipid evolved as a complex regulator of multiple physiological systems, and when dysregulated, contribute to numerous diseases.
Advances:
Extracellular spatial gradients of S1P, demonstrated using S1P reporters, are tightly regulated and control fundamental processes such as hematopoietic cell trafficking, immune cell fate and vascular integrity. The gradients are formed by location-specific function of metabolic enzymes, S1P transporters and chaperones. Such physiological S1P gradients are altered in diseases, thus contributing to conditions such as inflammation, autoimmunity and vascular dysfunction. S1P complexed to chaperone proteins, for example high density lipoprotein-bound apolipoprotein M, mediate unique modes of receptor activation resulting in biased receptor signaling and specific biological outcomes. S1PRs are also regulated tightly by endocytic mechanisms and receptor modulators that enhance or inhibit signal strength and duration. Various signaling mechanisms of this simple lysolipid mediator has helped reveal its multiple actions in the immune system, which include adaptive immune cell localization in various compartments (egress versus retention), fate switching, survival and activation that influences both cell-mediated and humoral immunity. In the cardiovascular system, high expression of multiple S1PR isoforms in various cell types regulate development, homeostasis and physiology. Current S1PR-targeted drugs which aim to tame autoimmunity exhibit considerable cardiovascular adverse events. In the central nervous system, widespread application of S1PR-targeted drugs in autoimmune neuroinflammatory diseases has stimulated research that revealed the broad but poorly understood effects of S1P signaling in neurodevelopment, the neurovascular unit, neurons and glia. Furthermore, in addition to the involvement of pathological S1P signaling in acute ischemic conditions of various organs, chronic dysregulated S1P signaling has been implicated in fibrotic diseases of lung, heart, liver and kidney.
Outlook:
Considerable challenges remain to fully harness the new knowledge in S1P pathobiology to translational utility in clinical medicine. Approaches that mimic S1P chaperones, S1P neutralizing agents, modulation of transporters, biased agonists and antagonists of S1PR isotypes, and sphingolipid metabolic enzyme modulators provide viable pathways to therapy. Focusing on the immune system, such approaches may widen the autoimmunity therapeutic landscape and provide new directions in cancer and chronic inflammatory diseases. For cardiovascular diseases, ischemic conditions as well as chronic heart failure are likely candidates for future translational efforts. Although further work is needed, S1P-targeted approaches may also be useful in regenerative therapies for the aging and diseased myocardium. The central nervous system (CNS)-targeted efforts may cross into neurodegenerative diseases, given the success with S1PR-targeted drugs in reducing brain atrophy in multiple sclerosis. Other potential applications include approaches in pain management and neurodevelopmental disorders. Such strategies, although challenging, are greatly helped by findings from basic research on S1P pathobiology as well as pharmacological and clinical insights derived from the application of S1P-targeted therapeutics.
Figure 0. S1PR signaling regulates multiorgan pathophysiological processes.
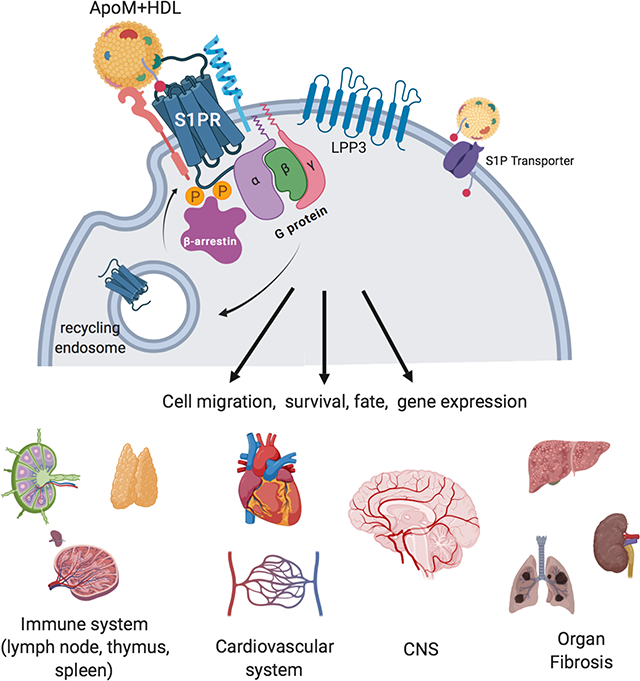
Extracellular S1P gradients created by transporters, chaperones (ApoM+HDL) and metabolic enzymes (LPP3) interact with S1PRs on the cell surface. Receptor activity, transmitted via G proteins, is regulated by multiple mechanisms including β-arrestin coupling, endocytosis and receptor modulators. The resultant cellular changes influence multiple organ systems in physiology and disease.
Acknowledgements:
Funding: This work is supported by NIH grant R35 HL135821 (TH), Fondation Leducq transatlantic network grant (SphingoNet)(TH) and a postdoctoral fellowship from the American Heart Association (AC). AC and TH both wrote and edited the manuscript. AC prepared figures 1–5 and compiled Table I. Competing interests: TH received a research grant from the ONO Pharmaceutical Inc., is an inventor in the patents and patent applications related to ApoM-Fc, ApoM+-HDL and S1P receptor modulators, and has consulted for the following commercial entities (Abbott Inc., Pfizer Inc., Sandoz Inc., Astellas Inc., Novartis Inc., Trevana Inc. and Arena Pharmaceuticals Inc.).
References:
- 1.Hla T, Genomic insights into mediator lipidomics. Prostaglandins Other Lipid Mediat 77, 197–209 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Shimizu T, Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol 49, 123–150 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Proia RL, Hla T, Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 125, 1379–1387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoffel W, Sticht G, LeKim D, Metabolism of sphingosine bases. IX. Degradation in vitro of dihydrospingosine and dihydrospingosine phosphate to palmitaldehyde and ethanolamine phosphate. Hoppe Seylers Z Physiol Chem 349, 1745–1748 (1968). [DOI] [PubMed] [Google Scholar]
- 5.Ghosh TK, Bian J, Gill DL, Intracellular calcium release mediated by sphingosine derivatives generated in cells. Science 248, 1653–1656 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Zhang H et al. , Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol 114, 155–167 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH, International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 62, 579–587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez SE et al. , Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hait NC et al. , Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strub GM et al. , Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25, 600–612 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kajimoto T et al. , Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter. Sci Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panneer Selvam S et al. , Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci Signal 8, ra58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etemadi N et al. , TRAF2 regulates TNF and NF-kappaB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendelson K, Lan Y, Hla T, Evans T, Maternal or zygotic sphingosine kinase is required to regulate zebrafish cardiogenesis. Dev Dyn 244, 948–954 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendelson K et al. , The ceramide synthase 2b gene mediates genomic sensing and regulation of sphingosine levels during zebrafish embryogenesis. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong Y et al. , Sphingosine kinases are not required for inflammatory responses in macrophages. The Journal of biological chemistry 288, 32563–32573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hannun YA, Obeid LM, Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9, 139–150 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Murata N et al. , Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J 352 Pt 3, 809–815 (2000). [PMC free article] [PubMed] [Google Scholar]
- 19.Schwab SR et al. , Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 309, 1735–1739 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Christoffersen C et al. , Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A 108, 9613–9618 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagida K, Hla T, Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu Rev Physiol 79, 67–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lovric S et al. , Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest 127, 912–928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos-Cortez RL et al. , Autosomal-Recessive Hearing Impairment Due to Rare Missense Variants within S1PR2. Am J Hum Genet 98, 331–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkmann V et al. , Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9, 883–897 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Lander AD, Morpheus unbound: reimagining the morphogen gradient. Cell 128, 245–256 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Maceyka M, Spiegel S, Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawahara A et al. , The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323, 524–527 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Kupperman E, An S, Osborne N, Waldron S, Stainier DY, A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature 406, 192–195 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Osborne N et al. , The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr Biol 18, 1882–1888 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuhara S et al. , The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest 122, 1416–1426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendoza A et al. , The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep 2, 1104–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramos-Perez WD, Fang V, Escalante-Alcalde D, Cammer M, Schwab SR, A map of the distribution of sphingosine 1-phosphate in the spleen. Nat Immunol 16, 1245–1252 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang V et al. , Gradients of the signaling lipid S1P in lymph nodes position natural killer cells and regulate their interferon-γ response. Nat Immunol 18, 15–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vu TM et al. , Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 550, 524–528 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Chen J et al. , Spinster homolog 2 (spns2) deficiency causes early onset progressive hearing loss. PLoS Genet 10, e1004688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang C et al. , S1P transporter SPNS2 regulates proper postnatal retinal morphogenesis. FASEB J, fj201701116R (2018). [DOI] [PubMed] [Google Scholar]
- 37.Fleming JK, Wojciak JM, Measuring Sphingosine-1-Phosphate/Protein Interactions with the Kinetic Exclusion Assay. Methods Mol Biol 1697, 1–8 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Galvani S et al. , HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal 8, ra79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen PM et al. , Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J 30, 2351–2359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding BS et al. , HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P. JCI Insight 1, e87058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurano M et al. , Apolipoprotein M Protects Lipopolysaccharide-Treated Mice from Death and Organ Injury. Thromb Haemost, (2018). [DOI] [PubMed] [Google Scholar]
- 42.Blaho VA et al. , HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 523, 342–346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee MH et al. , S1P in HDL promotes interaction between SR-BI and S1PR1 and activates S1PR1-mediated biological functions: calcium flux and S1PR1 internalization. J Lipid Res 58, 325–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venkataraman K et al. , Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res 102, 669–676 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnon TI et al. , GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science 333, 1898–1903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thangada S et al. , Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med 207, 1475–1483 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willinger T, Ferguson SM, Pereira JP, De Camilli P, Flavell RA, Dynamin 2-dependent endocytosis is required for sustained S1PR1 signaling. J Exp Med 211, 685–700 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nomachi A et al. , Moesin controls clathrin-mediated S1PR1 internalization in T cells. PLoS One 8, e82590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oo ML et al. , Engagement of S1P₁-degradative mechanisms leads to vascular leak in mice. J Clin Invest 121, 2290–2300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kono M et al. , Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. The Journal of clinical investigation 124, 2076–2086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kono M et al. , Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nat Commun 8, 1163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Studer E et al. , Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 55, 267–276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feistritzer C, Riewald M, Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 105, 3178–3184 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Singleton PA, Dudek SM, Ma SF, Garcia JG, Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem 281, 34381–34393 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Shiow LR et al. , CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Kempf A et al. , The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS Biol 12, e1001763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hisano Y et al. , Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J Exp Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laidlaw BJ, Gray EE, Zhang Y, Ramirez-Valle F, Cyster JG, Sphingosine-1-phosphate receptor 2 restrains egress of gammadelta T cells from the skin. J Exp Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanson MA et al. , Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaengel K, Genove G, Armulik A, Betsholtz C, Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29, 630–638 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Jung B et al. , Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 23, 600–610 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee MJ et al. , Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Paik JH et al. , Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev 18, 2392–2403 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiong Y, Yang P, Proia RL, Hla T, Erythrocyte-derived sphingosine 1-phosphate is essential for vascular development. J Clin Invest 124, 4823–4828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gazit SL et al. , Platelet and Erythrocyte Sources of S1P Are Redundant for Vascular Development and Homeostasis, but Both Rendered Essential After Plasma S1P Depletion in Anaphylactic Shock. Circ Res 119, e110–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burg N, Swendeman S, Worgall S, Hla T, Salmon JE, Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol 70, 1879–1889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swendeman SL et al. , An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perségol L et al. , Small dense HDLs display potent vasorelaxing activity, reflecting their elevated content of sphingosine-1-phosphate. J Lipid Res 59, 25–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeng Y, Adamson RH, Curry FR, Tarbell JM, Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol 306, H363–372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cantalupo A et al. , Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat Med 21, 1028–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cantalupo A et al. , S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 70, 426–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Książek M, Baranowska U, Chabowski A, Baranowski M, Arteriovenous Sphingosine-1-Phosphate Differences Across Selected Organs of the Rat. Cell Physiol Biochem 45, 67–77 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Skoura A et al. , Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J Clin Invest 117, 2506–2516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Caballero S et al. , Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp Eye Res 88, 367–377 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simón MV, Prado Spalm FH, Politi LE, Rotstein NP, Sphingosine-1-Phosphate Is a Crucial Signal for Migration of Retina Müller Glial Cells. Invest Ophthalmol Vis Sci 56, 5808–5815 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Fukui H et al. , S1P-Yap1 signaling regulates endoderm formation required for cardiac precursor cell migration in zebrafish. Dev Cell 31, 128–136 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Clay H et al. , Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev Biol 418, 157–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma A et al. , Stage-specific Effects of Bioactive Lipids on Human iPSC Cardiac Differentiation and Cardiomyocyte Proliferation. Sci Rep 8, 6618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ochi R, Momose Y, Oyama K, Giles WR, Sphingosine-1-phosphate effects on guinea pig atrial myocytes: Alterations in action potentials and K+ currents. Cardiovasc Res 70, 88–96 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Levkau B et al. , High-density lipoprotein stimulates myocardial perfusion in vivo. Circulation 110, 3355–3359 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Vessey DA et al. , Role of sphingosine kinase activity in protection of heart against ischemia reperfusion injury. Med Sci Monit 12, BR318–324 (2006). [PubMed] [Google Scholar]
- 82.Santos-Gallego CG et al. , Sphingosine-1-Phosphate Receptor Agonist Fingolimod Increases Myocardial Salvage and Decreases Adverse Postinfarction Left Ventricular Remodeling in a Porcine Model of Ischemia/Reperfusion. Circulation 133, 954–966 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Zhang Y et al. , Endothelial Nogo-B regulates sphingolipid biosynthesis to promote pathological cardiac hypertrophy during chronic pressure overload. JCI Insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Camm J, Hla T, Bakshi R, Brinkmann V, Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J 168, 632–644 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Arnon TI, Horton RM, Grigorova IL, Cyster JG, Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature 493, 684–688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tedford K et al. , The opposing forces of shear flow and sphingosine-1-phosphate control marginal zone B cell shuttling. Nat Commun 8, 2261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muppidi JR et al. , Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature 516, 254–258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chongsathidkiet P et al. , Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24, 1459–1468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Skon CN et al. , Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mackay LK et al. , Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol 194, 2059–2063 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Drouillard A et al. , Human Naive and Memory T Cells Display Opposite Migratory Responses to Sphingosine-1 Phosphate. J Immunol 200, 551–557 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Drouillard A et al. , S1PR5 is essential for human natural killer cell migration toward sphingosine-1 phosphate. J Allergy Clin Immunol, (2017). [DOI] [PubMed] [Google Scholar]
- 93.Teijaro JR et al. , S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-α autoamplification. Proc Natl Acad Sci U S A 113, 1351–1356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang Y et al. , S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 359, 114–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Michaud J, Im DS, Hla T, Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J Immunol 184, 1475–1483 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van der Weyden L et al. , Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 541, 233–236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mendoza A et al. , Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature 546, 158–161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jaigirdar SA et al. , Sphingosine-1-Phosphate Promotes the Persistence of Activated CD4 T Cells in Inflamed Sites. Front Immunol 8, 1627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garris CS et al. , Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nature immunology 14, 1166–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Claes N et al. , Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One 9, e111115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eken A et al. , S1P. Sci Rep 7, 12905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Degagné E et al. , Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest 124, 5368–5384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liang J et al. , Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23, 107–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV, Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163, 1064–1078 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yanagida K et al. , Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci U S A 114, 4531–4536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cannon RE, Peart JC, Hawkins BT, Campos CR, Miller DS, Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci U S A 109, 15930–15935 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pfeilschifter W et al. , Activation of sphingosine kinase 2 is an endogenous protective mechanism in cerebral ischemia. Biochem Biophys Res Commun 413, 212–217 (2011). [DOI] [PubMed] [Google Scholar]
- 108.Kim GS et al. , Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat Commun 6, 7893 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rolland WB et al. , Fingolimod reduces cerebral lymphocyte infiltration in experimental models of rodent intracerebral hemorrhage. Exp Neurol 241, 45–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fu Y et al. , Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A 111, 18315–18320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cruz-Orengo L et al. , Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J Clin Invest 124, 2571–2584 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dusaban SS, Chun J, Rosen H, Purcell NH, Brown JH, Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J Neuroinflammation 14, 111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choi JW et al. , FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108, 751–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fischer I et al. , Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS One 6, e23905 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liddelow SA et al. , Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ceccom J et al. , Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol Commun 2, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Couttas TA et al. , Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol Commun 2, 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Taguchi YV et al. , Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J Neurosci 37, 9617–9631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Di Pardo A, Maglione V, The S1P Axis: New Exciting Route for Treating Huntington’s Disease. Trends Pharmacol Sci 39, 468–480 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Zhang L et al. , Extracellular α-synuclein induces sphingosine 1-phosphate receptor subtype 1 uncoupled from inhibitory G-protein leaving β-arrestin signal intact. Sci Rep 7, 44248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aytan N et al. , Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci Rep 6, 24939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT, Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 23, 266–280 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thangada S et al. , Treatment with the immunomodulator FTY720 (fingolimod) significantly reduces renal inflammation in murine unilateral ureteral obstruction. J Urol 191, 1508–1516 (2014). [DOI] [PubMed] [Google Scholar]
- 124.Schwalm S et al. , Sphingosine Kinase-2 Deficiency Ameliorates Kidney Fibrosis by Up-Regulating Smad7 in a Mouse Model of Unilateral Ureteral Obstruction. Am J Pathol 187, 2413–2429 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Ghosh M et al. , Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS One 13, e0194053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bajwa A et al. , Sphingosine Kinase 2 Deficiency Attenuates Kidney Fibrosis. J Am Soc Nephrol 28, 1145–1161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sato M et al. , Sphingosine kinase-1, S1P transporter spinster homolog 2 and S1P2 mRNA expressions are increased in liver with advanced fibrosis in human. Sci Rep 6, 32119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang L et al. , Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis is involved in liver fibrosis-associated angiogenesis. J Hepatol 59, 114–123 (2013). [DOI] [PubMed] [Google Scholar]
- 129.Ikeda H et al. , Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J Lipid Res 50, 556–564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stelling A et al. , The tumor suppressive TGF-β/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood, (2018). [DOI] [PubMed] [Google Scholar]
- 131.Yang L et al. , Sphingosine 1-Phosphate Receptor 2 and 3 Mediate Bone Marrow-Derived Monocyte/Macrophage Motility in Cholestatic Liver Injury in Mice. Sci Rep 5, 13423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Milara J et al. , Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 67, 147–156 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Sobel K et al. , Sphingosine 1-phosphate (S1P) receptor agonists mediate pro-fibrotic responses in normal human lung fibroblasts via S1P2 and S1P3 receptors and Smad-independent signaling. J Biol Chem 288, 14839–14851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao J et al. , Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS One 13, e0197604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Murakami K et al. , Knock out of S1P3 receptor signaling attenuates inflammation and fibrosis in bleomycin-induced lung injury mice model. PLoS One 9, e106792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dyckman AJ, Modulators of Sphingosine-1-phosphate Pathway Biology: Recent Advances of Sphingosine-1-phosphate Receptor 1 (S1P. J Med Chem 60, 5267–5289 (2017). [DOI] [PubMed] [Google Scholar]
- 137.Kappos L et al. , Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 391, 1263–1273 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Jaillard C et al. , Edg8/S1P5: an oligodendroglial receptor with dual function on process retraction and cell survival. J Neurosci 25, 1459–1469 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rankovic Z, Brust TF, Bohn LM, Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26, 241–250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Viscusi ER et al. , A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain 157, 264–272 (2016). [DOI] [PubMed] [Google Scholar]
- 141.Skoura A et al. , Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol 31, 81–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.MacLennan AJ et al. , An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur J Neurosci 14, 203–209 (2001). [DOI] [PubMed] [Google Scholar]
- 143.Billich A et al. , Partial deficiency of sphingosine-1-phosphate lyase confers protection in experimental autoimmune encephalomyelitis. PLoS One 8, e59630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pyne S, Adams DR, Pyne NJ, Sphingosine Kinases as Druggable Targets. Handb Exp Pharmacol, (2018). [DOI] [PubMed] [Google Scholar]
- 145.Schnute ME et al. , Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J 444, 79–88 (2012). [DOI] [PubMed] [Google Scholar]
- 146.Kharel Y et al. , Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J Pharmacol Exp Ther 355, 23–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]