

Abstract
Keeping a balance between DNA methylation and demethylation balance is central for mammalian development and cell function, particularly in the hematopoietic system. In various mammalian cells, Tet methylcytosine dioxygenase 2 (Tet2) catalyzes oxygen transfer to a methyl group of 5-methylcytosine (5mC), yielding 5-hydroxymethylcytocine (5hmC). Tet2 mutations drive tumorigenesis in several blood cancers as well as in solid cancers. Here I discuss recent studies that elucidate mechanisms and biological consequences of Tet2 dysregulation in blood cancers. I focus on recent findings concerning Tet2 involvement in lymphoid and myeloid cell development and its functional roles, which may be associated with tumorigenesis. I also discuss how Tet2 activities are modulated by microRNAs, metabolites, and other interactors, including vitamin C and 2-hydroxyglutarate (2-HG), and review the clinical relevance and potential therapeutic applications of Tet2 targeting. Finally, I propose key unanswered hypotheses regarding Tet2 in the cancer-immunity cycle.
Subject terms: Tumour immunology, Tumour immunology
Shuai Jiang discusses the regulation and functional role of the Tet2 enzyme, a regulator of lymphoid and myeloid cell development frequently mutated in haematopoietic cancers. He highlights how Tet2 activity may be modulated to influence cancer immune responses.
Introduction
DNA methylation/demethylation is dynamically coordinated throughout hematopoietic differentiation, and Tet proteins play crucial roles in adjusting gene expression levels through balancing DNA methylation in hematopoiesis and during immune-cell activation and expansion1. The first identified gene of the Tet family, Tet1, acts as a fusion regulator in cases of acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL)2. Two other Tet genes, Tet2 and Tet3, were later identified based on sequence homology with Tet13. In mammalian cells, all Tet family members, Tet1, Tet2, and Tet3, catalyze the successive oxidation of 5mC, yielding 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC)4.
The Tet2 gene is subjected to frequent somatic mutations in an extensive range of hematopoietic cancers, including myeloid and lymphoid cancer, and several solid cancers5,6. DNA modification by Tet2 is fundamental for gene control in both cancer cells and immune-cell subtypes5,7,8. Tet2 protein and its downstream effectors-5hmC/5mC-mediated DNA modification are constitutional in immune cells, including T cells, B cells, and macrophages in both physiological and pathological conditions9 (Fig. 1). In blood cancer cells, Tet2 loss is the primary cause of 5mC generation10, whereas loss of functional roles of all three Tets is robustly linked to solid cancer progression11. Tet2 is one of the commonly mutated genes in hematopoietic cancers12. In engineered mouse models, Tet2 knockout (KO), or double KO (DKO) of Tet2 and Tet3, causes myeloid or lymphoid cell spread and the progression of fully infiltrating aggressive tumors13.
Fig. 1. Tet2 in the cancer-immunity crosstalk.

Tet2 loss causes an accumulation of 5mC, which promotes B-cell development and B-cell function. Tet2 loss also promotes CD4+ T-cell differentiation and M1 macrophage responses, which can modulate cancer cell activities. In contrast, cancer cells that carry Tet2 mutations counteract positive immune-cell responses, including T-cell and macrophages.
This review discusses Tet2-related profiles in hematopoietic cancers, different subtypes of adaptive and innate immune cells, and Tet2 regulation by various interactors. I particularly emphasize the potential applications of targeting Tet2 to influence normal and malignant cancer-immunity crosstalk.
Roles of Tet2 in hematopoiesis, immune-cell lineages, and hematopoietic malignancies
Tet2 in hematopoiesis
Tet2 epigenetically controls gene expression by modulating methylation-driven gene silencing and is expressed in diverse populations of mouse and human hematopoietic cells14. Tet2 mRNA is expressed in hematopoietic cell subpopulations, including progenitors and mature immune cells, displaying detectable levels of 5hmC and 5mC15. In mice, Tet2 deletion enhances the Lin−Sca-1+c-Kit+ (LSK) cell subpopulation, which exhibits boosted hematopoietic repopulation competence and biased cell differentiation toward monocyte/macrophage lineages, followed by myeloid cancer progression16. In vitro tissue culture work supports that RNA interference (RNAi)-mediated Tet2 silencing in murine hematopoietic precursors can alter their differentiation toward monocyte/macrophage lineages17. In mice, Tet2 is pivotal for modulating normal hematopoiesis, and negatively influences hematopoietic stem cell (HSC) homeostasis and differentiation. Tet2 loss reshapes the HSC niche and HSC proliferation/differentiation potential in vitro14, whereas Tet2-KO HSCs show elevated ability to reconstitute hematopoiesis in vivo17. Tet2 deletion results in reduced genomic 5hmC levels and expands the hematopoietic progenitor cell population in a cell-intrinsic manner16. Tet2-KO HSCs are capable of multilineage reconstitution and show a competitive advantage over wild-type HSCs, promoting increased hematopoiesis into myeloid and lymphoid lineages15. Tet2 disorder also results in DNA hypermethylation of enhancers in granulocyte–monocyte progenitor (GMP) and embryonic stem (ES) cells18. Overall, Tet2 plays vital roles in modulating HSC expansion and function, presumably by controlling 5hmC levels in genes crucial for HSC self-renewal, proliferation, and differentiation, which warrants future examination of the detailed mechanisms.
Tet2 in myeloid lineages and myeloid malignancies
In humans, Tet2 mutations occur in ~10–30% of myeloid cancers, including myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML), and myeloproliferative neoplasm (MPN)19. Tet2 mutations may be caused by splice site mutations, out-of-frame insertions or deletions and base substitutions. Tet2-KO mice recapitulate characteristics of patients with myeloid cancer, indicating that Tet2 plays a functional role as a tumor suppressor to sustain hematopoietic cell homeostasis. Tet2-KO mice show a phenotype that is similar to CMML, including splenomegaly and enhanced white blood cell (WBC) counts with unequal numbers of monocytes14. Moreover, Tet2-KO mice develop an extensive spectrum of myeloid cancers, which occur within the first year of life in ~30% of these animals17. Mechanistically, Tet2 blocks leukemic transformation by keeping enhancers free of aberrant DNA methylation that would lead to enhanced stem cell expansion and leukemogenesis20. RNAi-mediated Tet2 silencing reduces 5hmC levels in cord blood CD34+ cells and promotes progenitor differentiation toward the granulomonocytic lineage18. CD34+ stem cells from MPN patients with Tet2 mutations reconstitute hematopoiesis in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice and display skewed differentiation toward myeloid lineages15.
Tet2 in lymphoid lineages and lymphoid malignancies
Tet2 mutations are also common in both T-cell and B-cell lymphomas, with a 40–70% incidence observed among patients with peripheral T-cell lymphoma (PTCL), particularly in subsets of angioimmunoblastic T-cell lymphoma (AITL)21. Adult T-cell leukemia/lymphoma (ATLL) progression accompanies genomic 5hmC loss, suggesting that Tet2 downregulation is the vital mechanism underlying 5hmC regulation during ATLL progression22. Tet2-KO mice develop B-cell tumors, and B-cell conditional Tet2 knockout (cKO) mice show B1-cell subset abnormalities and B-cell tumor development after long latency23. Tet2 deletion may prompt alterations of peripheral B-cell subsets23. Certain Tet2-KO cancers have AID-mediated somatic mutations23. Future work is required to investigate whether and how dysregulation of Tet2 impacts on B-cell activation upon various stimuli. Young Tet2-KO mice exhibit a moderately enhanced B1a subset population in the spleen23. On the other hand, aging Tet2-KO mice expand clonal CD19+B220low immunoglobulin (Ig)M+ B-cell subpopulations, which are transplantable and display similarities to chronic lymphocytic leukemia (CLL), including sensitivity to ibrutinib-induced B-cell receptor signaling blockage24. Further investigations are needed to examine the molecular signaling pathways controlling B1a-cell development and proliferation in young adult Tet2-KO mice.
In mice, attenuated Tet2 function results in T-cell lymphoma with follicular helper T-cell-like (Tfh) features25. Compared with control mice, middle-aged Tet2 gene trap mice (Tet2gt/gt) show Tfh-like cell over-expansion in the spleen25. Aging Tet2gt/gt mice eventually generate T-cell lymphoma with Tfh-like features25. In future research, it will be intriguing to examine how Tet2 works in Tfh cells. A mouse model with conditional Tet2-KO (cKO) in Tfh cells would be valuable for in vivo and in vitro analyses.
Both Tet1 and Tet3 are expressed in hematopoietic cells, without alterations in Tet2-KO cells, demonstrating that Tet1/Tet3 is inadequate to compensate for Tet2 deletion. It will be interesting to dissect their physiological and pathological roles in various immune-cell subsets. Tet2 and Tet3 may cooperate to inhibit abnormal hematopoiesis, including hematopoietic transformation. Further investigations are necessary to test whether and how Tet2 works together with other Tet proteins in distinct blood cancer cells. Deletion of Tet2 in hematopoietic cells or various immune-cell subsets might prompt a specific and progressive enhancer hypermethylation feature linked to altered gene expression, enhanced cell accumulation rate, and leukemia progression, which also warrants future investigation. A better understanding of Tet proteins’ physiological and pathological functional roles may highlight potential novel avenues to develop epigenetic therapies to treat hematological cancers.
Intriguingly, in mice, the combined deletion of Tet1 and Tet2 apparently boosts B-cell cancer, without influencing myeloid cancers26. Tet1 and Tet2 are simultaneously downregulated in B-cell acute lymphoblastic leukemia26. Although double (DcKO) or triple (TcKO) conditional KO mice would be valuable, it might be challenging to conditionally KO all three Tet proteins in certain immune-cell sub-settings. Identifying Tet2-targeted genes/molecules in myelopoiesis and lymphomagenesis will be crucial for future studies aimed to uncover the molecular mechanisms through which Tet2 modulates hematopoiesis and serves as a tumor-suppressor gene in myelopoiesis and T/B-cell lymphomas. It is intriguing to hypothesize that inactivating somatic Tet2 mutations might latently remain in hematopoietic cells for a long period, possibly even years. Specifically targeting Tet2 or its downstream molecular signaling pathways might offer novel therapeutic strategies for blood cancer prevention.
Roles of Tet2 in anticancer immune responses
Tet2 in B-cell and anticancer responses
Tet2 is also essential for B-cell development and function27. In mice, Tet2 and Tet3 KO at an early B-cell stage halts the transition of pro-B cells to pre-B cells in the bone marrow (BM), and diminishes the rearrangement of Igk locus28. Tet2/Tet3 DKO pro-B cells exhibit enhanced CpG methylation levels at the Ig kappa 3ʹ and distal enhancers29. Tet2/Tet3 modulates the kappa-chain expression level, and the DNA modification status of the Ig kappa locus, both in vivo and in vitro29. In mice, Tet2 deletion causes myeloid leukemia after a long duration. Compared to Tet2 deficiency, Tet1/Tet2 deletion results in B-cell lymphoma with delayed disease progression26. The Tet2/Tet3 deletion in developing B cells leads to B-cell lymphoma development. These mice caused the pathological disorders within six months of age, which is earlier than in Tet1/Tet2-KO mice (~20 months)7. Moreover, regulatory B cells (Bregs) play central roles in boosting inflammation and carcinogenesis30, but the evidence is lacking regarding whether and how Tet2 protein functions in Bregs both in vitro and in vivo. Continued efforts to investigate the probable anticancer impact of Tet2 in Bregs will guide novel Tet2-based immune therapy. Additionally, in future studies, it would be useful to use B-cell-specific conditional (such as CD19-cKO) Tet2 cKO mice to examine whether and how Tet2 epigenetically affects antibody production in vivo.
Tet2 in T-cell and anticancer responses
Tet2 deletion augments CD8+ T-cell memory differentiation. Deletion of Tet2 advocates early gain of a memory CD8+ T-cell fate, in a cell-intrinsic manner, without affecting antigen-driven cell proliferation or effector behavour31. After secondary stimulation, Tet2-KO memory CD8+ T cells possess remarkable pathogen control capacity, both before and after re-challenge. CD8+ T cells with Tet2 conditional KO exhibit full effector functional role upon acute viral infection, and Tet2 deletion enhances memory CD8+ T-cell formation31. CD8+ cytotoxic T lymphocytes (CTLs) are favored immune cells for treating cancer progression32. Programmed death‐1 receptor (PD‐1) ligand (PD‐L1) and CTL‐associated antigen 4 (CTLA‐4) are checkpoint receptors that can be targeted to re-activate anticancer CD8+ T-cell responses in cancer-immune therapy33. It would be interesting to study whether Tet2 might be involved in CTLA-4 or PD-1 pathway-mediated immune checkpoint inhibition for cancer treatment. Further studies are required to explore whether and how Tet2 acts in CD8+ CTLs, and how it cooperates with PD-L1/CTLA-4 to modulate anticancer CD8+ T-cell responses.
Tet2flox/floxCD4Cre+ mice have intact thymic and peripheral T-cell subpopulations31. Tet2 enhances DNA demethylation and cytokine gene activation, for instance, IL17a, in CD4+ T cells. In autoimmune diseases, Tet2 modulates T-cell cytokine production in vivo, and specifically influences IL-10, IFN-g, and IL-17 expression levels34. Tet2 depletion in CD4+ T cells is linked to reduced cytokine expression, and diminished p300 recruitment34. Moreover, Tet2/Tet3 DcKO CD4+ T cells induce pathological diseases in healthy mouse35. Tet2 enhances typical cytokine gene expression level in Th1 and Th17 cells in vitro, and modulates Th1 and Th17 cell differentiation34.
Tet2/Tet3 loss in regulatory T cells (Tregs) contributes to effector behave phenotypes. Treg-cell-specific Tet2/Tet3 double conditional KO mice (Tet2flox/flox/Tet3flox/floxFoxp3Cre+ DcKO mice) show severe inflammation, resulting in inflammatory diseases, and Tregs from these DcKO mice exhibit adjusted expression levels of Treg-typical cytokine genes and dysregulation of genes involved in cancer progression35. Overall, these studies reveal that both Tet2 and Tet3 maintain Treg stability and homeostasis. Tet2/Tet3 DcKO in Tregs enhances T-cell activation. Moreover, Tet2/Tet3 DcKO Tregs show an altered cell surface feature and fair hypermethylation of conserved noncoding sequence 2 (CNS2)35. Wild-type (WT) Tregs are not able to regulate autoimmunity progression in Tet2-deficient mice35. Double Tet1/Tet2 deletion also causes Treg inactivation, differentiation, and even autoimmune diseases36. Notably, Tregs hamper anticancer immune surveillance in a healthy person, and prevent effective antitumor immunity in cancer patients, boosting tumor progression37. Thus, methods for specific targeting Tet2/Tet3 in Treg cells might be valuable for modulating anticancer immune responses.
Tet2 in iNKT cell and anticancer responses
Tet2 and Tet3 also work together to restrict invariant natural killer T cells (iNKT cells) proliferation and cell-lineage differentiation38. Tet2/Tet3 DcKO mice exhibit impressive iNKT cell proliferation, even at earlier stages. These iNKT cells produce large amounts of cytokines, thus eliciting strong immune responses that prompt other immune-cell subsets responses, and can activate anticancer cytotoxicity38. In Tet2/Tet3 DcKO mice, the enhanced iNKT cell numbers and concomitantly higher IL-4 secretion give rise to innate-like CD8+ T cells38. Tet2/Tet3 deletion yields converted numbers of iNKT cells and significantly influenced gene expression levels, thus changing the identity and functional role of each cell subpopulation9. Tet2/Tet3 DcKO iNKT cells show enhanced proliferative abilities, which can explain their in vivo spread38. Future immunotherapy methods specifically targeting Tet2/Tet3 to modulate iNKT cell activity could have interdependent consequences and employ strong anticancer immune responses.
Tet2 in innate immune-cell and anticancer responses
Tet2 is further essential for innate immune-cell functional roles9. Compared to other Tets, Tet2 is the most profoundly expressed in murine macrophage differentiation. Tet2 is essential for controlling inflammation through recruiting Hdac2, inhibiting IL-6 levels40. Tet2-KO macrophages and dendritic cells (DCs) produce more IL-6 in response to bacterial activation in vitro and in vivo. Thus, compared to wild-type (WT) mice, Tet2-KO mice show increased susceptibility to endotoxin-induced shock, DSS-induced colitis40 and Salmonella infections41. Tet2 transcription is induced by LPS activation, likely in a nuclear factor κβ (NF-κβ)-dependent way42. In vitro, Tet2 loss in BMDMs does not influence early LPS-induced gene activities but enhances arginase 1 (Arg1) mRNA level during later activation stages. Tet2 deletion does not dramatically alter alternative macrophage (M2) gene expression levels in response to IL-4 stimuli39. Tet2 expression is sustained in differentiated murine macrophages, and Tet2 deletion does not negatively alter markers of macrophage differentiation42. Resting Tet2-KO peritoneal macrophages reveal atypical induction of LPS-associated genes, and the Tet2-deficiency peritoneal macrophage features are linked to inflammation across the whole body40.
M1 macrophages are essential for tumor cell eradication43. Thus, modulating Tet2 expression levels in M1 macrophages might contribute to cancer-immune therapy (Fig. 1). Over a 20-week time-course, certain Tet2-KO strains show strong defects in myelopoiesis, leading to features similar to human CMML7. Acute inducible Tet2/Tet3 DKO in HSCs causes the rapid emergence of aggressive myeloid leukemia. Tet2 maintains the immunosuppressive functional role of tumor-infiltrating myeloid cells, promoting the advancement of melanoma39 and Tet2 deletion in myeloid cells reduces melanoma tumor burden in vivo. In tumor-associated macrophages (TAMs), Tet2 expression is activated along the IL-1R/MyD88 axis39. Myeloid-specific Tet2 loss causes higher numbers of tumor-infiltrating T cells39. During melanoma growth, Tet2 expression is enhanced in myeloid-derived suppressor cells (MDSCs) and TAMs39. Further, Tet2 preserves immunosuppressive gene expression levels in TAMs. Mice with myeloid cell-specific Tet2 deletion exhibit diminished tumor growth, and enhanced tumor-infiltrating T cells; moreover, T-cell depletion suppresses the attenuated tumor progression39. Whether Tet2 plays a similar or different role in myeloid cell-mediated tumor growth in cancers other than melanoma awaits further investigation.
Here, I have summarized our current understanding of the functional roles of Tet proteins in B cells, various types of T-cell subsets, and M1/M2 macrophages in both physiological and pathological conditions. Further study is essential to investigate how all Tet proteins and 5hmC/5mC modulate DNA modification, gene expression, and transcriptional networks in various types of innate and adaptive immune cells, and to dissect the underlying molecular mechanisms and potential for cancer-immune therapy in this field.
Tet2 regulation at the cancer-immunity interface
Numerous regulatory mechanisms have either directly or indirectly been demonstrated to regulate Tet2 activities or expression levels in immune-cell subsets and cancer cells (Fig. 2). Mutated IDH1/2 enzymes, which are often found in tumors, produce the Tet2 protein inhibitor D-2HG, causing DNA hypermethylation44. Vitamin C interacts with the core catalytic domain of Tet2, directly boosting its catalytic activities4. Additionally, Tet2 expression can be directly altered at the post-transcriptional level41,45. Numerous microRNAs (miRs) post-transcriptionally inhibit Tet expression and certain miRs work together to control the expression of Tet genes. Tet2 genes contain conserved elements in the three prime untranslated regions (3′-UTR) predicted to be directly regulated by miR-2246. High-throughput screening has validated that the Tet2 expression level in hematopoietic cells is regulated by multiple miRs, including miR-7, miR-125b, miR-29b/c, miR-26, and miR-10147. Moreover, Tet2 can be directly and indirectly modulated by the miR let-7adf in macrophages41. O-GlcNAc transferase (OGT) transfers a single O linked-GlcNAc moiety to serine/threonine residues in multiple proteins48. OGT co-localizes with Tet2 in the genome, mainly at GC-rich promoters and transcriptional start sites (TSS) containing CpG islands, and reduces Tet2’s enzymatic activities by enhancing its nuclear export49. In the context of metabolic pressure, Tet2 can also be modulated by alpha-ketoglutaric acid (α-KG)50. Tet2 deletion through CRISPR-CAS9 causes globally enhanced DNA methylation at enhancers, thus blocking the estrogen responses51, indicating that Tet2 acts as a transcriptional coactivator in the estrogen responses. Fe2+ and HIF-1a also support to maintain Tet2 activities52. Thus, various cofactors (e.g., Fe2 + , HIF-1a and OGT) and noncoding RNAs (such as miRs) can affect Tet2 functional roles, constituting an additional level of regulation. Moreover, the epigenetic regulator CXXC5 and Tet2 are essential in the fight against viral infections. CXXC5 enrolls Tet2 to modulate TLR7/9-elicited IFN responses, and to influence DNA methylation, in human plasmacytoid DCs53. RUNX1 alters DNA demethylation by recruiting Tet2 and other enzymes with demethylation functional roles to its attaching sites in various hematopoietic cells54. All of these interactors, and other small-molecule activators of Tet2, would be helpful methods for stabilizing in vitro-generated immune cells or modified tumor cells for potential clinical utilization.
Fig. 2. Potential therapeutic strategies related to cancer-immunity, based on interactors, specifically targeting Tet2 activities in immune and cancer cells.
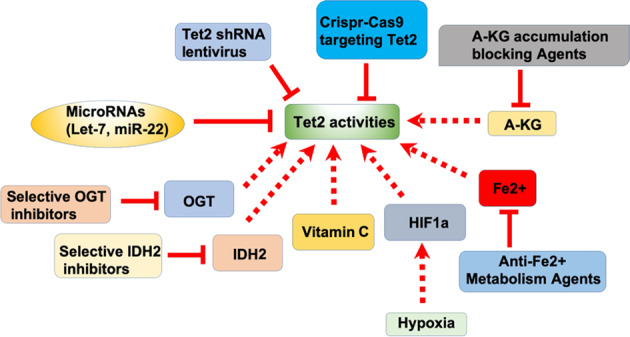
The Tet2 activities might be enhanced by vitamin C and hypoxia treatment through HIF-1a. The Tet2 activities might be halted by blocking a-KG accumulation, selective OGT inhibition, selective IDH2 inhibition, using anti-Fe2+ agent, or directly suppressed by microRNAs (e.g., Let-7 and miR-22), Tet2 shRNA lentivirus, and Crispr/cas9 targeting in cancer and immune cells.
Although many Tet2 interactors have been identified in both immune and cancer cells, several principal questions are waiting for answers. For instance, how do these interactors assist Tet2 dysregulation-mediated cancer transformation as well as anticancer immune responses? Further work is needed to demonstrate the underlying molecular mechanisms. It will also be crucial to determine the relationship between Tet2 interactors in Tet2 dysregulation of the progression of non-hematopoietic cancers and blood cancers. Additionally, it is crucial to investigate whether and how these interactors influence 5hmC quantification, which is a helpful diagnostic and/or prognostic tool in cancer treatment. Manipulation of the activity of Tet2 interactors via small molecules or other modulators would be useful for future cancer-immune therapy.
Targeting Tet2 for potential cancer-immune therapy
There may be several potential ways of specifically targeting Tet2 activity in immune-cell subpopulations and cancer cells (Fig. 2). In humans, functional loss of Tet2 plays an extensive potentiating role in immunotherapy against B-cell cancer, which utilizes T cells bearing the anti-CD19 chimeric antigen receptor (CAR). The insertional mutation of one copy of a Tet2 allele causes dysregulation of Tet2 activities, which apparently couples with the other Tet2 allele mutation, ultimately yielding strong antitumor properties to the expanded CAR-T cells55. Research on Tet2 loss in immune-cell responses and tumor development unveils the significance of DNA methylation in various biological activities, and in the advancement of hematological malignancies and solid tumors. With regards to potential clinical usage, it is hard to precisely target Tet2 for treating cancer progression, due to its inactivation in various cancer cell types. However, Tet2 is still expressed to certain levels in some immune-cell subsets and may be modulated in various manner (Fig. 2). For instance, fumarate hydratase (FH) and succinate dehydrogenase (SDH) inhibitors might be developed to prevent the global effectiveness of metabolic Tet2 inactivation in CD8+ T cells. Notably, the oncometabolite 2-hydroxyglutarate (2HG), generated by IDH mutation in immune cells, could also halt Tet2 activities in immune cells, (e.g., CD4+ T cells and M1 macrophages), which would trigger less-positive antitumor immune responses. Thus, it might also be valuable for future preclinical studies to investigate the restoration of Tet2 in these immune cells, for instance, by directly adding vitamin C or a-KG. Future work will focus on the accurate targeting of Tet2 in the context of different specific cell types, among both cancer and immune cells.
Investigations in both mouse models and human clinical samples have demonstrated that Tet2 loss is not essential to cause cancer, but rather influences cancer progression. This may be by reason for the behavior of Tet2 in various types of immune-cell subsets, and it might explain why some patients with Tet2 mutations stay healthy, whereas others develop either myeloid or lymphoid cancer. Future studies are needed to examine therapies targeting cytosine modifications, DNA methyltransferase inhibitors, and IDH inhibitors, which might be valuable for future cancer-immune therapy.
Conclusions and current challenges
Collectively, Tet2 is critical to the cancer-immunity crosstalk, which is essential for both cancer development and anticancer immune responses (Fig. 1). It appears that the downregulation of 5hmC depends on certain specific cell types. It is expected that elevating Tet2 activities using specific compounds that enhance or potentiate Tet2 functional roles may be broadly useful in cancer treatment and for the modulation of certain anticancer immune-cell responses. To further interpret the underlying molecular mechanisms, it is critical to elucidate the functional roles of Tet2/5hmC regulators, and the different ways of modulating the acquisition/removal of 5hmC in various cell types. This will require additional insights into how Tet2 works, and how it is modulated in various types of mammalian cells, including both cancer and immune cells. The toughest challenge might be determining how to precisely modulate Tet2 in specific cells where it plays either positive or negative roles for antitumor therapy. Future work must also explore how to utilize Tet2 interactors to maintain positive antitumor immune responses and halt negative immune responses against cancers. There is still a lot to grasp regarding the physiological and pathological roles of Tet2 in other aspects (e.g., metabolism) in both immune and cancer cells. The elucidation of these aspects and molecular mechanisms would be an exciting avenue for further study.
Acknowledgements
The author apologizes to the researchers whose work was not cited because of space limitation. Dr. Shuai Jiang is supported by Louisiana State University (LSU) FWCC Legacy Funds (149741373A), Intramural Research Council Support (110251225A), and by the Micro Seed Funds.
Competing interests
The author declares no competing interests. S.J. is an Editorial Board Member for Communications Biology, but was neither involved in the editorial review of nor the decision to publish this article.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017;18:517–534. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 2.Lorsbach RB, et al. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 3.Koivunen P, Laukka T. The TET enzymes. Cell. Mol. Life Sci. 2018;75:1339–1348. doi: 10.1007/s00018-017-2721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ko M, et al. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015;263:6–21. doi: 10.1111/imr.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan F, Weeks O, Yang F-C, Xu M. The TET2 interactors and their links to hematological malignancies. IUBMB Life. 2015;67:438–445. doi: 10.1002/iub.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y-P, et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019;129:4316–4331. doi: 10.1172/JCI129317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsagaratou A, Lio C-WJ, Yue X, Rao A. TET methylcytosine oxidases in T cell and B cell development and function. Front. Immunol. 2017;8:220. doi: 10.3389/fimmu.2017.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics. 2014;104:352–357. doi: 10.1016/j.ygeno.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lio C-WJ, Rao A. TET enzymes and 5hmC in adaptive and innate immune systems. Front. Immunol. 2019;10:210. doi: 10.3389/fimmu.2019.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The ten-eleven translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28:485–496. doi: 10.1038/leu.2013.337. [DOI] [PubMed] [Google Scholar]
- 11.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 function in hematopoietic malignancies, immune regulation, and DNA repair. Front. Oncol. 2019;9:210. doi: 10.3389/fonc.2019.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.An J, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat. Commun. 2015;6:10071. doi: 10.1038/ncomms10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko M, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl Acad. Sci. USA. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan F, et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat. Commun. 2017;8:15102. doi: 10.1038/ncomms15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pronier E, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–2555. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tefferi A, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen KD, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29:910–922. doi: 10.1101/gad.260174.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandell RF, Boddicker RL, Feldman AL. Genetic landscape and classification of peripheral T cell lymphomas. Curr. Oncol. Rep. 2017;19:28. doi: 10.1007/s11912-017-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marçais A, et al. Adult T cell leukemia aggressivenness correlates with loss of both 5-hydroxymethylcytosine and TET2 expression. Oncotarget. 2017;8:52256–52268. doi: 10.18632/oncotarget.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mouly E, et al. B-cell tumor development in Tet2-deficient mice. Blood Adv. 2018;2:703–714. doi: 10.1182/bloodadvances.2017014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quivoron C, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Muto H, et al. Reduced TET2 function leads to T-cell lymphoma with follicular helper T-cell-like features in mice. Blood Cancer J. 2014;4:e264. doi: 10.1038/bcj.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, et al. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep. 2015;13:1692–1704. doi: 10.1016/j.celrep.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominguez PM, et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 2018;8:1632–1653. doi: 10.1158/2159-8290.CD-18-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lio C-W, et al. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. eLife. 2016;5:e18290. doi: 10.7554/eLife.18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orlanski S, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc. Natl Acad. Sci. USA. 2016;113:5018–5023. doi: 10.1073/pnas.1604365113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B cells in anti-tumor immunity. Int. Immunol. 2015;27:521–530. doi: 10.1093/intimm/dxv034. [DOI] [PubMed] [Google Scholar]
- 31.Carty SA, et al. The loss of TET2 promotes CD8+ T cell memory differentiation. J. Immunol. 2018;200:82–91. doi: 10.4049/jimmunol.1700559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J. Cell. Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 33.Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front. Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ichiyama K, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015;42:613–626. doi: 10.1016/j.immuni.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue X, Lio C-WJ, Samaniego-Castruita D, Li X, Rao A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat. Commun. 2019;10:2011. doi: 10.1038/s41467-019-09541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang R, et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity. 2015;43:251–263. doi: 10.1016/j.immuni.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080–2089. doi: 10.1111/cas.14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsagaratou A, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat. Immunol. 2017;18:45–53. doi: 10.1038/ni.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan W, et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity. 2017;47:284–297.e5. doi: 10.1016/j.immuni.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang S, Yan W, Wang SE, Baltimore D. Dual mechanisms of posttranscriptional regulation of Tet2 by Let-7 microRNA in macrophages. Proc. Natl Acad. Sci. USA. 2019 doi: 10.1073/pnas.1811040116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017;55:56–70.e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Pan X-Q. The mechanism of the anticancer function of M1 macrophages and their use in the clinic. Chin. J. Cancer. 2012;31:557–563. doi: 10.5732/cjc.012.10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kernytsky A, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. 2015;125:296–303. doi: 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scherm MG, et al. miRNA142-3p targets Tet2 and impairs Treg differentiation and stability in models of type 1 diabetes. Nat. Commun. 2019;10:5697. doi: 10.1038/s41467-019-13587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song SJ, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87–101. doi: 10.1016/j.stem.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng J, et al. An extensive network of TET2-targeting microRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017;18:452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao M, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018;4:eaau6986. doi: 10.1126/sciadv.aau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fischer AP, Miles SL. Silencing HIF-1α induces TET2 expression and augments ascorbic acid induced 5-hydroxymethylation of DNA in human metastatic melanoma cells. Biochem. Biophys. Res. Commun. 2017;490:176–181. doi: 10.1016/j.bbrc.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 53.Ma S, et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J. Exp. Med. 2017;214:1471–1491. doi: 10.1084/jem.20161149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki T, et al. RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv. 2017;1:1699–1711. doi: 10.1182/bloodadvances.2017005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fraietta JA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]