

Abstract
A convergent sequence to access the indole alkaloid (±)-melokhanine E in 12-steps (8-step longest linear sequence) and an 11% overall yield is reported. The approach utilizes two cyclopropane moieties as reactive precursors to a 1,3-dipole and imine species to enable stereoselective construction of the core scaffold through a formal [3 + 2] cycloaddition. The natural product was evaluated for its antimicrobial activity based on isolation reports; however, no activity was observed. The reported efforts serve as a synthetic platform to prepare an array of alkaloids bearing this core structural motif.
Graphical Abstract:

New natural product skeletons with antimicrobial activity are of significant interest as there is a pressing need for the development of novel antimicrobial agents.1–3 Melokhanine E (1, Figure 1) and a series of related compounds were isolated in 2016 from Melodinus khasianus, a subtropical plant used in traditional Chinese medicine as a treatment for rheumatic heart disease and meningitis.4 These indole alkaloids possess a 6/5/5/6/6 pentacyclic structure and are reported to exhibit potent antimicrobial activity against Gram-negative pathogens P. aeruginosa and E. coli as well as Gram-positive pathogens such as E. faecalis. Within the family, melokhanine E is of particular interest due to its superior broad-spectrum activity (MIC = 2 μM against P. aeruginosa and 5 μM against E. faecalis).4 A key structural motif of the melokhanines is a core piperidine heterocycle bearing C3 quaternary substitution. This structural element is found in an array of bioactive natural products and represents a significant synthetic challenge (2–5, Figure 1).5,6 We therefore sought to develop a synthetic route to access melokhanine E (1) to confirm its antibacterial activity while developing innovative solutions to access its pentacyclic core. During the course of our efforts, a 14-step synthesis of 1 was reported as part of studies developing rearrangement approaches toward this broad class of natural products.7 Herein we describe a short synthetic sequence to stereoselectively access 1, confirm its chemical structure, and explore its potential as an antimicrobial lead.8
Figure 1.

Representative biologically active natural products containing a piperidine C3 all-carbon quaternary center.
A number of strategies have been developed to install the quaternary substituted piperidine motif found in many alkaloids. Pandey and co-workers have utilized a Birch reduction/alkylation followed by auxiliary removal9 as well as an approach utilizing a Johnson-Claisen rearrangement that provided greater than 99% ee.10 One of the most successful and broadly used strategies was developed by the Stoltz group, which employs racemic 3,3-disubstituted piperidines and chiral palladium catalysis to obtain enantioenriched 3,3-disubstituted piperidines with good yields and up to 99% enantiomeric excess.11–13 While high-yielding and stereoselective, these approaches yield products at nonideal oxidation states for their direct advancement to the target natural products. The approach to the piperidine scaffold presented herein is inspired by Waser and co-workers’ efforts on push-pull cyclopropane opening and subsequent cycloadditions, allowing for direct access to piperidines at the desired oxidation states.14–16
Inspired by Carreira’s utilization of a regioisomeric version of our oxindole 1,3-dipole for intermolecular cycloadditions, we set out to devise a concise retrosynthesis of melokhanine E featuring a similar oxindole 1,3-dipole-based intramolecular cycloaddition (Figure 2).17–21 Our retrosynthetic approach to 1 therefore began by recognizing that intramolecular formal [3 + 2] cycloaddition within biscyclopropane 7 would enable all relative stereochemistry in the molecule to be set in a single transformation based on the configuration of the quaternary piperidine moiety. We envisioned that the stereoselectivity of this transformation would derive from sterically favored exo approach of the revealed oxindole 1,3-dipole functionality into the imine as well as kinetically favored attack of the 1,3-dipole anti- to the ethyl group. We initially proposed that the kinetic preference for anti- addition into the imine would derive from the kinetically favored chairlike transition state that would be formed upon addition onto the antiface of the ring, a process that is likely supplemented by steric predisposition to the antiface of attack. Thus, a late stage intramolecular cycloaddition of an appropriately functionalized 1,3-dipole tethered to an imine or iminium derivative, represented by 6 (Figure 2), enables an extremely rapid approach to the core of 1. Then 6 was envisioned to arise from coupling of spirocyclic cyclopropane 8 with cyclopropanated piperidine 9, followed by subsequent Lewis acid/thermal opening of both cyclopropanes to generate the requisite reactive functionality in situ. Both cyclopropane precursors 8 and 9 are available through literature protocols, with 9 arising from cyclopropanation of enecarbamate 10.14,22
Figure 2.

Retrosynthetic access to melokhanine E (1).
The synthesis of coupling partners 8 and 9 was achieved via modification of literature procedures (Figure 3).14,22 To target the spirocyclic cyclopropane 8, anthranilic acid (11) was reacted with bromo-lactone 12 to provide amino acid 13 in 72% yield. Activation of the carboxylic acid functionality in the presence of base and heat forges the spirocyclic bicycle 14 in 61% yield. Heating this compound in the presence of NaCl enables a Krapcho decarboxylation-ring contraction sequence that reveals the desired alpha-keto cyclopropane 15 in 73% yield. Finally, methanolysis of 15 affords 8 in four steps and 32% overall yield (Figure 3a).22 Synthesis of the desired fused cyclopropane-piperidine coupling partner 9 begins with an alkylation-protection of delta-valerolactam (16) to generate 17 (61%, two steps). Reduction of the lactam with NaBH4 followed by dehydration of the resulting hemiaminal with H2SO4 affords Boc-protected enamine 10 (82%, two steps). Copper(I) triflate-mediated cyclopropanation of enecarbamate 10 then affords fused ester 18 in 73% yield, which saponifies under basic conditions and is simultaneously epimerized to the more stable exoisomer 9 (Figure 3b).
Figure 3.

(a) Synthesis of spirocyclic cyclopropane 8; (b) synthesis of fused bicyclic cyclopropane 9.
At this stage, we began to explore coupling conditions to unite 8 and 9 through the amide linkage to set the stage for our key intramolecular cycloaddition. The coupling of indolinone 8 and piperidine 9 proved to be more challenging than anticipated. We postulate that the difficulty of this amide bond formation is a result of steric bulk on both coupling partners as well as deactivation of the nucleophile via resonance with the 3-keto functionality of the indolinone. After exploring various conditions, we discovered that an acid chloride-based coupling approach proved most fruitful, with triphosgene providing our desired acid chloride species with minimal substrate decomposition. Our initial attempt with triphosgene provided 16% yield of the desired coupled product 7 after 16 h at room temperature (Table 1). We found that increased temperature allowed for shorter reaction time and higher yield; however, increased reaction time at elevated temperature decreased the overall yield, likely due to slow decomposition of the substrates or product. At this stage, we elected to take these conditions and press forward with the synthesis, having access to 7 in 45% yield. It should be noted that upon isolation and storage 7 can ring-open to the hydrated hemiaminal piperidine motif; however, this has no consequence on the yield of the subsequent cycloaddition chemistry.
Table 1.
Optimization of Coupling Conditions for 7
 | ||||
|---|---|---|---|---|
| entry | time (h) | temp (°C) | solvent | yield (%) |
| 1 | 19 | rt | CH2Cl2 | 16 |
| 2 | 41 | rt | CH2Cl2 | 26 |
| 3 | 3.5 | 40 | CH2Cl2 | 45 |
| 4 | 6 | 40 | CH2Cl2 | 31 |
| 5a | 6 | 40 | CH2Cl2 | 33 |
| 6a | 19 | 60 | THF | 9 |
| 7 | 52 | 40 | THF | 13 |
Lutidine was used in place of collidine.
With coupled biscyclopropane 7 in hand, we sought to explore the generation of our key cycloaddition intermediates via cyclopropane opening. To this end, 7 was subjected to cycloaddition conditions utilizing MgI2 to promote spirocycle opening, and thermolysis of the Boc-carbamate to reveal the reactive imine intermediate.17–21 We envision a sequence of Boc-deprotection/cyclopropane opening and MgI2-promoted cyclopropanation opening to generate the reactive 1,3 dipole 20, which undergoes stepwise imine addition/nitrogen alkylation to generate the formal [3 + 2] cycloaddition product (Figure 4). In our first attempt (0.43 equiv MgI2, THF, sealed vessel, 125 °C), we obtained 15% of the desired natural product 1 as a single diastereomer with no other related melokhanine products observed. To our delight, performing the reaction in toluene at 200 °C under microwave heating provided 88% yield of (±)-melokhanine E, again as a single detectible diastereomer (1, Figure 4). Interestingly, in the absence of MgI2, the reaction still proceeds, albeit at an exceptionally slow rate (29% yield after 8.5 h at 200 °C).
Figure 4.
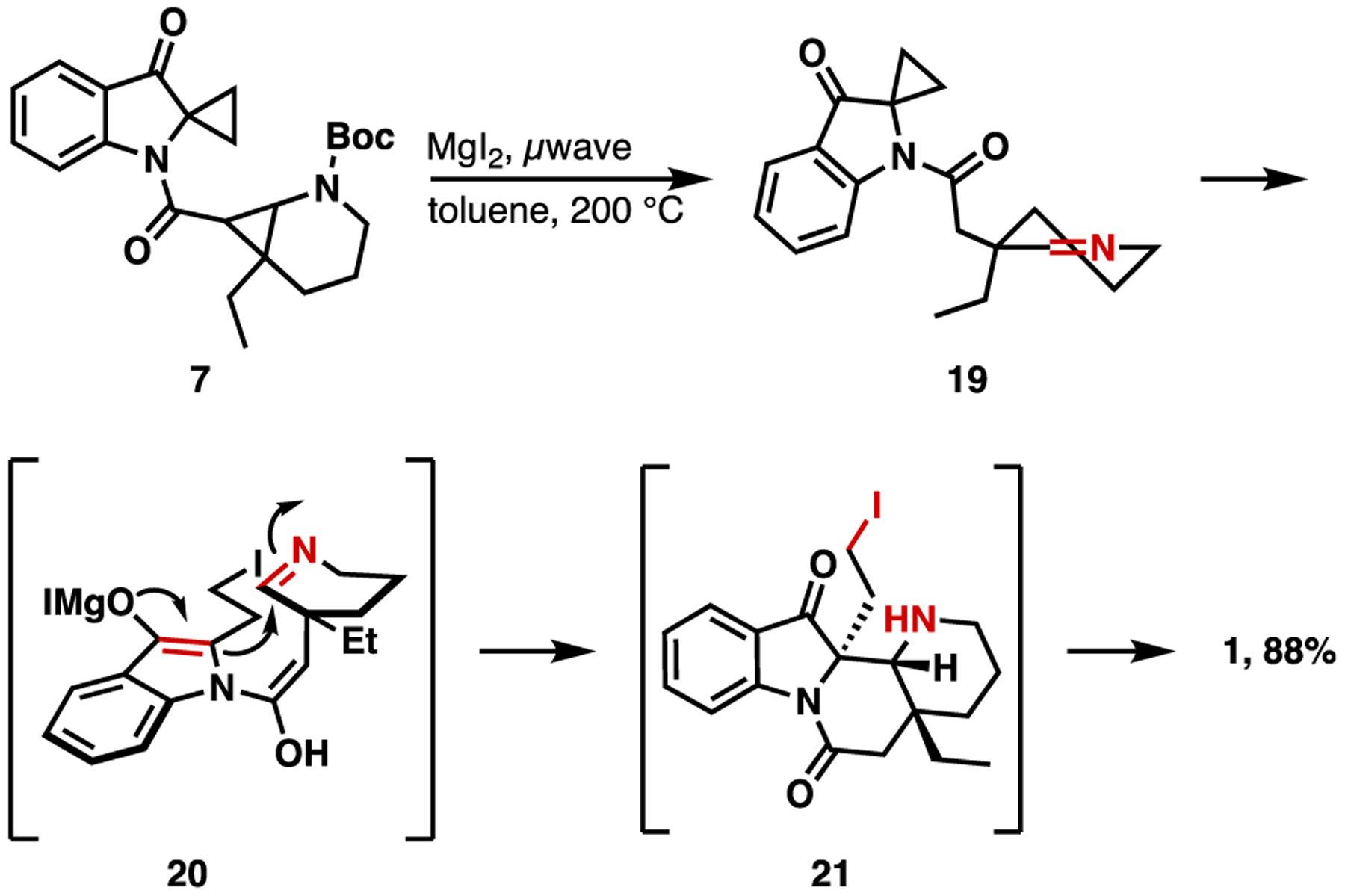
Formal [3 + 2] cycloaddition to synthesize (±)-melokhanine E (1).
With the racemic natural product in hand, we next sought to confirm the biological activity reported in Luo and co-workers’ isolation report.4 In the isolation report, Luo and co-workers reported impressive antibiotic against Gram-negative bacteria (P. aeruginosa MIC 2 μM, 0.62 μg/mL) as well as Gram-positive bacteria (E. faecalis MIC 5 μM, 1.56 μg/mL). We explored the activity of 1 against both the pathogens reported in the isolation report as well as the ATCC numbers reported23 and found in all cases the activity of the natural product was >256 μg/mL. Disappointed by these initial results, we have adjusted the assay conditions to nonstandard protocols (see Supporting Information for biological protocols and Table S1 for full data) and have explored alternate growth media but have not yet uncovered conditions that provide the reported levels of antimicrobial activity. Going forward, access to the enantiopure natural product will enable us to rule out the unlikely possibility that the unnatural enantiomer has a negative impact on the natural products’ antimicrobial activity.
In conclusion, a concise, stereoselective synthesis of (±)-melokhanine E has been reported in eight steps (longest linear sequence) and in 11% overall yield. The key step of our synthesis is a MgI2 promoted ring opening/cyclization that successfully provides the natural product core in excellent yield and as a single detectable diastereomer. The stereochemical control of this transformation relies on the piperidine C3 all-carbon quaternary center, presenting an opportunity to expand this approach to a number of related natural products with validated biological activities. The current work is focused on developing an asymmetric synthesis of 9 and extending this chemistry to the bisleuconothine family of natural products5,24,25 (5, Figure 1), and these efforts will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the NIH (R01GM117570) for partial support of this work and to NC State University for support of our program. Mass spectrometry data, NMR data, and X-ray data were obtained at the NC State Molecular, Education, Technology and Research Innovation Center (METRIC). Kaylib Robinson (NC State) is acknowledged for the training to conduct the antimicrobial assays, and Dr. Vincent Lindsay (NC State) is acknowledged for helpful discussion throughout the project.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.9b04546.
Experimental details, spectroscopic and analytical data of all new compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.9b04546
The authors declare no competing financial interest.
REFERENCES
- (1).Wright PM; Seiple IB; Myers AG The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem., Int. Ed 2014, 53 (34), 8840–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rossiter SE; Fletcher MH; Wuest WM Natural Products as Platforms To Overcome Antibiotic Resistance. Chem. Rev 2017, 117 (19), 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Abouelhassan Y; Garrison AT; Yang H; Chávez-Riveros A; Burch GM; Huigens RW Recent Progress in Natural-Product-Inspired Programs Aimed To Address Antibiotic Resistance and Tolerance. J. Med. Chem 2019, 62, 7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cheng G-G; Li D; Hou B; Li X-N; Liu L; Chen Y-Y; Lunga P-K; Khan A; Liu Y-P; Zuo Z-L; et al. Melokhanines A–J, Bioactive Monoterpenoid Indole Alkaloids with Diverse Skeletons From Melodinus Khasianus. J. Nat. Prod 2016, 79 (9), 2158–2166. [DOI] [PubMed] [Google Scholar]
- (5).Hirasawa Y; Shoji T; Arai T; Nugroho AE; Deguchi J; Hosoya T; Uchiyama N; Goda Y; Awang K; Hadi HA; et al. Bisleuconothine A, an Eburnane-Aspidosperma Bisindole Alkaloid from Leuconotis Griffithii. Bioorg. Med. Chem. Lett 2010, 20 (6), 2021–2024. [DOI] [PubMed] [Google Scholar]
- (6).Sears JE; Boger DL Total Synthesis of Vinblastine, Related Natural Products, and Key Analogues and Development of Inspired Methodology Suitable for the Systematic Study of Their Structure-Function Properties. Acc. Chem. Res 2015, 48 (3), 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li G; Piemontesi C; Wang Q; Zhu J Stereoselective Total Synthesis of Eburnane-Type Alkaloids Enabled by Conformation-Directed Cyclization and Rearrangement. Angew. Chem., Int. Ed 2019, 58 (9), 2870–2874. [DOI] [PubMed] [Google Scholar]
- (8).Pierce JG; Cholewczynski AE; Williams PC Stereocontrolled Synthesis of Melokhanine E via an Intramolecular Formal [3 + 2] Cycloaddition. ChemRxiv. 2019, DOI: 10.26434/chemrxiv.10022858.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pandey G; Prasanna KC Iminium Ion Cascade Reaction in the Total Synthesis of (+)-Vincadifformine. Org. Lett 2011, 13 (17), 4672–4675. [DOI] [PubMed] [Google Scholar]
- (10).Pandey G; Khamrai J; Mishra A Generation of All-Carbon Quaternary Stereocenters at the C-3 Carbon of Lactams via [3,3]-Sigmatropic Rearrangement and Revision of Absolute Configuration: Total Synthesis of (−)-Physostigmine. Org. Lett 2018, 20 (1), 166–169. [DOI] [PubMed] [Google Scholar]
- (11).Behenna DC; Liu Y; Yurino T; Kim J; White DE; Virgil SC; Stoltz BM Enantioselective Construction of Quaternary NHeterocycles by Palladium-Catalysed Decarboxylative Allylic Alkylation of Lactams. Nat. Chem 2012, 4 (2), 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Numajiri Y; Pritchett BP; Chiyoda K; Stoltz BM Enantioselective Synthesis of A-Quaternary Mannich Adducts by Palladium-Catalyzed Allylic Alkylation: Total Synthesis of (+)-Sibirinine. J. Am. Chem. Soc 2015, 137 (3), 1040–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pritchett BP; Kikuchi J; Numajiri Y; Stoltz BM Enantioselective Pd-Catalyzed Allylic Alkylation Reactions of Dihydropyrido[1,2-a]Indolone Substrates: Efficient Syntheses of (−)-Goniomitine, (+)-Aspidospermidine, and (−)-Quebrachamine. Angew. Chem., Int. Ed 2016, 55 (43), 13529–13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).De Simone F; Gertsch J; Waser J Catalytic Selective Cyclizations of Aminocyclopropanes: Formal Synthesis of Aspidospermidine and Total Synthesis of Goniomitine. Angew. Chem., Int. Ed 2010, 49 (33), 5767–5770. [DOI] [PubMed] [Google Scholar]
- (15).De Simone F; Saget T; Benfatti F; Almeida S; Waser J Formal Homo-Nazarov and Other Cyclization Reactions of Activated Cyclopropanes. Chem. - Eur. J 2011, 17 (51), 14527–14538. [DOI] [PubMed] [Google Scholar]
- (16).Frei R; Staedler D; Raja A; Franke R; Sasse F; Gerber-Lemaire S; Waser J Total Synthesis and Biological Evaluation of Jerantinine E. Angew. Chem., Int. Ed 2013, 52 (50), 13373–13376. [DOI] [PubMed] [Google Scholar]
- (17).Alper PB; Meyers C; Lerchner A; Siegel DR; Carreira EM Facile, Novel Methodology for the Synthesis of Spiro[Pyrrolidin-3,3′-oxindoles]: Catalyzed Ring Expansion Reactions of Cyclopropanes by Aldimines. Angew. Chem., Int. Ed 1999, 38 (21), 3186–3189. [PubMed] [Google Scholar]
- (18).Fischer C; Meyers C; Carreira EM Efficient Synthesis of (±)-Horsfiline through the MgI2-Catalyzed Ring-Expansion Reaction of a Spiro[Cyclopropane-1,3′-indol]-2′-one. Helv. Chim. Acta 2000, 83 (6), 1175–1181. [Google Scholar]
- (19).Lerchner A; Carreira EM First Total Synthesis of (±)-Strychnofoline via a Highly Selective Ring-Expansion Reaction. J. Am. Chem. Soc 2002, 124 (50), 14826–14827. [DOI] [PubMed] [Google Scholar]
- (20).Meyers C; Carreira EM Total Synthesis of (−)-Spiro-tryprostatin B. Angew. Chem., Int. Ed 2003, 42 (6), 694–696. [DOI] [PubMed] [Google Scholar]
- (21).Marti C; Carreira EM Total Synthesis of (−)-Spiro-tryprostatin B: Synthesis and Related Studies. J. Am. Chem. Soc 2005, 127 (32), 11505–11515. [DOI] [PubMed] [Google Scholar]
- (22).Kawada M; Kawano Y; Sugihara H; Takei S; Imada I Spirocyclopropane Compounds. I. Synthesis and Reactivity of Spiro [Cyclopropane-1, 2’-[2H] Indol]-3′(1’H)-Ones. Chem. Pharm. Bull 1981, 29 (7), 1900–1911. [DOI] [PubMed] [Google Scholar]
- (23). There is a discrepancy between the bacteria code identifiers and the strains reported; both the strain reported, and the code identifier were used in these studies. See Supporting Information for full MIC data and the SI for assay protocols.
- (24).Wong C; Seki A; Horiguchi K; Shoji T; Arai T; Nugroho A; Hirasawa Y; Sato F; Kaneda T; Morita H Bisleuconothine A Induces Autophagosome Formation by Interfering with AKT-MTOR Signaling Pathway. J. Nat. Prod 2015, 78 (7), 1656–1662. [DOI] [PubMed] [Google Scholar]
- (25).Nugroho A; Zhang W; Hirasawa Y; Tang Y; Wong C; Kaneda T; Hadi HA; Morita H Bisleuconothines B–D, Modified Eburnane-Aspidosperma Bisindole Alkaloids from Leuconotis Griffithii. J. Nat. Prod 2018, 81 (11), 2600–2604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.