

Abstract
Coumarin-hydroxamic acid derivatives 7a–k were herein designed with a dual purpose: as antiproliferative agents and fluorescent probes. The compounds were synthesized in moderate yields (30–87%) through a simple methodology, biological evaluation was carried out on prostate (PC3) and breast cancer (BT-474 and MDA-MB-231) cell lines to determine the effects on cell proliferation and gene expression. For compounds 7c, 7e, 7f, 7i and 7j the inhibition of cancer cell proliferation was similar to that found with the reference compound at a comparable concentration (10 μM), in addition, their molecular docking studies performed on histone deacetylases 1, 6 and 8 showed strong binding to the respective active sites. In most cases, antiproliferative activity was accompanied by greater levels of cyclin-dependent kinase inhibitor p21, downregulation of the p53 tumor suppressor gene, and regulation of cyclin D1 gene expression. We conclude that compounds 7c, 7e, 7f, 7i and 7j may be considered as potential anticancer agents, considering their antiproliferative properties, their effect on the regulation of the genes, as well as their capacity to dock to the active sites. The fluorescent properties of compound 7j and 7k suggest that they can provide further insight into the mechanism of action.
Keywords: coumarins, hydroxamic acids, HDAC, gene expression, fluorescent probe, docking analysis
1. Introduction
Coumarins are part of a flavonoid group that acts as a secondary metabolite in plants. These heterocyclic compounds are known to form part of a wide variety of natural and synthetic compounds showing diverse pharmacological activity. Such compounds include anti-inflammatory [1], antioxidant [2], hepatoprotective [3], antithrombotic [4], antiviral [5], antimicrobial [6,7,8], antituberculosis [9,10], anti-carcinogenic [11], antidepressant [12,13], antihyperlipidemic [14] and anticholinesterase agents [15,16], and they are usually associated with low toxicity.
Coumarins have been extensively examined as bioactive agents [17,18], today they are one of the most versatile types of compounds for anticancer drug design and discovery, as evidenced by several reports [19]. According to an in vitro study against renal cell carcinoma, for example, coumarin (Figure 1a) and 7-hydroxycoumarin (Figure 1b) are potent cytotoxic and cytostatic agents [20]. A common strategy for the optimization of antiproliferative activity is the functionalization of the coumarin core [21,22] (Figure 1c,d). A study of coumarins with stilbene revealed that the substitution pattern is the key to their potency. The shifting of trans-vinylbenzene from the C-4 to C-3 position in the coumarin ring leads to a two-fold decrease in activity [23] (Figure 1e). For the discussed above, the synthesis and characterization of coumarin based compounds is a relevant topic to be addressed. Even that, only a few recent studies include a computational characterization of coumarin based compounds as a HDAC Inhibitor. Regarding with this, Zhang and coworkers in recent research indicate the effectiveness of having a coumarin moiety as the main structure for its modification and application as anticancer agents, in combination with an hydroxamic acid functionalization to potentialize the activity (Figure 2) [24,25,26]. This kind of information is a strong background to embrace the hypothesis of the present work.
Figure 1.

Library of structures with a coumarin core: (a) coumarin, (b) 7-hydroxycoumarin, (c) Siedel’s cytostatic coumarin, (d) Wu’s coumarin cytostatic agent, and (e) coumarin-stilbene.
Figure 2.

Examples for structures of coumarin and hydroxamic acids moieties that indicates the effectiveness in the application as anticancer agents.
Histone deacetylase (HDAC) is an amidohydrolase which deacetylates the histone lysine residues for chromatin remodeling and thus plays a vital role in the epigenetic regulation of gene expression, due to its over expression in several forms of cancer, HDAC is considered as a potential anticancer drug target. HDAC inhibitors are natural or synthetic small molecules that can alter the acetylation status to regulate various cellular events such as cell survival, differentiation and apoptosis in tumor cells and thus exhibit anticancer activity [27,28]. The application of coumarins, a well-known fluorophore [29,30,31], as fluorescent probes, has been described in various contexts, including the real-time (RT) detection of histone deacetylase (HDAC) activity by an acetyl-lysine functionalized mimic (Figure 3a) [32], as well as the monitoring of HDAC activity by K4(Ac)-CCB (Figure 3b) which consists of the histone H3 peptide containing acetyl-Lys and a coumarin fluorophore with a carbonate ester [33] and by a scriptaid derivative (Figure 3c). Unlike K4(Ac)-CCB, the scriptaid derivative bears the pharmacophore structure of HDAC inhibitors and has shown potential anticancer activity, as observed in the visualization of its distribution in MDA-MB-231 cells. Other reports have also described the use of coumarins as fluorescent probes and at the same time as HDAC inhibitors. Jung, M. et al. [34,35] developed probes with distinct structures that can access the catalytic site of HDAC in order to inhibit sirtuins (Figure 3d). Similarly, a coumarin-SAHA compound (Figure 3e) [36] based in the FDA approved structure suberoylanilide hydroxamic acid (SAHA) (Figure 4a) [37], was employed as a fluorescent probe for determining the Kd and Koff of HDAC8 enzyme-inhibitor complexes. Moreover, it was found that coumarin-thiophene derivatives induce the selective inhibition of HeLa cells (IC50 = 9.43 μM) [38]. In these studies, coumarin compounds used as fluorescent probes have provided insights into the mechanisms of action of HDACs. In addition, in a recent study Abdizadeh et al. [39] addresses the construction of a QSAR model to evaluate the biological activity of coumarin based benzamides as HDACs inhibitors. We think that the interdisciplinary work combining the synthesis, biological evaluation, and computational characterization of coumarin based compounds is a relevant issue.
Figure 3.

Fluorescent probes for the detection of HDAC activity. (a) Coumarin structure for real-time detection of HDAC activity, (b) K4(Ac)-CCB structure for monitoring of HDAC activity, (c) Scriptaid derivative (d) Coumarin probe that can access the catalytic site of HDAC, (e) Coumarin-SAHA derivatives.
Figure 4.

Structure of SAHA (C14H20N2O3) (a) and the synthesized test compounds (b).
There are few descriptions in the literature of coumarin derivatives that target HDACs in order to function simultaneously as a fluorescent probe and an antiproliferative agent. The aim of the current contribution was to design coumarin derivatives with this dual function. Two strategies were employed for the design of HDAC inhibitors: to improve the binding property of the capping group and to explore the influence of a flexible linker on binding (by the integration of an amide group containing an aliphatic linker of different chain sizes at the C-3 position). Hence, the principle characteristics of the present test compounds are similar to those of SAHA (Figure 4a). Evaluation was made of the effects of the derivatives on the proliferation of cancer cells as well as on the regulation of the p21, p53 and cyclin D1 (CDK1) genes. In addition, the subcellular location was established with two of these fluorescent analogues.
2. Results and Discussion
2.1. Chemistry
Based on the results of our previous studies [40,41], we envisioned the design of a compound with a dual purpose, serving as a fluorescent probe and an inhibitor of HDAC enzymes. For the former objective, the coumarin core was included in the compounds. For the latter, this core was structurally modified to improve ligand binding to the active site of the HDAC enzymes. To increase the interaction of the coumarin derivative with the external surface of the active site of the enzyme, the aromatic ring was substituted with different groups having distinct electronic characteristics.
The synthetic route for the new compounds is outlined in Scheme 1. Compounds 3a–f were prepared in high yield by the condensation of Meldrum’s acid (2) and aldehydes 1a–f [42,43]. After a recrystallization process, the carboxylic acids underwent a coupling reaction with various hydrochloride amine salts, and then with carbonyldiimidazole (CDI) to provide the esters 5a–k. The hydrolysis of this series with lithium hydroxide gave the terminal carboxylic acids 6a–k. The latter compounds were converted into hydroxamic acids by another coupling reaction with hydroxylamine hydrochloride and CDI to afford 7a–k (Scheme 1, Table 1). The structure of the target compounds was confirmed by mass spectroscopy (HRSM) and NMR. Thus, a simple methodology was developed for the synthesis of the proposed molecules. It was tolerant to different functional substituents on the coumarin core. Water and filtration could be used as the only form of purification at all stages. These are attractive characteristics that facilitate the application of the methodology.
Scheme 1.

Reagent and reaction conditions. (a) Aldehyde (1 mmol), Meldrum’s acid (1.2 mmol), H2O, reflux, 5–6 h, 80–90%. (b) Compounds 3a–f (1 mmol), amine hydrochloride (1.2 mmol), DMAP (5 mol%), CDI (1.1 mmol), DMF, rt, overnight, 46–90%. (c) Compounds 5a–k (1 mmol), LiOH (7 mmol), THF/H2O (1/2), rt, 12 h, 50–93%; (d) Compounds 6a–k (1 mmol), NH2OH/HCl (1.2 mmol), DMAP (5% mmol), CDI (1.1 mmol), DMF, rt, overnight, 30–87%.
Table 1.
Results of the synthesis of coumarins 7a–k as well as data on absorption and emission.
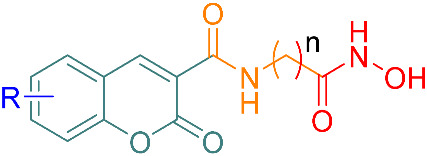
| Product | R | n | Yield (%) | λab (nm) | λem (nm) |
|---|---|---|---|---|---|
| 7a | H- | 1 | 50 | 297 | 406 |
| 7b | H- | 2 | 45 | 303 | 405 |
| 7c | H- | 5 | 50 | 329 | 405 |
| 7d | 6-MeO- | 3 | 30 | 363 | 458 |
| 7e | 6-MeO- | 5 | 87 | 365 | 458 |
| 7f | 8-EtO- | 5 | 48 | 311 | 486 |
| 7g | 6-Br- | 1 | 46 | 294 | 424 |
| 7h | 6-Br- | 2 | 45 | 295 | 415 |
| 7i | 6-Br- | 5 | 65 | 283 | 424 |
| 7j | 7-Et2N- | 2 | 82 | 419 | 470 |
| 7k | 7-Et2N- | 5 | 82 | 419 | 470 |
Absorbance and emission studies on 7a–k were first carried out in MeOH before testing other solvents. Solubility turned out to be the limiting factor. In general, the peak of absorption at a concentration of 1 × 10−6 M was found between 283–419 nm. The emission peak at the same concentration and with the same solvent was observed at 405–470 nm (Table 1).
2.2. Biological Evaluation
Several HDAC inhibitors, such as SAHA have shown potent antiproliferative activities against various cancer cell lines, including human breast and prostate cancer cells [44,45,46]. Therefore, the latter two cancer cell lines were employed to evaluate the antiproliferative effect of compounds 7a–k. The cells were incubated with different concentrations of SAHA or the newly synthesized compounds for 3–6 days. Depending on the cell line and the antiproliferative response, some of them were then assessed with the SRB assay. As expected, SAHA inhibited the proliferation of all cancer cell lines in a dose-dependent manner. Although the effect of compounds 7a–k varied, most of them demonstrated powerful antiproliferative activity against the cell lines (Supplementary Material Figure S1). The growth inhibitory effect was tested at 10 μM, revealing SAHA-like activity for 7c, 7e, 7f, 7i and 7j, and less potent activity for 7b and 7h (Table 2).
Table 2.
Growth inhibitory effects (GI%) exerted by compounds 7a–k on three cancer cell lines.
| Compound | BT-474 | MDA-MB-231 | PC3 |
|---|---|---|---|
| SAHA | 98.22 ± 1.82 * | 97.54 ± 1.59 * | 87.86 ± 7.85 * |
| 7a | 9.90 ± 7.79 | 17.39 ± 15.15 | 11.31 ± 7.65 |
| 7b | 40.06 ± 18.94 * | 47.81 ± 11.06 * | 22.89 ± 19.30 |
| 7c | 95.98 ± 2.61 * | 91.61 ± 2.49 * | 68.82 ± 22.14 * |
| 7d | 33.24 ± 15.25 | 16.57 ± 8.78 | 8.97 ± 16.21 |
| 7e | 82.08 ± 25.72 * | 92.28 ± 5.43 * | 71.02 ± 11.55 * |
| 7f | 91.88 ± 12.54 * | 100.08 ± 0.29 * | 88.62 ± 14.78 * |
| 7g | 6.73 ± 7.40 | 14.00 ± 7.13 | 5.10 ± 8.93 |
| 7h | 61.83 ± 11.82 * | 64.30 ± 17.82 * | 32.64 ± 14.31 |
| 7i | 93.47 ± 4.29 * | 93.64 ± 3.27 * | 75.85 ± 16.43 * |
| 7j | 70.84 ± 18.62 * | 78.50 ± 5.79 * | 53.02 ± 13.35 * |
| 7k | 7.05 ± 13.26 | 0.00 ± 3.9 | 7.69 ± 21.95 |
Data are expressed as the mean ± SD of the percentage of growth inhibition, based on determinations in triplicate in at least three different experiments. * p < 0.05 vs. vehicle.
Greater sensitivity to antiproliferative activity was shown by the breast versus prostate cancer cells. Zhao et al. obtained similar results where coumarin-containing hydroxamate HDAC inhibitors were more potent to inhibit MDA-MB-231 cell proliferation compared with lung adenocarcinoma cell lines [26]. The compounds exhibiting the strongest antiproliferative activity were 7c, 7e, 7f and 7i, substituted with H, 6-MeO, 8-EtO and 6-Br, respectively. Interestingly, a common structural characteristic of the most active compounds 7c, 7e, 7f and 7i, is that they all possess the same side chain, comprising from five methylene groups, whereas the aromatic ring coumarin substituents vary. The compounds unable to modify cell growth were 7a, 7d, 7g and 7k, substituted with H, 6-MeO, 6-Br and 7-Et2N, respectively. Consequently, compounds 7c, 7e, 7f, 7i and 7j (7-Et2N) could possibly be used as antineoplastic agents.
2.2.1. Effects of SAHA Analogues on the Expression of Cell Cycle Regulatory Genes
SAHA suppresses growth and induces cell cycle arrest and apoptosis of human breast and prostate cancer cells. This occurs in part by the regulation of the proteins involved in the cell cycle and apoptosis, such as cyclin-dependent kinase (CDK) inhibitors p21, p53 and cyclin D1 (CD1) [45,46,47,48]. Therefore, the effect of compounds 7a–k on the expression of cell cycle regulatory proteins was examined.
Accordingly, breast and prostate cancer cells were treated in the presence and absence of the compounds having demonstrated the best antiproliferative activity (7c, 7e, 7f, 7i and 7j) and one that did not change cell growth (7d). In both the BT-474 and PC3 cell lines, most of the compounds produced an effect similar to SAHA, significantly increasing p21 gene expression and diminishing p53 and CD1 mRNA levels compared to the vehicle (Table 3). These results were not found in cells treated with 7i. As expected, p21 and CD1 gene expression was not affected by 7d, but mRNA levels of p53 were significantly decreased.
Table 3.
The effect of compounds 7a–k, when tested against two cancer cell lines, on the expression of genes related to apoptosis and the cell cycle.
| BT-474 | |||
| Compound | p21 | p53 | CD1 |
| SAHA | 17.39 ± 3.70 * | 0.12 ± 0.08 * | 0.02 ± 0.00 * |
| 7c | 3.81 ± 1.08 * | 0.57 ± 0.27 * | 0.51 ± 0.09 * |
| 7d | 1.47 ± 0.41 | 0.47 ± 0.08 * | 0.92 ± 0.06 |
| 7e | 5.47 ± 2.01 * | 0.08 ± 0.00 * | 0.57 ± 0.07 * |
| 7f | 23.65 ± 6.69 * | 0.51 ± 0.22 * | 0.40 ± 0.36 |
| 7i | 0.44 ± 0.02 * | 0.74 ± 0.21 | 0.91 ± 0.05 |
| 7j | 26.56 ± 0.44 * | 0.57 ± 0.24 * | 0.32 ± 0.09 * |
| PC3 | |||
| Compound | p21 | p53 | CD1 |
| SAHA | 7.44 ± 2.24 * | 0.12 ± 0.14 * | 0.04 ± 0.01 * |
| 7c | 3.48 ± 0.55 * | 0.25 ± 0.17 * | 0.61 ± 0.13 |
| 7d | 1.41 ± 0.27 | 0.49 ± 0.23 * | 0.64 ± 0.19 |
| 7e | 3.90 ± 0.43 * | 0.18 ± 0.11 * | 0.30 ± 0.02 * |
| 7f | 3.60 ± 0.78 * | 0.16 ± 0.20 * | 0.32 ± 0.22 * |
| 7i | 2.07 ± 0.66 | 0.42 ± 0.39 | 0.90 ± 0.30 |
| 7j | 3.80 ± 2.33 | 0.16 ± 0.06 * | 0.43 ± 0.27 * |
BT-474 and PC3 cells were incubated in the presence (10 μM) or absence of SAHA or compounds 7c–f, i–j for 24 h. Subsequently, mRNA was extracted, and real-time PCR (qPCR) was performed. Data are expressed as the mean ± SD of the genes/GAPDH, using the normalized ratio of triplicate determinations for mRNA from at least two different experiments. Vehicle-treated cells were arbitrarily given a value of one. * p < 0.05 vs. vehicle.
Overall, the antiproliferative activity of the compounds correlated well with gene regulation in the BT-474 and PC3 cancer cell lines. The exception was 7i, which displayed growth inhibition comparable to that of SAHA at 10 μM (Table 2) but it did not change the expression of cell cycle regulatory genes. In this compound, due the enhanced lipophilicity character from Br substituent on the coumarin ring could modify its capacity to enter the nucleus of the cell, thus not allowing it to modulate genetic expression. Nevertheless, it showed activity against HDACs located in the cytoplasm or plasma membrane, such as HDAC3 [49,50].
According to the current findings, 7c, 7e, 7f, 7i and 7j likely influence the enzymatic activity of HDACs [45,47,51], considering the antiproliferative properties of the compounds, their effect on the regulation of the p21, p53 and cyclin D1 genes, as well as their ability to dock at the active sites of HDAC1, HDAC6 and HDAC8. However, future research is needed to directly analyze the inhibition induced by the compounds on the enzymatic activity of HDACs.
2.2.2. Novel Fluorescent SAHA Analogues
Due to the inherent fluorescent properties of coumarins, an analysis of the excitability and subcellular location of compounds 7a–k was carried out. The relative fluorescence emitted was evaluated after BT-474, PC3 and MDA-MB-231 cell lines were incubated with various concentrations of the compounds for 72–144 h. SAHA was used as the non-fluorescent negative control. Of the 11 compounds evaluated, the relative fluorescence was only significantly increased for 7j and 7k, compared to vehicle-treated cells (Figure 5a–c). Indeed, a significant fluorescence emission intensity was shown by 7j as of 0.1 μM in MDA-MB-231 cells (Figure 5b). Data are expressed as the mean ± SD of determinations in triplicate from at least three distinct experiments. The value of vehicle-treated cells was arbitrarily set at one.
Figure 5.

Relative fluorescence emission intensity and subcellular location of compounds 7j and 7k. (a–c) Breast and prostate cancer cells were treated in the presence (at different concentrations) and absence of the two compounds, followed by spectrofluorometric quantification. The value of vehicle-treated cells was set to one. * p < 0.05 vs vehicle. (d–f) Fluorescence microscopy of 7j and 7k in the three cell lines. Blue = 7j, 7k. Red = propidium iodide (PI). Blue/red = merged image of 7j, 7k and PI. Representative images from two independent experiments are shown.
The cells with a significant difference in relative fluorescence were exposed to 10 μM of 7j, 7k or SAHA and confocal microscopy images were taken (Figure 5d–f). Subsequently, to visualize the cellular penetration and subcellular location of 7j and 7k, confocal microscopy images were obtained for the three cell lines. There was a significant increase in fluorescent emission intensity (indicating cellular uptake), especially for compound 7j (Figure 5d–f). With intracellular cytoplasmic localization and colocalization experiments using propidium iodide, a dramatic change was observed in the color of the nucleus in BT-474 cells (Figure 5d) and to a lesser extent in MDA-MB-231 and PC3 cells (Figure 5e,f), indicating that 7j had penetrated the nucleus.
Since the fluorescence of 7k was found in the cytoplasm but not the nucleus, this compound apparently did not penetrate the nuclear envelope of any of the three cell lines herein examined. Hence, the lack of antiproliferative activity of 7k could be due to its inability to reach the nucleus as well as its limited cellular uptake. Compound 7j is expected to be a valuable tool for future studies on the mechanism of action of HDAC inhibitors, given its fluorescent properties and antiproliferative activity.
2.3. Molecular Docking
The overexpression of HDAC1 in the BT-474 [52,53] and PC3 cell lines [44] is reported to promote the development of cancer. In the MDA-MB-231 cell line, contrarily, HDAC6 and HDAC8 play a critical role in proliferation [54]. Accordingly, such isoforms are ideal biological targets for docking experiments. Compounds 7c, 7e, 7f, 7i and 7j were docked into the active site of HDAC1, HDAC6 and HDAC8 to gain insight into the ligand-receptor binding mode.
The total interaction energies are shown in Table 4. There is not a direct correlation between the energies and the biological activity. Nevertheless, the molecular coupling in the active site of the protein was successfully found, validating the docking methodology. We observe that the best pose of SAHA docked on HDAC8 displays a RMSD of about 0.51 Å, when it is compared with the co-crystallized structure. In addition, we used the study of the interactions between drugs and HDACs to get insight about the interaction mechanism.
Table 4.
Interaction energies (ΔG) of molecules docked into the active site of HDACs isoforms.
| Ligand | HDAC1 (kcal/mol) | HDAC6 (kcal/mol) | HDAC8 (kcal/mol) |
|---|---|---|---|
| SAHA | −86.16 | −55.33 | −20.42 |
| 7c | −89.78 | −73.66 | −25.20 |
| 7e | −118.77 | −68.80 | −31.56 |
| 7f | −111.23 | −79.28 | −30.17 |
| 7i | −108.99 | −68.82 | −29.20 |
| 7j | −113.59 | −80.73 | −48.89 |
Regarding HDAC1, docking calculations demonstrated that four of the five compounds (7c, 7e, 7f and 7i) have a binding mode like SAHA. For 7j, the hydroxamic group inside the pocket is coordinated with the zinc atom in monodentate geometry. For all the other ligands, including SAHA, there is a bidentate chelation effect. The coumarin rings of all ligands cap the active site. For 7c and 7j, the coumarin rings expose the carbonyl group of the moiety to the enzyme, giving rise to acceptor hydrogen bond interactions. For 7e, 7f and 7i, the carbonyl group of the moiety is positioned opposite to the enzyme surface (Figure 6).
Figure 6.

The binding modes for SAHA and compounds 7c, 7e, 7f, 7i and 7j at the active site of HDAC1, according to molecular docking calculations. The ligands are illustrated with a tube model. Oxygen atoms are depicted in red, the bromine atom in dark red, carbon atoms in gray, nitrogen atoms in purple and the zinc atom in pink.
The docking simulations at the active site of HDAC6 evidence a potential T-stacking interaction between the phenylalanine ring and the coumarin ring of all ligands (7c, 7e, 7f, 7i, 7j and SAHA), with distances of about 5.5 Å. A bidentate coordination with the zinc atom was exhibited by four of these compounds and a monodentate coordination by 7j. For all these compounds, the coumarin ring exposes the carbonyl group moiety in a very similar conformation (Figure 7).
Figure 7.

Potential T-stacking interactions between the coumarin ring of studied compounds (SAHA, 7c, 7e, 7f, 7i and 7j center) and the phenylalanine residue (structure in blue labeled as F620) in the active site of HDAC6. T-stacking interactions are displayed as red lines, distances between interacting aromatic moieties are showed in Ångstroms (Å). Zn atom is presented as a pink sphere.
Finally, when docking the five compounds at the active site of HDAC8, all the ligands except 7j displayed a binding mode comparable to that of the reference drug. The fundamental similarity is the coordination between the hydroxamic group and the zinc atom. Compounds 7c and 7i had the strongest acceptor hydrogen bonds (and the largest quantity of the same) with histidine and tyrosine, and such bonds were formed at the bottom of the pocket. For 7c, 7e and 7f, acceptor hydrogen bonds with lysine were detected higher up in the pocket, and for 7e and 7f an extra acceptor hydrogen-bond was observed with tyrosine outside the pocket. Contrarily, the halogen in 7i promoted the opposite orientation of the coumarin rings, thus avoiding hydrogen bond interactions with lysine. For 7j, on the other hand, there were two acceptor hydrogen bonds with tyrosine, two with lysine and one with histidine. However, it showed a monodentate coordination with the zinc atom (Figure 8). The binding properties of 7f agree with its antiproliferative activity displayed by the biological test. It has several desirable properties that are common among HDAC inhibitors, including stacking interactions, a bidentate coordination with the atomic center, and a considerably number of hydrogen-bond interactions.
Figure 8.

Hydrogen-bond interactions (blue lines) for SAHA, 7c, 7e, 7f, 7i, 7f and 7j in the pocket of HDAC8. Zinc atom displayed as a pink sphere.
It is important to compare the binding properties of tested molecules with the ones of coumarin and hydroxycoumarins (3, 4, 5, 6 and 7-hydroxycoumarins). The docking between coumarin and hydroxycoumarins and HDAC1, HDAC6 and HDAC8, revealed unions mainly of π-staking type. The aromatic groups of the amino acids of the enzymatic active site interact with the coumarin ring of both ligands via a π-staking interaction (Figure 9). An essential characteristic of HDAC inhibitors studied in this work is the coordination with the Zn (II) cofactor. Therefore, the docking experiments suggest that 4- and 7-hydroxycoumarins do not bind the cofactor at all, an example for this is displayed in Figure 9 lower panel. We detected that, for only some cases, there is a possible monodentate coordination with Zn (II) for coumarin and hydroxycoumarins. From these results, we conclude that SAHA and its analogues possess a pharmacological activity that coumarin and hydroxycoumarins cannot display by itself, since SAHA and analogs exhibit bidentate coordination between the hydroxamic functional group and Zn (II). Hence, the functionalization of the coumarin ring enlarges the possibility of chelation with the cofactor Zn (II). Such fact increases the possibilities of cancer cell growth inhibition. A detailed information of the binding energies of coumarin and hydroxycoumarins is located in the Supplementary Material.
Figure 9.

Potential interactions between the coumarin and 4-hydroxycoumarin and the residue in the active site of HDAC1, HDAC6 and HDAC8. The stacking interactions are show as a red line, hydrogen bond is blue dotted line and the coordination bond with Zinc is the green line.
2.4. ADMET Properties for SAHA Analogues
The Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties are relevant for the characterization of a drug candidate. In the present contribution, the determination of the physicochemical and pharmacokinetic properties of SAHA and analogues (7c, 7e, 7f, 7i and 7j) was carried out through a free web tool to evaluate pharmacokinetics: SwissADME [55]. In general, SAHA and its analogs display a large polar surface area (PSA) and good solubility in water. Regarding the partition coefficient between n-octanol and water (log Po/w), the classic descriptor of lipophilicity, SAHA and analogs are within the optimal range (−0.7, +5.0) (Table 5). Besides, the prediction for passive human gastrointestinal absorption (HIA) was found to be high, in contrast to the permeability of the blood-brain barrier (BBB) which was found negative for SAHA and analogues (Table 6). On the other hand, SAHA is a substrate for P-glycoprotein (P-gp), the same behavior is exhibited by 7e, 7f, 7i and 7j, except for the 7c. The binding of a drug with P-gp is relevant, since this protein can export molecules to reduce its intracellular concentration below the effective cytotoxic threshold [56]. The above discussion can be related with the in vitro observations of a treatment with 7c, in which the sensitivity of BT-474 breast cancer cells was higher than the rest of the analogues. As a matter of fact, in such, culture P-gp is overexpressed and for this reason it is capable to disperse all SAHA analogues except for the 7c.
Table 5.
Physicochemical parameter values of compounds SAHA and 7a–k.
| Compound | PSA (Å2) | Log P | Solubility (mg/mL) | Solubility Class |
|---|---|---|---|---|
| SAHA | 108.64 | 1.92 | 0.509 | Soluble |
| 7c | 91.57 | 3.41 | 0.0665 | Soluble |
| 7e | 117.87 | 1.75 | 0.636 | Soluble |
| 7f | 117.87 | 1.75 | 0.636 | Soluble |
| 7i | 107.89 | 1.77 | 0.38 | Soluble |
| 7j | 98.74 | 1.52 | 1.04 | Soluble |
Table 6.
Pharmacokinetic parameter of compounds SAHA and 7a–k.
| Compound | HIA | BBB | Pgp Substrate | CYP1A2 Inhibitor |
|---|---|---|---|---|
| SAHA | High | No | Yes | No |
| 7c | High | No | No | Yes |
| 7e | High | No | Yes | No |
| 7f | High | No | Yes | No |
| 7i | High | No | Yes | No |
| 7j | High | No | Yes | No |
Therefore, it can be inferred that the new SAHA analogs exhibit good absorption, favorable permeability to the interior of the cell and low toxicity to the cytochrome P450 enzyme. It should be noted that in vitro and in vivo studies indicate that SAHA is not metabolized by the cytochrome p450 system, rather is metabolized by glucuronidation, hydrolysis and beta-oxidation [57]. In addition, SAHA is eliminated via renal [58].
3. Materials and Methods
3.1. Chemistry
All solvents and reagents were commercially available and used without further purification. Melting points were determined on a RY-1 MP apparatus. Absorption spectra were recorded on a Perkin-Elmer Lambda 50 apparatus (Perkin-Elmer Inc., Waltham, MA, USA), and Emission spectra on a FL-7000 FL spectrometer (Hitachi Inc., Chiyoda, Tokio, Japon). ESI-HRMS spectra were obtained on a Maxis Impact ESI-QTOF-MS spectrometer (Bruker Corporation 40 Manning Road Billerica, MA 01821, USA) and a Bruker Daltonics mass spectrometer (Bruker Corporation 40 Manning Road Billerica, MA 01821, USA). 1H-NMR and 13C-NMR spectra were acquired in CDCl3 or DMSO-d6 solutions with the following NMR spectrometers: a Bruker Avance III HD NMR spectrometer [Bruker Corporation 40 Manning Road Billerica, MA 01821, USA] with a Bruker Ascent 400 MHz magnet or a Bruker Avance III HD with a Bruker Ultra Shield 500 MHz HD magnet. Spectra were recorded at 25 °C with TMS and solvent signals allotted as internal standards. Chemical shifts are expressed in ppm (δ) and J-values in hertz.
3.1.1. General Procedure for the Preparation of Coumarin-3-Carboxylic Acids (3a–f)
In a round-bottom flask, a solution of the corresponding salicylaldehyde (1 mmol) and Meldrum’s acid (1.2 mmol) was stirred in water at reflux for 5–6 h. The precipitate was obtained by filtration. The purification was carried out by recrystallization with MeOH and cold water [42,43].
2-oxo-2H-chromene-3-carboxylic acid (3a), white solid (82% yield); mp = 183–184 °C; IR (KBr): νmax/cm−1 = 1746, 1685, 1614, 1569; UV-Vis (MeOH): λmax/nm = 296; 1H-NMR (500 MHz, CDCl3): δ 12.26 (s, 1H, H-3a), 8.96 (s, 1H, H-4), 7.80 (dd, J = 14, 7.7 Hz, 2H, H-5,8), 7.51 (t, J = 8.9 Hz, 2H, H-6,7); 13C-NMR (125 MHz, CDCl3): δ 164.10, 162.42, 154.59, 151.52, 135.79, 130.52, 126.28, 118.48, 117.23, 114.90; HRMS (ESI) m/z calcd. for C10H6O4: [M + H]+ 191.0339, found 191.0342.
6-methoxy-2-oxo-2H-chromene-3-carboxylic acid (3b), yellowish solid (90% yield); mp = 202–205 °C; IR (KBr): νmax/cm−1 = 1761, 1607, 1622, 1574; UV-Vis (MeOH): λmax/nm = 295; 1H-NMR (500 MHz, CDCl3): δ 12.37 (s, 1H, H-3a), 8.91 (s, 1H, H-4), 7.43 (d, J= 9.1 Hz, 1H, H-8), 7.36 (dd, J = 9.1, 2.8 Hz, 1H, H-7), 7.12 (d, J = 2.8 Hz, 1H, H-5), 3.91 (s, 2H, H-6a); 13C-NMR (125 MHz, CDCl3): δ 164.22, 162.55, 157.25, 151.22, 149.22, 124.44, 118.90, 118.34, 115.00, 111.01, 56.05; HRMS (ESI) m/z calcd. for C11H8O5: [M + H]+ 221.0444, found 221.0446.
6-bromo-2-oxo-2H-chromene-3-carboxylic acid (3c), white solid (85% yield); mp = 195–197 °C; IR (KBr): νmax/cm−1 = 1737, 1716, 1690, 1609; UV-Vis (MeOH): λmax/nm = 291; 1H-NMR (500 MHz, CDCl3): δ 12.06 (s, 1H, H-3a), 8.87 (s, 1H, H-4), 7.90 (d, J = 2.1 Hz, 1H, H-8), 7.87 (dd, J = 8.8, 2.2 Hz, 1H, H-7), 7.40 (d, J = 8.8 Hz, 1H, H-5); 13C-NMR (125 MHz, CDCl3): δ 163.41, 161.82, 153.33, 150.09, 138.42, 132.45, 119.82, 118.97, 116.01; HRMS (ESI) m/z calcd. for C10H5BrO4: [M + H]+ 268.9444, found 270.9428.
8-ethoxy-2-oxo-2H-chromene-3-carboxylic acid (3d), greenish solid (80% yield); mp = 186–188 °C; IR (KBr): νmax/cm−1 = 1756, 1674; UV-Vis (MeOH): λmax/nm = 309; 1H-NMR (500 MHz, CDCl3): δ 12.32 (s, 1H, H-3a), 8.91 (s, 1H, H-4), 7.38 (t, J = 7.9 Hz, 1H, H-6), 7.30 (d, J = 7.9 Hz, 2H, H-5,7), 4.24 (q, J = 7.0 Hz, 2H, H-8a), 1.54 (t, J = 7.0 Hz, 3H, H-8b); 13C-NMR (125 MHz, CDCl3): δ 163.89, 162.53, 151.78, 146.81, 144.31, 126.16, 121.23, 119.19, 118.17, 114.90, 65.32, 14.64; HRMS (ESI) m/z calcd. for C12H10O5: [M + H]+ 235.0601, found 235.0604.
7-(diethylamino)-2-oxo-2H-chromene-3-carboxylic acid (3e), white solid (85% yield); mp = 195–197 °C; IR (KBr): νmax/cm−1 = 1737, 1716, 1690, 1609; UV-Vis (MeOH): λmax/nm= 291; 1H-NMR (500 MHz, CDCl3): δ 12.06 (s, 1H, H-3a), 8.87 (s, 1H, H-4), 7.90 (d, J = 2.1 Hz, 1H, H-8), 7.87 (dd, J = 8.8, 2.2 Hz, 1H, H-7), 7.40 (d, J= 8.8 Hz, 1H, H-5); 13C-NMR (125 MHz, CDCl3): δ 163.41, 161.82, 153.33, 150.09, 138.42, 132.45, 119.82, 118.97, 116.01; HRMS (ESI) m/z calcd. for C10H5BrO4: [M + H]+ 268.9444, found 270.9428.
7-hydroxy-2-oxo-2H-chromene-3-carboxylic acid (3f), brownish solid (90% yield); mp= 263–264 °C; IR (KBr): νmax/cm−1 = 3126, 1712, 1683, 1619; UV-Vis (MeOH): λmax/nm = 351; 1H-NMR (500 MHz, DMSO-d6): δ 11.87 (s, 1H, H-7a), 8.64 (s, 1H, H-4), 7.70 (d, J = 8.6 Hz, 1H, H-5), 6.82 (dd, J = 8.6, 2.1 Hz, 1H, H-6), 6.70 (d, J = 2.0 Hz, 1H, H-8); 13C-NMR (125 MHz, DMSO-d6): δ 164.66, 164.39, 158.12, 157.42, 149.87, 132.44, 114.46, 112.83, 111.06, 102.24; HRMS (ESI) m/z calcd. for C10H6O5: [M + H]+ 207.0288, found 207.0291.
3.1.2. General Procedure for the Preparation of Coumarin-3-Carboxamides (5a–k)
In a dry round-bottom flask purged with N2, the corresponding coumarin (1 mmol), 4-(dimethylamino)pyridine (DMAP) (5% mol), and carbonyldiimidazole (CDI) (1.1 mmol) were dissolved in DMF. After 30 min of stirring at room temperature, hydrochloride amine salt was added and stirring continued overnight. Water was added when the reaction was completed. The resulting precipitate was obtained by filtration and purified by recrystallization with MeOH and cold water.
Methyl (2-oxo-2H-chromene-3-carbonyl)glycinate (5a), white solid (84% yield); mp = 181–183 °C; IR (KBr): νmax/cm−1 = 3328, 3054, 1749, 1710, 1655; UV-Vis (MeOH): λmax/nm= 300; 1H-NMR (500 MHz, CDCl3): δ 9.25 (s, 1H, H-3a), 8.91 (s, 1H, H-4), 7.77–7.56 (m, 2H, H-5,7), 7.50–7.31 (m, 2H, H-6,8), 4.26 (d, J = 5.6 Hz, 2H, H-3b), 3.80 (s, 3H, H-3c); 13C-NMR (125 MHz, CDCl3): δ 169.73, 161.91, 161.24, 154.55, 148.77, 134.30, 129.90, 125.35, 118.52, 117.96, 116.72, 52.42, 41.69; HRMS (ESI) m/z calcd. for C13H11NO5: [M + H]+ 262.0710, found 262.0718.
Ethyl 3-(2-oxo-2H-chromene-3-carboxamido)propanoate (5b), white solid (60% yield); mp = 128–130 °C; IR (KBr): νmax/cm−1 = 3348, 3055, 1726, 1706, 1647, 1612; UV-Vis (MeOH): λmax/nm = 297; 1H-NMR (500 MHz, CDCl3): δ 9.14 (s, 1H, H-3a), 8.90 (s, 1H, H-4), 7.81-7.56 (m, 2H, H-6,8), 7.39 (m, 2H, H-5,7), 4.20 (q, J = 7.1 Hz, 2H, H-3d), 3.76 (q, J = 6.3 Hz, 2H, H-3b), 2.66 (t, J = 6.4 Hz, 2H, H-3c), 1.29 (t, J = 7.1 Hz, 3H, H-3e); 13C-NMR (125 MHz, CDCl3): δ 171.77, 161.59, 161.26, 154.47, 148.36, 134.06, 129.80, 125.27, 118.61, 118.39, 116.65, 60.84, 35.44, 34.21, 14.20; HRMS (ESI) m/z calcd. for C15H15NO5: [M + H]+ 290.1023, found 290.1029.
Methyl 6-(2-oxo-2H-chromene-3-carboxamido)hexanoate (5c), white solid (85% yield); mp = 88–90 °C; IR (KBr): νmax/cm−1 = 3321, 3054, 1742, 1721, 1706, 1607; UV-Vis (MeOH): λmax/nm = 298; 1H-NMR (500 MHz, CDCl3): δ 8.91 (s, 1H, H-4), 8.83 (s, 1H, H-3a), 7.73–7.63 (m, 2H, H-6,8), 7.39 (m, 2H, H-5,7), 3.67 (s, 3H, H-3g), 3.47 (dd, J = 13.1, 6.9 Hz, 2H, H-3b), 2.34 (t, J = 7.5 Hz, 2H, H-3f), 1.68 (tt, J = 14.8, 7.4 Hz, 4H, H-3c,3e), 1.49–1.36 (m, 2H, H-3d); 13C-NMR (125 MHz, CDCl3): δ 174.02, 161.47, 154.39, 148.26, 133.98, 129.79, 125.28, 118.59, 116.61, 51.51, 39.66, 33.91, 29.09, 26.52, 24.58; HRMS (ESI) m/z calcd. for C17H19NO5: [M + H]+ 318.1336, found 318.1345.
Ethyl 4-(6-methoxy-2-oxo-2H-chromene-3-carboxamido)butanoate (5d), yellowish solid (81% yield); mp = 131–132 °C; IR (KBr): νmax/cm−1 = 3325, 2981, 2948, 1734, 1698; UV-Vis (MeOH): λmax/nm= 297; 1H-NMR (500 MHz, CDCl3): δ 8.92 (s, 1H, H-3a), 8.86 (s, 1H, H-4), 7.34 (d, J = 9.1 Hz, 1H, H-8), 7.24 (dd, J = 9.1, 2.9 Hz, 1H, H-7), 7.08 (d, J = 2.9 Hz, 1H, H-5), 4.15 (q, J = 7.1 Hz, 2H, H-3e), 3.88 (s, 3H, H-6a), 3.51 (dd, J = 13.2, 6.8 Hz, 2H, H-3b), 2.41 (t, J = 7.4 Hz, 2H, H-3d), 1.98 (p, J = 7.2 Hz, 2H, H-3c), 1.26 (t, J = 7.1 Hz, 3H, H-3f); 13C-NMR (125 MHz, CDCl3): δ 172.95, 161.64, 156.61, 149.01, 148.14, 122.57, 118.98, 118.54, 117.73, 110.67, 60.48, 55.92, 39.12, 31.72, 24.79, 14.22; HRMS (ESI) m/z calcd. for C17H19NO6: [M + H]+ 334.1285, found 334.1304.
Methyl 6-(6-methoxy-2-oxo-2H-chromene-3-carboxamido)hexanoate (5e), pale yellowish solid (90% yield); mp = 101–102 °C; IR (KBr): νmax/cm−1 = 3310, 2939, 2859, 1745, 1717, 1697, 1658; UV-Vis (MeOH): λmax/nm = 297; 1H-NMR (500 MHz, CDCl3): δ 8.88 (s, 1H, H-3a), 8.86(s, 1H, H-4), 7.33 (d, J = 9.1 Hz, 1H, H-8), 7.24 (dd, J = 9.1, 2.9 Hz, 1H, H-7), 7.08 (d, J = 2.9 Hz, 1H, H-5), 3.88 (s, 3H, H-6a), 3.67 (s, 3H, H-3g), 3.46 (dd, J = 13.1, 6.9 Hz, 2H, H-3b), 2.34 (t, J = 7.5 Hz, 2H, H-3f), 1.67 (tt, J = 15.1, 7.5 Hz, 4H, H-3c,3e), 1.52–1.36 (m, 2H, H-3d); 13C-NMR (125 MHz, CDCl3): δ 173.99, 161.55, 156.59, 148.97, 148.02, 122.49, 119.00, 118.63, 117.69, 110.65, 55.91, 51.50, 39.64, 33.90, 29.09, 26.52, 24.58; HRMS (ESI) m/z calcd. for C18H21NO6: [M + H]+ 348.1442, found 348.1480.
Methyl 6-(8-ethoxy-2-oxo-2H-chromene-3-carboxamido)hexanoate (5f), white solid (68% yield); mp= 98–99 °C; IR (KBr): νmax/cm−1 = 3343, 1718, 1655, 1613; UV-Vis (MeOH): λmax/nm = 311; 1H-NMR (500 MHz, CDCl3): δ 8.88 (s, 1H, H-4), 8.86 (s, 1H, H-3a), 7.27 (d, J = 7.6 Hz, 1H, H-6), 7.25 (t, J = 3.9 Hz, 1H, H-7), 7.18 (dd, J = 7.7, 1.4 Hz, 1H, H-5), 4.22 (q, J = 7.0 Hz, 2H, H-8a), 3.67 (s, 3H, H-3g), 3.46 (dd, J = 13.2, 6.9 Hz, 2H, H-3b), 2.34 (t, J = 7.5 Hz, 2H, H-3f), 1.67 (tt, J = 14.7, 7.5 Hz, 4H, H-3c, 3e), 1.52 (t, J = 7.0 Hz, 3H, H-8b), 1.47–1.39 (m, 2H, H-3d); 13C-NMR (125 MHz, CDCl3): δ 173.96, 161.52, 161.17, 148.46, 146.41, 144.31, 125.07, 120.84, 119.44, 118.62, 116.73, 65.16, 51.45, 39.65, 33.91, 29.09, 26.52, 24.59, 14.68; HRMS (ESI) m/z calcd. for C19H23NO6: [M + H]+ 362.1598, found 362.1620.
Methyl (6-bromo-2-oxo-2H-chromene-3-carbonyl)glycinate (5g), white solid (46% yield); mp = 160–162 °C; IR (KBr): νmax/cm−1 = 3337, 3048, 1740, 1724, 1655; UV-Vis (MeOH): λmax/nm= 290; 1H-NMR (500 MHz, CDCl3): δ 9.18 (s, 1H, H-3a), 8.82 (s, 1H, H-4), 7.83 (d, J = 2.3 Hz, 1H, H-5), 7.76 (dd, J = 8.8, 2.3 Hz, 1H, H-7), 7.31 (d, J = 8.8 Hz, 1H, H-8), 4.25 (d, J = 5.6 Hz, 2H, H-3b), 3.80 (s, 3H, H-3c); 13C-NMR (125 MHz, CDCl3): δ 169.63, 161.39, 160.59, 153.32, 147.40, 137.00, 131.92, 119.96, 119.00, 118.45, 118.01, 52.48, 41.73; HRMS (ESI) m/z calcd. for C13H10NO5: [M + H]+ 339.9742, found 339.9802.
Ethyl 3-(6-bromo-2-oxo-2H-chromene-3-carboxamido)propanoate (5h), white solid (58% yield); mp = 148–149 °C; IR (KBr): νmax/cm−1 = 3383, 3053, 1734, 1717, 1657; UV-Vis (MeOH): λmax/nm = 292; 1H-NMR (500 MHz, CDCl3): δ 9.09 (s, 1H, H-3a), 8.82 (s, 1H, H-4), 7.83 (d, J = 2.0 Hz, 1H, H-5), 7.74 (dd, J = 8.8, 2.1 Hz, 1H, H-7), 7.29 (d, J = 8.8 Hz, 1H, H-8), 4.20 (q, J = 7.1 Hz, 2H, H-3d), 3.75 (q, J = 6.2 Hz, 2H, H-3b), 2.66 (t, J = 6.3 Hz, 2H, H-3c), 1.29 (t, J = 7.1 Hz, 3H, H-3e); 13C-NMR (125 MHz, CDCl3): δ 171.76, 161.06, 160.58, 153.22, 147.00, 136.74, 131.84, 120.07, 119.43, 118.36, 117.90, 60.87, 35.49, 34.11, 14.19; HRMS (ESI) m/z calcd. for C15H14BrNO5: [M + H]+ 368.0128, found 368.0132.
Methyl 6-(6-bromo-2-oxo-2H-chromene-3-carboxamido)hexanoate (5i), white solid (77% yield); mp = 147–149 °C; IR(KBr): νmax/cm−1 = 3336, 3051, 1734, 1715, 1658; UV-Vis (MeOH) λmax/nm = 292; 1H-NMR (500 MHz, CDCl3): δ 8.84 (s, 1H, H-4), 8.75 (s, 1H, H-3a), 7.84 (d, J = 2.2 Hz, 1H, H-5), 7.74 (dd, J = 8.8, 2.3 Hz, 1H, H-7), 7.30 (d, J = 8.8 Hz, 1H, H-8), 3.67 (s, 3H, H-3g), 3.46 (dd, J = 13.1, 7.0 Hz, 2H, H-3b), 2.34 (t, J = 7.5 Hz, 2H, H-3f), 1.67 (tt, J = 15.0, 7.5 Hz, 4H, H-3c,3e), 1.50–1.37 (m, 2H, H-3d); 13C-NMR (125 MHz, CDCl3): δ 173.98, 160.91, 160.84, 153.16, 146.89, 136.67, 131.83, 120.14, 119.56, 118.33, 117.92, 51.52, 39.75, 33.89, 29.05, 26.50, 24.57; HRMS (ESI) m/z calcd. for C17H18BrNO5: [M + H]+ 396.0441, found 396.0448.
Ethyl 3-(7-(diethylamino)-2-oxo-2H-chromene-3-carboxamido)propanoate (5j), yellow solid (40% yield); mp = 134–136 °C; IR (KBr): νmax/cm−1 = 3444, 3320, 2940, 1739, 1702; UV-Vis (MeOH): λmax/nm = 419; 1H-NMR (500 MHz, CDCl3): δ 9.08 (s, 1H, H-3a), 8.69 (s, 1H, H-4), 7.42 (d, J = 9.0 Hz, 1H, H-5), 6.64 (dd, J = 9.0, 2.1 Hz, 1H, H-6), 6.49 (d, J = 1.8 Hz, 1H, H-8), 4.19 (q, J = 7.1 Hz, 4H, H-7a), 3.73 (q, J = 6.3 Hz, 2H, H-3d), 3.45 (q, 2H, H-3b), 2.64 (t, J = 6.5 Hz, 2H, H-3c), 1.29 (t, J = 7.2 Hz, 3H, H-3e), 1.24 (t, J = 7.2 Hz, 6H, H-7b); 13C-NMR (125 MHz, CDCl3): δ 171.94, 163.25, 162.61, 157.67, 152.55, 148.09, 131.13, 110.21, 109.91, 108.35, 96.59, 60.72, 45.07, 35.25, 34.49, 14.20, 12.42. HRMS (ESI) m/z calcd. for C19H24N2O5: [M + H]+ 361.1758, found 361.1780.
Methyl 6-(7-(diethylamino)-2-oxo-2H-chromene-3-carboxamido)hexanoate (5k), yellow solid (61% yield); mp = 90–91 °C; IR (KBr): νmax/cm−1 = 3367, 2978, 1735, 1688, 1650; UV-Vis (MeOH): λmax/nm = 418; 1H-NMR (500 MHz, CDCl3): δ 8.80 (s, 1H, H-3a), 8.70 (s, 1H, H-4), 7.43 (d, J = 8.9 Hz, 1H, H-5), 6.64 (dd, J = 8.9, 2.3 Hz, 1H, H-6), 6.50 (d, J = 2.0 Hz, 1H, H-8), 3.67 (s, 3H, H-3g), 3.49–3.38 (m, 6H, H-7a,3b), 2.33 (t, J = 7.5 Hz, 2H, H-3f), 1.66 (qd, J = 15.0, 7.4 Hz, 4H, H-3c,3e), 1.51–1.36 (m, 2H, H-3d), 1.24 (t, J = 7.1 Hz, 6H, H-7b); 13C-NMR (125 MHz, CDCl3): δ 174.08, 163.08, 162.80, 157.60, 152.47, 148.01, 131.08, 110.43, 109.91, 108.40, 96.56, 51.49, 45.06, 39.42, 33.97, 29.28, 26.58, 24.66. HRMS (ESI) m/z calcd. for C21H28N2O5: [M + H]+ 389.2071, found 389.2102.
3.1.3. General Procedure for the Preparation of N-(2-Oxo-2H-Chromene-3-Carboxamide) Acids (6a–k)
In a round-bottom flask, the corresponding ester (1 mmol) was added and dissolved in 10 mL of THF:H2O (1:2). The mixture was stirred at room temperature before adding LiOH (7 mmol) and stirring was continued overnight. Upon completion of the reaction, HCl was slowly added until a precipitate appeared, which was filtered and purified by recrystallization with MeOH and cold water to afford the desired product. Diethylamine derivatives were synthesized by using the Mravljak, J. et al. methodology [59].
(2-oxo-2H-chromene-3-carbonyl)glycine (6a), white solid (93% yield); mp = 246–247 °C; IR (KBr): νmax/cm−1 = 3315, 2967, 2686, 1759, 1712, 1638; UV-Vis (MeOH): λmax/nm = 297; 1H-NMR (400 MHz, DMSO-d6): δ 12.78 (s, 1H, H-3c), 8.99 (s, 1H, H-3a), 8.86 (s, 1H, H-4), 7.95 (d, J = 7.4 Hz, 1H, H-8), 7.71 (t, J = 7.5 Hz, 1H, H-7), 7.46 (d, J = 8.1 Hz, 1H, H-5), 7.39 (t, J = 7.0 Hz, 1H, H-6), 4.00 (s, 2H, H-3b); 13C-NMR (100 MHz, DMSO-d6): δ 171.27, 161.63, 160.84, 154.46, 148.58, 134.77, 130.89, 125.64, 118.89, 118.66, 116.64, 41.97; HRMS (ESI) m/z calcd. for C12H9NO5: [M + H]+ 248.0553, found 248.0561.
3-(2-oxo-2H-chromene-3-carboxamido)propanoic acid (6b), white solid (70% yield); mp = 199–200 °C; IR (KBr): νmax/cm−1 = 3323, 3068, 1725, 1706, 1635; UV-Vis (MeOH): λmax/nm = 297; 1H-NMR (400 MHz, DMSO-d6): δ 12.34 (s, 1H, H-3d), 8.85 (s, 1H, H-3a), 8.82 (s, 1H, H-4), 7.93 (d, J = 7.4 Hz, 1H, H-8), 7.69 (t, J = 7.4 Hz, 1H, H-7), 7.44 (d, J = 8.2 Hz, 1H, H-5), 7.38 (t, J = 7.1 Hz, 1H, H-4), 3.47 (d, J = 5.0 Hz, 2H, H-3b), 2.46 (d, J = 7.7 Hz, 2H, H-3c); 13C-NMR (100 MHz, DMSO-d6): δ 173.58, 161.46, 160.86, 154.35, 148.14, 134.58, 130.76, 125.59, 119.08, 118.92, 116.59, 35.55, 34.13; HRMS (ESI) m/z calcd. for C13H11NO5: [M + H]+ 262.0710, found 262.0719.
6-(2-oxo-2H-chromene-3-carboxamido)hexanoic acid (6c), white solid (75% yield); mp = 145–146 °C; IR (KBr): νmax/cm−1 = 3357, 1710, 1694, 1527; UV-Vis (MeOH): λmax/nm = 299; 1H-NMR (500 MHz, DMSO-d6): δ 12.01 (s, 1H, H-3g), 8.85 (s, 1H, H-4), 8.69 (t, J = 5.6 Hz, 1H, H-3a), 7.99 (d, J = 7.4 Hz, 1H, H-8), 7.88–7.65 (m, 1H, H-7), 7.51 (d, J = 8.3 Hz, 1H, H-5), 7.44 (t, J = 7.4 Hz, 1H, H-6), 3.39–3.29 (m, 2H, H-3b), 2.22 (t, J = 7.3 Hz, 2H, H-3f), 1.62–1.47 (m, 4H, H-3c,3e), 1.36–1.27 (m, 2H, H-3d); 13C-NMR (125 MHz, DMSO-d6): δ 174.86, 161.48, 160.85, 154.31, 147.72, 134.45, 130.67, 125.57, 119.62, 118.96, 116.58, 39.42, 34.04, 29.15, 26.43, 24.64; HRMS (ESI) m/z calcd. for C16H17NO5: [M + H]+ 304.1179, found 304.1184.
4-(6-methoxy-2-oxo-2H-chromene-3-carboxamido)butanoic acid (6d), light yellow solid (81% yield); mp = 200–203 °C; IR (KBr): νmax/cm−1 = 3311, 1700, 1531; UV-Vis (MeOH) λmax/nm = 298; 1H-NMR (500 MHz, DMSO-d6): δ 12.12 (s, 1H, H-3e), 8.78 (s, 1H, H-4), 8.74 (t, J = 5.8 Hz, 1H, H-3a), 7.52 (d, J = 2.9 Hz, 1H, H-5), 7.43 (d, J = 9.1 Hz, 1H, H-8), 7.32 (dd, J = 9.1, 3.0 Hz, 1H, H-7), 3.82 (s, 3H, H-6a), 3.36 (dd, J = 13.1, 6.7 Hz, 2H, H-3b), 2.30 (t, J = 7.4 Hz, 2H, H-3d), 1.78 (p, J = 7.2 Hz, 2H, H-3c); 13C-NMR (125 MHz, DMSO-d6): δ 174.54, 161.66, 160.88, 156.40, 148.78, 147.54, 122.29, 119.64, 119.35, 117.65, 112.24, 56.27, 39.00, 31.59, 24.99; HRMS (ESI) m/z calcd. for C15H15NO6: [M + H]+ 306.0972, found 306.0974.
6-(6-methoxy-2-oxo-2H-chromene-3-carboxamido)hexanoic acid (6e), light yellow solid (81% yield); mp = 142–144 °C; IR (KBr): νmax/cm−1 = 3322, 1721, 1702, 1575; UV-Vis (MeOH): λmax/nm = 298; 1H-NMR (500 MHz, DMSO-d6): δ 8.79 (s, 1H, H-4), 8.72 (s, 1H, H-3a), 7.52 (s, 1H, H-5), 7.42 (d, J = 9.2 Hz, 1H, H-8), 7.38-7.27 (m, 1H, H-7), 3.82 (s, 3H, H-6a), 3.32 (dd, J = 13.0, 6.7 Hz, 2H, H-3b), 2.23 (t, J = 7.3 Hz, 2H, H-3f), 1.61–1.49 (m, 4H, H-3c,3e), 1.40–1.30 (m, 2H, H-3d); 13C-NMR (125 MHz, DMSO-d6): δ 174.85, 161.45, 160.98, 156.41, 148.77, 147.61, 122.32, 119.52, 119.36, 117.66, 112.23, 56.26, 39.42, 34.04, 29.14, 26.44, 24.64; HRMS (ESI) m/z calcd. for C17H19NO6: [M + H]+ 334.1285, found 334.1288.
6-(8-ethoxy-2-oxo-2H-chromene-3-carboxamido)hexanoic acid (6f), white solid (83% yield); mp = 169–170 °C; IR (KBr): νmax/cm−1 = 3337, 1716, 1605, 1542; UV-Vis (MeOH): λmax/nm = 312; 1H-NMR (500 MHz, DMSO-d6): δ 12.02 (s, 1H, H-3g), 8.80 (s, 1H, H-4), 8.69 (t, J = 5.7 Hz, 1H, H-3a), 7.49 (d, J = 7.1 Hz, 1H, H-7), 7.40 (d, J = 7.6 Hz, 1H, H-5), 7.34 (t, J = 7.9 Hz, 1H, H-6), 4.20 (q, J = 6.9 Hz, 2H, H-8a), 3.32 (dd, J = 13.2, 6.7 Hz, 2H, H-3b), 2.22 (t, J = 7.3 Hz, 2H, H-3f), 1.59–1.49 (m, 4H, H-3c,3e), 1.42 (t, J = 7.0 Hz, 3H, H-8b), 1.38–1.29 (m, 2H, H-3d); 13C-NMR (125 MHz, DMSO-d6): δ 174.84, 161.45, 160.63, 147.96, 145.97, 143.74, 125.49, 121.55, 119.60, 117.31, 65.03, 39.43, 34.05, 29.15, 26.44, 24.64, 15.01; HRMS (ESI) m/z calcd. for C18H21NO6: [M + H]+ 348.1442, found 348.1452.
(6-bromo-2-oxo-2H-chromene-3-carbonyl)glycine (6g), white solid (83% yield); mp = 269–270 °C; IR (KBr): νmax/cm−1 = 3341, 1726, 1648, 1560; UV-Vis (MeOH) λmax/nm = 291; 1H-NMR (500 MHz, DMSO-d6): δ 12.84 (s, 1H, H-3c), 9.02 (t, J = 5.5 Hz, 1H, H-3a), 8.87 (s, 1H, H-4), 8.27 (d, J = 2.3 Hz, 1H, H-5), 7.90 (dd, J = 8.8, 2.3 Hz, 1H, H-7), 7.49 (d, J = 8.9 Hz, 1H, H-8), 4.07 (d, J = 5.5 Hz, 2H, H-3b). 13C-NMR (125 MHz, DMSO-d6): δ 171.16, 161.32, 160.33, 153.48, 147.25, 136.87, 132.70, 120.75, 119.79, 118.89, 117.14, 42.00. HRMS (ESI) m/z calcd. for C12H8NO5: [M + H]+ 325.9659, found 325.9668.
3-(6-bromo-2-oxo-2H-chromene-3-carboxamido)propanoic acid (6h), white solid (50% yield); mp = 251–252 °C; IR (KBr): νmax/cm−1 = 3380, 1705, 1657, 1533; UV-Vis (MeOH): λmax/nm = 292; 1H-NMR (500 MHz, DMSO-d6): δ 12.39 (s, 1H, H-3d), 8.88 (t, J = 5.8 Hz, 1H, H-3a), 8.84 (s, 1H, H-4), 8.25 (d, J = 2.2 Hz, 1H, H-5), 7.88 (dd, J = 8.8, 2.3 Hz, 1H, H-7), 7.47 (d, J = 8.9 Hz, 1H, H-8), 3.54 (q, J = 6.4 Hz, 2H, H-3b), 2.54 (t, J = 6.7 Hz, 2H, H-3c); 13C-NMR (125 MHz, DMSO-d6): δ 173.50, 161.16, 160.35, 153.38, 146.79, 136.70, 132.59, 120.80, 120.25, 118.85, 117.10, 35.62, 34.11; HRMS (ESI) m/z calcd. for C13H10NO5: [M + H]+ 339.9815, found 339.9817.
6-(6-bromo-2-oxo-2H-chromene-3-carboxamido)hexanoic acid (6i), white solid (93% yield); mp = 182–183 °C; IR (KBr): νmax/cm−1 = 3347, 1715, 1657, 1562; UV-Vis (MeOH): λmax/nm= 293; 1H-NMR (500 MHz, DMSO-d6): δ 12.01 (s, 1H, H-3g), 8.80 (s, 1H, H-4), 8.66 (t, J = 5.7 Hz, 1H, H-3a), 8.25 (d, J = 2.2 Hz, 1H, H-5), 7.88 (dd, J = 8.8, 2.3 Hz, 1H, H-7), 7.47 (d, J = 8.8 Hz, 1H, H-8), 3.31 (dd, J = 13.1, 6.7 Hz, 2H, H-3b), 2.22 (t, J = 7.4 Hz, 2H, H-3f), 1.59–1.47 (m, 4H, H-3c,3e), 1.37–1.28 (m, 2H, H-3d); 13C-NMR (125 MHz, DMSO-d6): δ 174.84, 161.18, 160.32, 153.33, 146.37, 136.59, 132.51, 120.83, 120.78, 118.85, 117.08, 39.47, 34.05, 29.11, 26.42, 24.64; HRMS (ESI) m/z calcd. for C16H16NO5: [M + H]+ 382.0212, found 382.0289.
3-(7-(diethylamino)-2-oxo-2H-chromene-3-carboxamido)propanoic acid (6j), yellow crystals (75% yield); mp = 202–205 °C; 1H-NMR (DMSO-d6, 500 MHz): δ 8.84 (t, J = 5.1 Hz, 1H), 8.65 (s, 1H), 7.66 (d, J = 8.9 Hz, 1H), 6.78 (d, J = 8.6 Hz, 1H), 6.59 (s, 1H), 3.54–3.50 (m, 2H), 3.47 (dd, J = 13.7, 6.8 Hz, 4H), 2.49 (t, J = 6.2 Hz, 2H), 1.14 (t, J = 6.9 Hz, 6H).
6-(7-(diethylamino)-2-oxo-2H-chromene-3-carboxamido)hexanoic acid (6k), yellow crystals (73% yield); mp = 150–152 °C; 1H-NMR (DMSO-d6, 500 MHz): δ 8.67–8.62 (m, 2H), 7.68 (d, J = 9.0 Hz, 1H), 6.80 (dd, J = 9.0, 2.2 Hz, 1H), 6.62 (d, J = 2.0 Hz, 1H), 3.48 (dd, J = 14.0, 7.0 Hz, 4H), 3.31–3.27 (m, 2H), 2.21 (t, J = 7.3 Hz, 2H), 1.51 (dq, J = 14.8, 7.4 Hz, 4H), 1.31 (dd, J = 15.2, 8.1 Hz, 2H), 1.14 (t, J = 7.0 Hz, 6H).
3.1.4. General Procedure for the Preparation of the Title Compounds (7a–k)
In a dry round-bottom flask purged with N2, the corresponding carboxylic acid (1 mmol), DMAP (5% mol) and CDI (1.1 mmol) were dissolved in DMF. After 30 min of stirring at room temperature, hydroxylamine hydrochloride was added and stirring continued overnight. Upon completion of the reaction, sodium bicarbonate was added. The resulting precipitate was obtained by filtration and purified by recrystallization with MeOH and cold water.
N-(2-(hydroxyamino)-2-oxoethyl)-2-oxo-2H-chromene-3-carboxamide (7a), white solid (50% yield); mp = 226–227 °C; IR (KBr): νmax/cm−1 = 3304, 3240, 3043, 1709, 1668; UV-Vis (MeOH): λmax/nm = 297; Em (MeOH): λmax/nm = 406; 1H-NMR (400 MHz, DMSO-d6): δ 10.68 (s, 1H), 9.04 (s, 1H), 8.90 (s, 1H), 8.00 (s, 1H), 7.76 (s, 1H), 7.61–7.35 (m, 2H), 3.92 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 165.66, 161.61, 160.84, 154.38, 148.16, 134.71, 130.84, 125.64, 118.92, 118.88, 116.62, 41.09; HRMS (ESI) m/z calcd. for C12H10N2O5: [M + Na]+ 285.0488, found 285.0505.
N-(3-(hydroxyamino)-3-oxopropyl)-2-oxo-2H-chromene-3-carboxamide (7b), white solid (45% yield); mp = 170–173 °C; IR (KBr): νmax/cm−1 = 3469, 3302, 3194, 1708, 1648; UV-Vis (MeOH) λmax/nm = 303.12; Em (MeOH): λmax/nm = 405; 1H-NMR (500 MHz, DMSO-d6): δ 8.88 (s, 1H), 8.87 (s,1H), 7.99 (d, J = 6.6 Hz, 1H), 7.76 (dd, J = 11.5, 4.2 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.45 (t, J = 7.2 Hz, 1H), 3.54 (dd, J = 12.7, 6.5 Hz, 2H), 2.29 (t, J = 6.7 Hz, 2H); 13C-NMR (125 MHz, DMSO-d6): δ 167.78, 161.45, 160.78, 154.36, 148.09, 134.59, 130.76, 125.60, 119.11, 118.91, 116.60, 116.56, 36.12, 32.39. HRMS (ESI) m/z calcd. for C13H12N2O5: [M + H]+ 277.0819, found 277.0860.
N-(6-(hydroxyamino)-6-oxohexyl)-2-oxo-2H-chromene-3-carboxamide (7c), white solid (50% yield); mp = 110–112 °C; IR (KBr): νmax/cm−1 = 3336, 3207, 1706, 1537; UV-Vis (MeOH): λmax/nm = 329.96; Em (MeOH): λmax/nm = 405; 1H-NMR (500 MHz, DMSO-d6): δ 8.84 (s, 1H), 8.68 (s, 1H), 7.97 (d, J = 6.3 Hz, 1H), 7.74 (s, 1H), 7.50 (d, J = 7.4 Hz, 1H), 7.44 (s, 2H), 3.31 (d, J = 3.4 Hz, 3H), 1.96 (s, 2H), 1.53 (s, 5H), 1.29 (s, 2H); 13C-NMR (125 MHz, DMSO-d6): δ 169.47, 161.48, 160.85, 154.30, 147.71, 134.46, 130.66, 125.58, 119.59, 118.94, 116.57, 39.49, 32.66, 29.17, 26.50, 25.31; HRMS (ESI) m/z calcd. for C16H18N2O5: [M + Na]+ 341.1114, found 341.1019.
N-(4-(hydroxyamino)-4-oxobutyl)-6-methoxy-2-oxo-2H-chromene-3-carboxamide (7d), light green solid (30% yield); mp = 154–156 °C; IR (KBr): νmax/cm−1 = 3531, 3241, 1706, 1554; UV-Vis (MeOH): λmax/nm = 363; Em (MeOH): λmax/nm = 458; 1H-NMR (500 MHz, DMSO-d6): δ 8.80 (s, 1H), 8.75 (s, 1H), 7.54 (d, J = 2.2 Hz, 1H), 7.44 (d, J = 9.1 Hz, 1H), 7.32 (dd, J = 9.1, 2.5 Hz, 1H), 3.82 (s, 3H), 3.32 (dd, J = 13.1, 6.6 Hz, 2H), 2.03 (t, J = 7.5 Hz, 2H), 1.82–1.72 (m, 2H); 13C-NMR (125 MHz, DMSO-d6): δ 169.09, 161.64, 160.89, 156.40, 148.79, 147.62, 122.35, 119.63, 119.37, 117.70, 112.25, 56.28, 39.22, 30.32, 25.72; HRMS (ESI) m/z calcd. for C15H16N2O6: [M + H]+ 321.1081, found 321.1132.
N-(6-(hydroxyamino)-6-oxohexyl)-6-methoxy-2-oxo-2H-chromene-3-carboxamide (7e), light green solid (87% yield); mp = 75–77 °C; IR (KBr): νmax/cm−1 = 3318, 2935, 1702, 1575; UV-Vis (MeOH): λmax/nm = 299; Em (MeOH): λmax/nm = 458; 1H-NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H), 8.81 (s, 1H), 8.72 (s, 1H), 7.54 (s, 1H), 7.44 (d, J = 9.0 Hz, 1H), 7.33 (d, J = 8.9 Hz, 1H), 3.82 (s, 3H), 3.31 (s, 2H), 2.21 (t, J = 7.2 Hz, 1H), 1.97 (d, J = 7.2 Hz, 1H), 1.53 (d, J = 6.3 Hz, 4H), 1.37–1.27 (m, 2H); 13C-NMR (125 MHz, DMSO-d6): δ 169.52, 161.48, 161.01, 156.43, 148.80, 147.65, 122.36, 119.60, 119.40, 117.70, 112.28, 56.30, 39.48, 32.65, 29.18, 26.50, 25.30; HRMS (ESI) m/z calcd. for C18H20N2O5: [M + Na]+ 371.1219 found 371.1194.
8-ethoxy-N-(6-(hydroxyamino)-6-oxohexyl)-2-oxo-2H-chromene-3-carboxamide (7f), light green solid (48% yield); mp = 167–169 °C; IR (KBr): νmax/cm−1 = 3335, 1933, 1716, 1539; UV-Vis (MeOH): λmax/nm= 311; Em (MeOH) λmax/nm= 486; 1H-NMR (500 MHz, DMSO-d6): δ 8.81 (s, 3H), 8.69 (s, 3H), 7.50 (d, J = 7.6 Hz, 3H), 7.41 (d, J = 8.0 Hz, 3H), 7.34 (t, J = 7.9 Hz, 3H), 4.20 (q, J = 6.8 Hz, 6H), 1.53 (s, 12H), 1.42 (t, J = 6.9 Hz, 11H), 1.32 (dd, J = 17.8, 7.6 Hz, 7H); 13C-NMR (125 MHz, DMSO-d6): δ 169.51, 161.47, 160.65, 147.99, 145.97, 143.72, 125.53, 121.55, 119.61, 117.32, 65.02, 34.06, 32.64, 29.19, 26.50, 25.30, 15.02; HRMS (ESI) m/z calcd. for C18H22N2O6: [M + H]+ 362.3820, found 362.3843.
6-bromo-N-(2-(hydroxyamino)-2-oxoethyl)-2-oxo-2H-chromene-3-carboxamide (7g), white solid (46% yield); mp = 202–205 °C; IR (KBr) νmax/cm−1 = 3417, 3272, 1712, 1537; UV-Vis (MeOH): λmax/nm = 294; Em (MeOH): λmax/nm = 424; 1H-NMR (500 MHz, DMSO-d6): δ 9.01 (d, J = 4.6 Hz, 1H), 8.86 (s, 1H), 8.28 (s, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 8.9 Hz, 1H), 3.92 (d, J = 5.2 Hz, 2H); 13C-NMR (125 MHz, DMSO-d6): δ 165.55, 161.34, 160.34, 153.43, 146.83, 136.83, 132.67, 120.78, 120.13, 118.91, 117.14, 41.13; HRMS (ESI) m/z calcd. for C12H9BrN2O5: [M + H]+ 339.9695, found 339.9711.
6-bromo-N-(3-(hydroxyamino)-3-oxopropyl)-2-oxo-2H-chromene-3-carboxamide (7h), white solid (45% yield); mp = 185–187 °C; IR (KBr): νmax/cm−1 = 3354, 3040, 2854, 1708, 1538; UV-Vis (MeOH): λmax/nm = 295; Em (MeOH): λmax/nm = 415; 1H-NMR (500 MHz, DMSO-d6): δ 8.86 (d, J = 5.4 Hz, 1H), 8.83 (s, 1H), 8.26 (s, 2H), 7.89 (dd, J = 8.8, 1.8 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 3.53 (dd, J = 12.6, 6.4 Hz, 3H), 2.28 (t, J = 6.7 Hz, 2H; 13C-NMR (125 MHz, DMSO-d6): δ 161.16, 160.30, 153.40, 146.78, 146.58, 136.72, 136.54, 132.57, 120.82, 118.86, 117.06, 36.19, 32.31; MS (ESI) m/z calcd. for C13H11BrN2O6: [M + H]+ 356.1440, found 356.1252.
6-bromo-N-(6-(hydroxyamino)-6-oxohexyl)-2-oxo-2H-chromene-3-carboxamide (7i), white solid (65% yield); mp = 202–205 °C; IR (KBr): νmax/cm−1 = 3344, 2934, 1724, 1559; UV-Vis (MeOH): λmax/nm = 283; Em (MeOH): λmax/nm = 424; 1H-NMR (500 MHz, DMSO-d6): δ 8.80 (d, J = 4.2 Hz, 2H), 8.67 (t, J = 5.7 Hz, 2H), 8.25 (dd, J = 6.4, 2.2 Hz, 2H), 7.88 (dd, J = 8.8, 2.3 Hz, 2H), 7.47 (dd, J = 8.6, 5.5 Hz, 2H), 3.30 (dd, J = 12.6, 6.4 Hz, 5H), 1.96 (t, J = 7.3 Hz, 4H), 1.52 (dt, J = 14.9, 7.4 Hz, 8H), 1.29 (dd, J = 14.7, 7.7 Hz, 4H); 13C-NMR (125 MHz, DMSO-d6): δ 169.41, 162.78, 161.19, 160.36, 153.32, 146.40, 136.61, 132.51, 120.79, 118.87, 117.09, 39.52, 36.25, 29.16, 26.48, 25.32; HRMS (ESI) m/z calcd. for C16H17BrN2O5: [M + H]+ 397.0321, found 397.1750.
7-(diethylamino)-N-(3-(hydroxyamino)-3-oxopropyl)-2-oxo-2H-chromene-3-carboxamide (7j), yellow solid (82% yield); mp = 126–128 °C; IR (KBr): νmax/cm−1 = 3231, 1699, 1616; UV-Vis (MeOH): λmax/nm = 419; Em (MeOH): λmax/nm = 468; 1H-NMR (500 MHz, DMSO-d6): δ 8.78 (t, J = 5.7 Hz, 1H), 8.65 (s, 2H), 7.67 (d, J = 9.0 Hz, 1H), 6.78 (d, J = 9.0 Hz, 1H), 6.60 (d, J = 1.3 Hz, 1H), 3.47 (q, J = 6.8 Hz, 4H), 2.25 (t, J = 6.6 Hz, 2H), 2.01 (t, J = 7.5 Hz, 2H), 1.13 (t, J = 6.9 Hz, 6H); 13C-NMR (125 MHz, DMSO-d6): δ 169.13, 167.85, 162.60, 162.05, 157.70, 152.87, 148.16, 132.02, 110.57, 108.12, 108.10, 96.34, 44.79, 12.77; HRMS (ESI) m/z calcd. for C17H21N3O5: [M + H]+ 348.1554, found 348.1570.
7-(diethylamino)-N-(6-(hydroxyamino)-6-oxohexyl)-2-oxo-2H-chromene-3-carboxamide (7k), yellow solid (82% yield); mp = 173–174 °C; IR (KBr): νmax/cm−1 = 3122, 1700, 1618; UV-Vis (MeOH): λmax/nm = 418; Em (MeOH): λmax/nm = 470; 1H-NMR (500 MHz, DMSO-d6): δ 8.78 (t, J = 5.7 Hz, 1H), 8.65 (s, 2H), 7.67 (d, J = 9.0 Hz, 1H), 6.78 (d, J = 9.0 Hz, 1H), 6.60 (d, J = 1.3 Hz, 1H), 3.47 (q, J = 6.8 Hz, 4H), 2.25 (t, J = 6.6 Hz, 2H), 2.01 (t, J = 7.5 Hz, 2H), 1.13 (t, J = 6.9 Hz, 6H); 13C-NMR (125 MHz, DMSO-d6): δ 169.13, 167.85, 162.60, 162.05, 157.70, 152.87, 148.16, 132.02, 110.57, 108.12, 108.10, 96.34, 44.79, 35.87, 32.68, 12.77. HRMS (ESI) m/z calcd. for C20H27N3O5: [M + H]+ 389.1951, found 389.1969.
3.2. Computational Methodology: Molecular Docking
Compounds 7a–k were docked on the X-ray structure of the HDAC homologue (downloaded with the PDB code 1T69), which was found co-crystalized with a SAHA molecule. During the molecular docking procedure, the types of protein-ligand interactions were carefully analyzed. This procedure was carried out by using the Molegro Virtual Docker V5 software suite, the search algorithm MolDock Simplex Evolution (with a mesh size of 0.3 Å), and the evaluation function MolDock Score. All bound water molecules and ligands were eliminated, and the polar hydrogen was added. The compounds were carefully monitored to analyze the types of ligand-protein interactions involved. All ligands were previously built on Spartan 08 [60,61] and optimized with the semi empirical AM1 method [62]. The output files were exported as pdb files to be used with the docking software. All flexible bonds were set free to proceed with the flexible computational experiments.
The crystal structures of human HDAC1, 6 and 8 were obtained from the protein data bank (PDB codes 5ICN, 5EDU and 1T69, respectively) [63,64,65]. The former was acquired in complex with the H4K16Hx peptide, with 14 structural errors that were detected and repaired. HDAC6 was obtained in complex with TSA, with 22 structural errors that were detected and repaired. HDAC8 was received in complex with SAHA, with 16 structural errors that were detected and repaired. Previous to docking, all water molecules were removed, and the polar hydrogen was added. After the calibration process, a root-mean-square deviation (RMSD) value of 1.89 Å was found for the H4K16Hx peptide on HDAC1, a value of 0.27 Å for TSA on HDAC6, and a value of 0.51 Å for SAHA on HDAC8. Molecular docking was carried out on the Molegro Virtual Docker V5 software suite with the search algorithm MolDock Simplex Evolution (with a grid of 0.2 Å) and the evaluation function MolDock Score [66], using a docking spherical zone defined by a 15 Å radius centered on the active site of each HDAC isoform.
3.3. Biological Assay
3.3.1. Cell Culture
Breast (BT-474, MDA-MB-231) and prostate (PC3) cancer cell lines (ATCC, Manassas, VA, USA) were cultured and maintained according to the indications of the supplier. The cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C.
3.3.2. Sulforhodamine B (SRB) Assay
The cells were seeded in 96-well culture plates at a density of 3000 cells/well and treated in the presence (at various concentrations) and absence of compounds 7a–k or SAHA. All biological assays were performed in triplicate. The cells were incubated for 72–144 h (depending on the cell line) at 37 °C in a humid environment with 95% air and 5% CO2. Cell proliferation was determined by using the SRB assay [67]. Briefly, the cells were fixed with 10% trichloroacetic acid at 4 °C for 1 h. The plates were then washed with tap water and air-dried. The cells were stained for 1 h with 0.4% SRB dissolved in 1% acetic acid. To remove unbound stain, plates were washed four times with 1% acetic acid and air-dried. The bound protein stain was solubilized with 10 mM of unbuffered Tris base [tris(hydroxymethyl) aminomethane]. Absorbance was measured at 492 nm in a microplate reader (BioTek, Winooski, VT, USA).
3.3.3. Fluorescence Microscopy and Spectrofluorometric Quantification
For relative fluorescence quantification, the cells were grown in suitable 96-well plates (Corning Costar Model 3904 assay plate; Corning, NY, USA) and treated as aforementioned. Subsequently, they were washed with phosphate-buffered saline and incubated in serum-free DME medium without phenol red. Fluorescence was read on a Biotek Synergy HT plate reader (420 nm excitation, 485 nm emission).
For confocal microscopy, the cells were grown in 2-well chamber slides (Lab-Tek II) at a density of 1 × 105 cells/well. Then the cells were exposed to 10 μM of 7j, 7k or SAHA for 48 h. After two washings with Hanks balanced salt solution (HBSS), the cells were fixed with methanol/water (80/20 v/v) at 4 °C for 30 min, followed by two washings with HBSS. To stain the nuclei, the cells were incubated for 1 h at room temperature in a solution of propidium iodide (PI) (3 μg/mL) and rinsed with HBSS. The chambers were removed, and cover slips were mounted onto slides with VECTASHIELD mounting medium (Vector Laboratories). Images were taken on an Olympus FV1000 confocal upright microscope with a 40X objective lens at excitation and emission wavelengths, respectively, of 536 and 617 nm for red fluorescence (PI) and 420 and 485 nm for fluorescence analogues.
3.3.4. Real Time RT-PCR
For gene expression analysis, the cells were incubated in the presence of SAHA, one of the compounds 7a–k (10 μM) or the vehicle only (DMSO) for 24 h. RNA was extracted with the Trizol reagent and subjected to reverse transcription with the RT transcriptor system. RT-PCR was carried out on a LightCycler 2.0 instrument from Roche (Roche Diagnostics, Mannheim, Germany) according to the following protocol: activation of Taq DNA polymerase and DNA denaturing at 95 °C for 10 min, then 45 amplification cycles consisting of 10 s at 95 °C, 30 s at 60 °C, and 1 s at 72 °C. The oligonucleotides employed were cyclin D1 (CCND1)-F, GAAGATCGTCGCCACCTG; CCND1-R, GACCTCCTCCTCGCACTTCT; p21-F, TCACTGTCTTGTACCCTTGTGC; p21-R, GGCGTTTGGAGTGGTAGAAA; p53-F, GTCCCAAGCAATGGATGATT; and p53-R, TCTGGACCTGGGTCTTCAGT. The gene expression of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), was used as an internal control. It was AGCCACATCGCTGAGACAC for GAPDH-F and GCCCAATACGACCAAATCC for GAPDH-R.
3.3.5. Statistical Analyses
Data are expressed as the mean ± standard deviation (SD). Statistical differences were determined by one-way ANOVA followed by the Holm–Sidak procedure on a specialized software package (SigmaStat Version 3.5, Jandel Scientific Systat Software, Inc., Richmond, VA, USA).
4. Conclusions
A new family of coumarin derivatives functionalized with SAHA-like hydroxamates is herein reported. The compounds show an important antiproliferative activity on two cell lines of breast cancer and one of prostate cancer. The antiproliferative effect of the reference compound (SAHA) and the test compounds was generated at comparable concentrations (10 μM). For instance, both 7i and SAHA generated a high percentage of antiproliferative activity at a concentration of 10 μM. Furthermore, this activity correlates with the gene regulation detected in two human cancer cell lines. The experiment with propidium iodide revealed the location of compound 7j in cancer cells, evidenced by the dramatic change in the color of the nucleus in BT-474 cells and to a lesser extent in MDA-MB-231 and PC3 cells. The insights gained presently should certainly be useful for designing derivatives as better fluorescence probes and inhibitors of HDAC enzymes. The docking studies suggest that HDAC1, 6 and 8 are probably inhibited by 7c, 7e, 7f, 7i and 7j, which efficiently blocked the active sites of these three isoforms.
Acknowledgments
The authors thank Bruce A. Larsen for proofreading and are appreciative of the Guanajuato National Laboratory (UG-UAA-CONACyT 299078) for their generous allocation of analytical and computing resources. Thanks to “Fundación para la Salud y la Educación Dr. Salvador Zubirán” and “FOINS-INCMNSZ” by postdoctoral fellowship to N.S.-M (A-307-7).
Supplementary Materials
The supplementary materials are available online.
Author Contributions
Conceptualization, Y.A., M.A.G.-R., R.G.-B. and M.A.V.; methodology, S.G., I.M.-S. and L.B.; software, M.A.G.-R. and J.R.; validation, J.R., N.S.-M. and D.O.-R.; formal analysis, Y.A., M.A.G.-R., R.G.-B. and M.A.V.; investigation, S.G., I.M.-S., R.G.-B. and M.A.V.; resources, J.R., R.G.-B. and M.A.V.; writing—original draft preparation, S.G., M.A.G.-R., R.G.-B. and M.A.V.; funding acquisition, Y.A. and M.A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CONACYT (grant A1-S-27694) and DAIP-UG (Grant 072/2020) for financial support. S.G. acknowledges CONACYT for a graduate scholarship (# 328700).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability: Samples of the compounds are available from the authors.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fylaktakidou K.C., Hadjipavlou-Litina D.J., Litinas K.E., Nicolaides D.N. Natural and Synthetic Coumarin Derivatives with Anti-Inflammatory / Antioxidant Activities. Curr. Pharm. Des. 2004;10:3813–3833. doi: 10.2174/1381612043382710. [DOI] [PubMed] [Google Scholar]
- 2.Saeedi M., Goli F., Mahdavi M., Dehghan G., Faramarzi M.A., Foroumadi A., Shafiee A. Synthesis and Biological Investigation of Some Novel Sulfonamide and Amide Derivatives Containing Coumarin Moieties. Iran. J. Pharm. Res. 2014;13:881–892. [PMC free article] [PubMed] [Google Scholar]
- 3.Atmaca M., Bilgin H.M., Obay B.D., Diken H., Kelle M., Kale E. The hepatoprotective effect of coumarin and coumarin derivates on carbon tetrachloride-induced hepatic injury by antioxidative activities in rats. J. Physiol. Biochem. 2011;67:569–576. doi: 10.1007/s13105-011-0103-5. [DOI] [PubMed] [Google Scholar]
- 4.Peng X.-M., Damu G.L.V., Zhou C.-H. Current Developments of Coumarin Compounds in Medicinal Chemistry. Curr. Pharm. Des. 2013;19:3884–3930. doi: 10.2174/1381612811319210013. [DOI] [PubMed] [Google Scholar]
- 5.Završnik D., Muratović S., Makuc D., Plavec J., Cetina M., Nagl A., De Clercq E., Balzarini J., Mintas M. Benzylidene-bis-(4-Hydroxycoumarin) and Benzopyrano-Coumarin Derivatives: Synthesis, 1H/13C-NMR Conformational and X-ray Crystal Structure Studies and In Vitro Antiviral Activity Evaluations. Molecules. 2011;16:6023–6040. doi: 10.3390/molecules16076023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S.-F., Yin Y., Wu X., Qiao F., Sha S., Lv P.-C., Zhao J., Zhu H.-L. Synthesis, molecular docking and biological evaluation of coumarin derivatives containing piperazine skeleton as potential antibacterial agents. Bioorg. Med. Chem. 2014;22:5727–5737. doi: 10.1016/j.bmc.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 7.Ostrov D.A., Hernández Prada J.A., Corsino P.E., Finton K.A., Le N., Rowe T.C. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob. Agents Chemother. 2007;51:3688–3698. doi: 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shamsa F., Foroumadi A., Shamsa H., Samadi N., Faramarzi M.A., Shafiee A. Synthesis and In-vitro Antibacterial Activities of Acetylanthracene and Acetylphenanthrene Derivatives of Some Fluoroquinolones. Iran. J. Pharm. Res. 2011;10:225–231. [PMC free article] [PubMed] [Google Scholar]
- 9.Patel R.V., Patel P.K., Kumari P., Rajani D.P., Chikhalia K.H. Synthesis of benzimidazolyl-1,3,4-oxadiazol-2ylthio-N-phenyl (benzothiazolyl) acetamides as antibacterial, antifungal and antituberculosis agents. Eur. J. Med. Chem. 2012;53:41–51. doi: 10.1016/j.ejmech.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 10.Keri R.S., Sasidhar B.S., Nagaraja B.M., Santos M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015;100:257–269. doi: 10.1016/j.ejmech.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Nair R.V., Fisher E.P., Safe S.H., Cortez C., Harvey R.G., DiGiovanni J. Novel coumarins as potential anticarcinogenic agents. Carcinogenesis. 1991;12:65–69. doi: 10.1093/carcin/12.1.65. [DOI] [PubMed] [Google Scholar]
- 12.Gu X., Zhou Y., Wu X., Wang F., Zhang C.-Y., Du C., Shen L., Chen X., Shi J., Liu C., et al. Antidepressant-like effects of auraptenol in mice. Sci. Rep. 2014;4:4433. doi: 10.1038/srep04433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capra J.C., Cunha M.P., Machado D.G., Zomkowski A.D.E., Mendes B.G., Santos A.R.S., Pizzolatti M.G., Rodrigues A.L.S. Antidepressant-like effect of scopoletin, a coumarin isolated from Polygala sabulosa (Polygalaceae) in mice: Evidence for the involvement of monoaminergic systems. Eur. J. Pharmacol. 2010;643:232–238. doi: 10.1016/j.ejphar.2010.06.043. [DOI] [PubMed] [Google Scholar]
- 14.Yuce B., Danis O., Ogan A., Sener G., Bulut M., Yarat A. Antioxidative and lipid lowering effects of 7,8-dihydroxy-3-(4-methylphenyl) coumarin in hyperlipidemic rats. Arzneimittelforschung. 2009;59:129–134. doi: 10.1055/s-0031-1296375. [DOI] [PubMed] [Google Scholar]
- 15.Orhan I.E., Gulcan H.O. Coumarins: Auspicious Cholinesterase and Monoamine Oxidase Inhibitors. Curr. Top. Med. Chem. 2015;15:1673–1682. doi: 10.2174/1568026615666150427113103. [DOI] [PubMed] [Google Scholar]
- 16.Brühlmann C., Ooms F., Carrupt P.-A., Testa B., Catto M., Leonetti F., Altomare C., Carotti A. Coumarins Derivatives as Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase. J. Med. Chem. 2001;44:3195–3198. doi: 10.1021/jm010894d. [DOI] [PubMed] [Google Scholar]
- 17.Katsori A.-M., Hadjipavlou-Litina D. Coumarin derivatives: An updated patent review (2012–2014) Expert Opin. Ther. Pat. 2014;24:1323–1347. doi: 10.1517/13543776.2014.972368. [DOI] [PubMed] [Google Scholar]
- 18.Borges F., Roleira F., Milhazes N., Santana L., Uriarte E. Simple Coumarins and Analogues in Medicinal Chemistry: Occurrence, Synthesis and Biological Activity. Curr. Med. Chem. 2005;12:887–916. doi: 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]
- 19.Thakur A., Singla R., Jaitak V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015;101:476–495. doi: 10.1016/j.ejmech.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Egan D., James P., Cooke D., O’Kennedy R. Studies on the cytostatic and cytotoxic effects and mode of action of 8-nitro-7-hydroxycoumarin. Cancer Lett. 1997;118:201–211. doi: 10.1016/S0304-3835(97)00331-5. [DOI] [PubMed] [Google Scholar]
- 21.Seidel C., Schnekenburger M., Zwergel C., Gaascht F., Mai A., Dicato M., Kirsch G., Valente S., Diederich M. Novel inhibitors of human histone deacetylases: Design, synthesis and bioactivity of 3-alkenoylcoumarines. Bioorg. Med. Chem. Lett. 2014;24:3797–3801. doi: 10.1016/j.bmcl.2014.06.067. [DOI] [PubMed] [Google Scholar]
- 22.Wu X.-Q., Huang C., Jia Y.-M., Song B.-A., Li J., Liu X.-H. Novel coumarin-dihydropyrazole thio-ethanone derivatives: Design, synthesis and anticancer activity. Eur. J. Med. Chem. 2014;74:717–725. doi: 10.1016/j.ejmech.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Belluti F., Fontana G., Bo L.D., Carenini N., Giommarelli C., Zunino F. Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: Identification of novel proapoptotic agents. Bioorg. Med. Chem. 2010;18:3543–3550. doi: 10.1016/j.bmc.2010.03.069. [DOI] [PubMed] [Google Scholar]
- 24.Ding J., Liu J., Zhang Z., Guo J., Cheng M., Wan Y., Wang R., Fang Y., Guan Z., Jin Y. Design, synthesis and biological evaluation of coumarin-based N-hydroxycinnamamide derivatives as novel histone deacetylase inhibitors with anticancer activities. Bioorg. Chem. 2020;101:104023. doi: 10.1016/j.bioorg.2020.104023. [DOI] [PubMed] [Google Scholar]
- 25.Yang F., Zhao N., Song J., Zhu K., Jiang C.-S., Shan P., Zhang H. Design, Synthesis and Biological Evaluation of Novel Coumarin-Based Hydroxamate Derivatives as Histone Deacetylase (Hdac) Inhibitors with Antitumor Activities. Molecules. 2019;24:2569. doi: 10.3390/molecules24142569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao N., Yang F., Han L., Qu Y., Ge D., Zhang H. Development of Coumarin-Based Hydroxamates as Histone Deacetylase Inhibitors with Antitumor Activities. Molecules. 2020;25:717. doi: 10.3390/molecules25030717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manal M., Chandrasekar M.J.N., Gomathi Priya J., Nanjan M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016;67:18–42. doi: 10.1016/j.bioorg.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Qiu X., Xiao X., Li N., Li Y. Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2017;72:60–72. doi: 10.1016/j.pnpbp.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Tasior M., Kim D., Singha S., Krzeszewski M., Ahn K.H., Gryko D.T. π-Expanded coumarins: Synthesis, Optical Properties and Applications. J. Mater. Chem. C. 2015;3:1421–1446. doi: 10.1039/C4TC02665A. [DOI] [Google Scholar]
- 30.Yan F., Sun X., Zu F., Bai Z., Jiang Y., Fan K., Wang J. Fluorescent probes for detecting cysteine. Methods Appl. Fluoresc. 2018;6:1–48. doi: 10.1088/2050-6120/aad580. [DOI] [PubMed] [Google Scholar]
- 31.Jung Y., Jung J., Huh Y., Kim D. Benzo[g]coumarin-Based Fluorescent Probes for Bioimaging Applications. J. Anal. Methods Chem. 2018;2018:5249765. doi: 10.1155/2018/5249765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rooker D.R., Buccella D. Real-time detection of histone deacetylase activity with a small molecule fluorescent and spectrophotometric probe. Chem. Sci. 2015;6:6456–6461. doi: 10.1039/C5SC02704G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baba R., Hori Y., Mizukami S., Kikuchi K. Development of a Fluorogenic Probe with a Transesterification Switch for Detection of Histone Deacetylase Activity. J. Am. Chem. Soc. 2012;134:14310–14313. doi: 10.1021/ja306045j. [DOI] [PubMed] [Google Scholar]
- 34.Hoffmann K., Brosch G., Loidl P., Jung M. A non-isotopic assay for histone deacetylase activity. Nucleic Acids Res. 1999;27:2057–2058. doi: 10.1093/nar/27.9.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heltweg B., Dequiedt F., Marshall B.L., Brauch C., Yoshida M., Nishino N., Verdin E., Jung M. Subtype Selective Substrates for Histone Deacetylases. J. Med. Chem. 2004;47:5235–5243. doi: 10.1021/jm0497592. [DOI] [PubMed] [Google Scholar]
- 36.Singh R.K., Mandal T., Balasubramanian N., Cook G., Srivastava D.K. Coumarin-suberoylanilide hydroxamic acid as a fluorescent probe for determining binding affinities and off-rates of histone deacetylase inhibitors. Anal. Biochem. 2011;408:309–315. doi: 10.1016/j.ab.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Yacoub N., Fecker L.F., Möbs M., Plötz M., Braun F.K., Sterry W., Eberle J. Apoptosis Induction by SAHA in Cutaneous T-Cell Lymphoma Cells Is Related to Downregulation of c-FLIP and Enhanced TRAIL Signaling. J. Investig. Dermatol. 2012;132:2263–2274. doi: 10.1038/jid.2012.125. [DOI] [PubMed] [Google Scholar]
- 38.Wittine K., Ratkaj I., Benci K., Suhina T., Mandić L., Ilić N., Pavelić S.K., Pavelić K., Mintas M. The novel coumarin[3,2-c]thiophene and its hydroxamic acid and ureido derivatives: Synthesis and cytostatic activity evaluations. Med. Chem. Res. 2016;25:728–737. doi: 10.1007/s00044-016-1523-0. [DOI] [Google Scholar]
- 39.Abdizadeh R., Hadizadeh F., Abdizadeh T. QSAR analysis of coumarin-based benzamides as histone deacetylase inhibitors using CoMFA, CoMSIA and HQSAR methods. J. Mol. Struct. 2020;1199:126961. doi: 10.1016/j.molstruc.2019.126961. [DOI] [Google Scholar]
- 40.Garcia S., Vazquez J.L., Renteria M., Aguilar-Garduño I.G., Delgado F., Trejo-Duran M., Garcia-Revilla M.A., Alvarado-Mendez E., Vazquez M.A. Synthesis and experimental-computational characterization of nonlinear optical properties of triazacyclopentafluorene-coumarin derivatives. Opt. Mater. (Amst.) 2016;62:231–239. doi: 10.1016/j.optmat.2016.09.065. [DOI] [Google Scholar]
- 41.Bahena L., Cervantes C., Soto-Arredondo K.J., Martínez-Alfaro M., Zarco N., García-Revilla M.A., Alcaraz-Contreras Y., Tirado L.P., Vázquez M.A., Robles J. In Silico, Synthesis and Biological Investigations of Pyrrolo[3,4-C]Pyrrole Hydroxamic Acid Derivatives as Potential Anticancer Agents. J. Mex. Chem. Soc. 2017;61:297–308. doi: 10.29356/jmcs.v61i4.460. [DOI] [Google Scholar]
- 42.Song A., Wang X., Lam K.S. A convenient synthesis of coumarin-3-carboxylic acids via Knoevenagel condensation of Meldrum’s acid with ortho-hydroxyaryl aldehydes or ketones. Tetrahedron Lett. 2003;44:1755–1758. doi: 10.1016/S0040-4039(03)00108-4. [DOI] [Google Scholar]
- 43.Maggi R., Bigi F., Carloni S., Mazzacani A., Sartori G. Uncatalysed reactions in water: Part 2. Preparation of 3-carboxycoumarins. Green Chem. 2001;3:173–174. doi: 10.1039/b101822c. [DOI] [Google Scholar]
- 44.Kim N.H., Kim S.-N., Kim Y.K. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem. Biophys. Res. Commun. 2011;404:915–921. doi: 10.1016/j.bbrc.2010.12.081. [DOI] [PubMed] [Google Scholar]
- 45.Huang L., Pardee A.B. Suberoylanilide hydroxamic acid as a potential therapeutic agent for human breast cancer treatment. Mol. Med. 2000;6:849–866. doi: 10.1007/BF03401823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler L.M., Agus D.B., Scher H.I., Higgins B., Rose A., Cordon-Cardo C., Thaler H.T., Rifkind R.A., Marks P.A., Richon V.M. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- 47.Patra N., De U., Kim T.H., Lee Y.J., Ahn M.Y., Kim N.D., Yoon J.H., Choi W.S., Moon H.R., Lee B.M., et al. A novel histone deacetylase (HDAC) inhibitor MHY219 induces apoptosis via up-regulation of androgen receptor expression in human prostate cancer cells. Biomed. Pharmacother. 2013;67:407–415. doi: 10.1016/j.biopha.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Knutson A.K., Welsh J., Taylor T., Roy S., Wang W.-L.W., Tenniswood M. Comparative effects of histone deacetylase inhibitors on p53 target gene expression, cell cycle and apoptosis in MCF-7 breast cancer cells. Oncol. Rep. 2012;27:849–853. doi: 10.3892/or.2011.1590. [DOI] [PubMed] [Google Scholar]
- 49.Kong Y., Jung M., Wang K., Grindrod S., Velena A., Lee S.A., Dakshanamurthy S., Yang Y., Miessau M., Zheng C., et al. Histone Deacetylase Cytoplasmic Trapping by a Novel Fluorescent HDAC Inhibitor. Mol. Cancer Ther. 2011;10:1591–1599. doi: 10.1158/1535-7163.MCT-10-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longworth M.S., Laimins L.A. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene. 2006;25:4495–4500. doi: 10.1038/sj.onc.1209473. [DOI] [PubMed] [Google Scholar]
- 51.Gui C.-Y., Ngo L., Xu W.S., Richon V.M., Marks P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA. 2004;101:1241–1246. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greer C.B., Tanaka Y., Kim Y.J., Xie P., Zhang M.Q., Park I.-H., Kim T.H. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015;13:1444–1455. doi: 10.1016/j.celrep.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De U., Kundu S., Patra N., Ahn M.Y., Ahn J.H., Son J.Y., Yoon J.H., Moon H.R., Lee B.M., Kim H.S. A New Histone Deacetylase Inhibitor, MHY219, Inhibits the Migration of Human Prostate Cancer Cells via HDAC1. Biomol. Ther. (Seoul) 2015;23:434–441. doi: 10.4062/biomolther.2015.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park S.Y., Jun J.A., Jeong K.J., Heo H.J., Sohn J.S., Lee H.Y., Park C.G., Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011;25:1677–1681. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- 55.Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holohan C., Van Schaeybroeck S., Longley D.B., Johnston P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 57.Herrera-Martínez M., Orozco-Samperio E., Montaño S., Ariza-Ortega J.A., Flores-García Y., López-Contreras L. Vorinostat as potential antiparasitic drug. Eur. Rev. Med. Pharmacol. Sci. 2020;24:7412–7419. doi: 10.26355/eurrev_202007_21909. [DOI] [PubMed] [Google Scholar]
- 58.Iwamoto M., Friedman E.J., Sandhu P., Agrawal N.G.B., Rubin E.H., Wagner J.A. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother. Pharmacol. 2013;72:493–508. doi: 10.1007/s00280-013-2220-z. [DOI] [PubMed] [Google Scholar]
- 59.Mravljak J., Ojsteršek T., Pajk S., Sollner Dolenc M. Coumarin-based dual fluorescent spin-probes. Tetrahedron Lett. 2013;54:5236–5238. doi: 10.1016/j.tetlet.2013.07.084. [DOI] [Google Scholar]
- 60.Shao Y., Molnar L.F., Jung Y., Kussmann J., Ochsenfeld C., Brown S.T., Gilbert A.T.B., Slipchenko L.V., Levchenko S.V., O’Neill D.P., et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006;8:3172–3191. doi: 10.1039/B517914A. [DOI] [PubMed] [Google Scholar]
- 61.Wavefunction, Inc. [(accessed on 30 October 2020)]; Available online: https://www.wavefun.com.
- 62.Rocha G.B., Freire R.O., Simas A.M., Stewart J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006;27:1101–1111. doi: 10.1002/jcc.20425. [DOI] [PubMed] [Google Scholar]
- 63.Watson P.J., Millard C.J., Riley A.M., Robertson N.S., Wright L.C., Godage H.Y., Cowley S.M., Jamieson A.G., Potter B.V.L., Schwabe J.W.R. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 2016;7:11262. doi: 10.1038/ncomms11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hai Y., Christianson D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Somoza J.R., Skene R.J., Katz B.A., Mol C., Ho J.D., Jennings A.J., Luong C., Arvai A., Buggy J.J., Chi E., et al. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure. 2016;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 66.Thomsen R., Christensen M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 67.Keepers Y.P., Pizao P.E., Peters G.J., van Ark-Otte J., Winograd B., Pinedo H.M. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur. J. Cancer Clin. Oncol. 1991;27:897–900. doi: 10.1016/0277-5379(91)90142-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.