

Abstract
Objectives: Adult mesenchymal stem cells (MSC) have been proven to be of benefit to the kidney in different experimental models of renal injuries. All studies have been performed in valuable rodent models, but the relevance of these results to large mammals and ultimately, to humans remains unknown. Therefore, the aim of this study was to investigate the effect of MSC transplantation in an alternative ovine large‐animal model of bilateral kidney ischaemia reperfusion injury.
Material and methods: Sheep were divided into three groups: one sham‐operated group and two groups submitted to renal bilateral ischaemia for 60 min. Animals with ischaemia reperfusion injury were treated with injection of autologous MSCs or with vehicle medium.
Results: The model sheep presented with renal histological manefestations that closely resembled lesions seen in patients. Transplanted MSCs were found in glomeruli but not in tubules and did not express glomerular cell markers (podocin, von Willebrand factor), but functional evaluation showed no beneficial effect of MSC infusion. Morphological and molecular analyses corroborated the functional results. MSCs did not repair kidney parenchyma and failed to modulate cell death and proliferation or cytokine release (tumour necrosis factor‐alpha, vascular endothelial growth factor alpha (VEGF‐α), Bcl‐2, caspase).
Conclusion: In this unique autologous large‐animal model, MSCs did not exhibit reparative or paracrine protective properties.
Introduction
Adult stem cells have been studied extensively and a growing body of evidence shows that adult bone marrow‐derived cells have the capacity to trans‐differentiate into a variety of somatic cell types (1, 2, 3, 4, 5). Therefore, adult stem cell‐based therapy has triggered a large interest as it may provide new therapeutic strategies to replace, repair or protect organs and tissues, restoring function in many diseases (6, 7, 8, 9, 10). In the kidney, recent studies have reported the beneficial effect of adult stem cell transplantation in different models of diseases. Exogenous extra‐renal stem cells improved renal function in renal experimental ischaemia‐, drug‐ or toxic‐induced injury (6, 7, 8, 9, 10); bone marrow‐derived cells also offered therapeutic benefit in an Alport syndrome model (11) and attenuated glomerular sclerosis in a further study (12). Together, these findings are promising and might soon be accompanied by an exhortation to launch clinical trials.
Nevertheless, all these studies have been performed in rodent models and relevance of these findings to larger mammals and ultimately, to humans remains unknown. While small animal systems offer invaluable insight into fundamental biological questions, there are numerous differences between rodents and humans that render translation of rodent data imperfect (13, 14). When it comes to cell‐based therapy, it has been shown that stem cell turnover is remarkably higher in small animals than in large ones (15). Such a difference in this fundamental ‘stemness’ property might account for failure in stem cell‐based clinical trials. In cardiology, after remarkably promising results in rodents, large‐animal models have tempered the results and sometimes uncovered unexpected side‐effects (16); furthermore, recent clinical trials seem to be disappointing (17, 18). Therefore, studies in large‐animal models are warranted in an attempt to better set the stage for future human studies.
Taking into account these considerations, the aim of this study was to investigate the effect of mesenchymal stem cells (MSC) in an alternative large‐animal model of acute renal failure (ARF). We therefore developed a model of bilateral ischaemia reperfusion injury (IRI) in the adult sheep. This model allowed us to perform autologous transplantation of MSCs. We first demonstrated that the model sheep present with a typical pattern of IRI lesions that closely mimics that of humans. As we sought to determine mechanisms underlying potential beneficial effect of MSCs, we investigated the fate of MSCs and performed extensive renal functional, morphological, cellular and gene expression evaluation.
Materials and methods
MSC isolation, culture, labelling and characterization
Culture MSCs were isolated from bone marrow harvested from each ewe in order to perform autologous transplantation. Bone marrow aspirations were performed under general anaesthesia on tibial tuberosities and humeral tuberculae, 2 weeks before grafting. A total of 20 ml of bone marrow aspirate was resuspended in four sterile 75‐cm2 flasks containing culture medium [30 ml of Dulbecco's modified Eagle's medium (DMEM), Gibco, Invitrogen, Grand Island, NY, USA] complete with foetal calf serum (DMEM 79%, foetal calf serum 20%, penicillin streptomycin 1%). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. Every 3 days, culture medium was completely replaced and non‐adherent cells were discarded. MSCs were recognized by their ability to proliferate in culture with attached, spindle‐shape morphology. When cells were more than 70% confluent, adherent cells were detached by 5 min in 0.25% trypsin at 37 °C containing 1 mm EDTA, and were replated to obtain 4–5 × 106 cells per flask. Cells were replated twice (first and second passages) and one hundred million MSCs were obtained in 2 weeks. MSCs were labelled before transplantation.
Labelling Two different chemical bound dyes were used: 5‐bromo‐2‐deoxyuridine (BrdUrd, Roche Diagnostics, Basel, Switzerland) and chloromethylbenzamido‐DiI derivative (cm‐DiI, Cell Tracker, Molecular Probes, Eugene, OR, USA). For BrdUrd labelling, a sterile BrdUrd stock solution was added to the culture medium 48 h before culture ending at a final concentration of 10 µmol/l, in order to allow integration of BrdUrd into DNA of dividing cells. For cm‐DiI labelling, sterile stock solution (50 µl of a 1 mg/ml cm‐DiI concentrated solution) was added to resuspended cell solution of 50 × 106 cells. The dye was allowed to remain in contact with MSCs for 5 min at room temperature and for 15 min at 4 °C. Cells were then rinsed at least three times in polyphosphate‐buffered solution (Invitrogen) to remove all excess (unbound) cm‐DiI. Before injection, a sample of MSCs was systematically collected in order to assess cell labelling efficacy: both cm‐DiI and BrdUrd showed very good staining efficiency in quality and quantity (approximately 95%).
MSC characterization by in vitro differentiation Sheep MSC phenotype was confirmed by the cells’ ability to differentiate into osteocytes, chondrocytes and adipocytes, as described in our previous study (19). Briefly, MSCs were forced towards osteoblast, chondroblast and adipocyte lineages using respective inductive media.
Osteogenesis Iscove's modified Dulbecco's medium (Invitrogen), 20% foetal calf serum, 100 UI/ml penicillin, 100 µg/ml streptomycin and 0.05 mmβ‐mercaptoethanol were used. Inductive reagents consisted in 10 mmβ‐glycerophosphate, 50 µg/ml ascorbic acid 2‐phosphate, and 10−9 m dexamethasone. After 3 weeks of inductive culture, calcium‐rich hydroxyapatite deposition in the extracellular matrix (as a marker of bone induction) was assessed by staining with Alizarin Red S (Sigma, St. Louis, MO, USA).
Chondrogenesis MSCs were centrifuged in a 15‐ml polypropylene tube (Falcon). Cells formed a micromass at the bottom of the tube and were cultured in 500 µl of chondrogenic medium that consisted of 50 µg/ml ascorbic acid 2‐phosphate and 1 ng/ml TGF‐β1 (Sigma). MSCs formed cell clumps after 3 weeks and were fixed in formalin, paraffin wax embedded and were cut into 3‐µm sections. Glycosaminoglycans were revealed after staining with Safranin O (Sigma).
Adipogenesis For osteogenesis, Iscove's medium was used, and inductive reagents were 5 µg/ml insulin (Sigma) and 10−9 m dexamethasone (Sigma). Adipogenesis was recognized after staining refractive intracellular lipid vacuoles with oil red O (Sigma).
Flow cytometry analysis Further identification of sheep MSCs was performed by cytofluorimetric analysis of cell surface markers using techniques previously described (20). The following antibodies were used: anti‐CD45 (monoclonal antihuman, BioSource International, Camarillo, CA, USA); anti‐CD44 (monoclonal anti‐porcine), anti‐CD58 and anti‐CD31 (monoclonal anti‐sheep, Fitzgerald Industries International Inc., Concord, MA, USA). Antigen location was detected by fluorescein isothiocyanate‐conjugated donkey anti‐mouse immunoglobulin G. Cells were also labelled with isotype control antibodies (Fitzgerald Industries International Inc.). Briefly, cells were trypsinized and aliquoted at a concentration of 0.5 × 106 cells/ml and were stained for 30 min with either conjugated specific antibodies or istotype‐matched control mouse immunoglobulin G, at recommended concentrations. Labelled cells were washed twice and resuspended in FACs buffer. The analysis was performed using an EPICS Altra flow cytometer (Beckman Coulter, Fullerton, CA, USA) after excitation at 488 nm. Maker expression was determined by ratio of mean fluorescence intensity of each specific antibody/control antibody. For each sample 10 000 events were collected. Results were analysed using the Kolmogorov–Smirnov statistical test. Differences between histograms were considered statistically significant when Kolmogorov–Smirnov was ≥ 0.2.
Ischaemia/reflow experiments and MSC transplantation
Fifteen adult female sheep were used in this study. All animals received veterinary care in compliance with the ‘Guide for the Care and Use of Laboratory Animals’ and the procedures described were approved by the institutional ethics committee. Animals were assigned into either of three groups. Group A (n = 5): sham‐operated animals: At day 0, these animals were submitted to the complete bilateral renal artery catheterization procedure, except that they had no renal occlusion, and no MSCs infusion. Animals received culture medium and were evaluated 7 days after the sham procedure. Group B (n = 5): animals submitted to IRI: At day 0, these animals were submitted to the complete renal artery catheterization procedure and were submitted to a 60‐min transient bilateral renal artery occlusion. Animals received culture medium and were sacrificed 7 days after the procedure. Group C (n = 5): animals submitted to IRI and grafted with MSCs immediately after damage. At day 0, MSCs injection was carried out immediately after balloon occlusion termination. Animals were euthanized 7 days after the graft.
IRI procedure Briefly, animals were anaesthetized (isoflurane in 100% O2), endotracheally intubated and placed on mechanical ventilation; electrocardiogram, invasive blood pressure, end tidal CO2 and body temperature were continuously monitored. The procedure was performed under fluoroscopy (Stenoscope®, General Electric, Fairfield, CT, USA). Animals received a 0.5‐mg/kg injection of intravenous heparin (Heparine, Choay®, Sanofi‐Aventis, Paris, France); two 7F introducers (Input Medtronic® AVE Medtronic, Tolochenaz, Switzerland) were placed percutaneously, respectively, into the left and right femoral arteries. Two 6F guiding catheters were positioned, respectively, into both renal arteries, and two 4.0‐mm diameter rapid exchange balloon catheters were advanced into the proximal zone of both renal arteries, and was inflated for 60 min.
MSC infusion After 60 min, balloon catheters were removed and 50 × 106 MSCs in 10‐ml culture medium were slowly infused into each renal artery simultaneously through the guiding catheters. Considering that the average weight of our sheep in group C was 53.6 ± 3.6 kg, animals were treated with approximately 1.5 million MSCs per kilogram. In groups A and B, culture medium was injected instead of MSCs. In all groups, renal arterial reflow was assessed after the IRI procedure and cell injection.
MSC tracking and differentiation analysis after transplantation
MSC tracking In group C, counting of positive glomeruli and tubules was performed in order to quantify the degree of engraftment. Tubules and glomeruli that had integrated labelled cells were considered positive (≥ 1 labelled cell per tubule or glomerulus section in 5 µm renal sections). Evaluation was performed over 10 high‐powered fields (×100) (Leica DMIL®, Leica) for each slide. Three slides of each kidney were randomly read. Percentage of positive tubules or glomeruli was expressed as number of positive glomeruli or tubules versus total number of glomeruli or tubules per microscope field.
Immunophenotypic analysis To identify the phenotype of MSCs that localized to the kidney, sections were stained for antibody revelation or lectin presence that identify specific nephron segments or glomerular cells. Co‐localization of these markers and cm‐DiI, was investigated to assess MSCs’ phenotype. Tubules were stained with an antibody against Tamm–Horsfall for Henle's loops identification, against Dolichos biflorus agglutinin (DBA) for distal and collecting duct identification, and against Lotus tetragonolobus (LTA) for proximal tubule identification. Glomeruli were stained with an antibody against podocin (kind gift from M‐C Gubbler, Inserm U574, Paris) for podocyte identification and against von Willebrand factor (Dako, Glostrup, Denmark) for endothelial cell identification. Experiments were performed on frozen sections (5 µm) which were fixed in ice‐cold acetone for 10 min. Negative controls were obtained for all experiments by replacing specific antisera with non‐immune sera. (i) Cm‐DiI/LTA: LTA (Vector Laboratories, Burlingame, CA, USA) conjugated to fluorescein (dilution 1 : 50) was incubated on sections for 2 h at 37 °C, (ii) Cm‐DiI/DBA: DBA (Vector Laboratories) conjugated to fluorescein (dilution 1 : 50) was incubated on them for 2 h at 37 °C, (iii) Cm‐DiI/Tamm–Horsfall: sections were incubated with anti‐human Tamm–Horsfall protein (monoclonal antibody, Cedarlane, Burlington, ON, Canada) at dilution of 1 : 100 for 2 h at 37 °C followed by anti‐mouse‐Ig‐fluorescein (Roche) at 1 : 50 dilution, (iv) cm‐DiI/podocin and cm‐DiI/von Willebrand factor: sections were incubated with anti‐podocin (1 : 500) or anti‐von Willebrand factor (1 : 500) for 2 h at 37 °C followed by anti‐rabbit‐Ig‐fluorescein isothiocyanate (1 : 100, Dako). cm‐DiI is a red fluorescent dye.
Evaluation of renal function
Blood urea nitrogen, creatinine and plasma renin activity Blood urea nitrogen (BUN), plasma creatinine concentration and plasma renin activity (PRA) were measured before, 10 min, 30 min, 1 day, 2 days and 7 days after renal ischaemia, by jugular vein blood sampling. PRA was measured using a routine radio‐immuno assay (REN‐CT2 kit®, Cis Bio International, Gif sur Yvette, France).
Urine analysis Urine was collected at the same time points as described above and electrolyte and creatinine concentrations were measured. The following ratios were calculated: Na/creatinine, Ca/creatinine.
Renal histology and injury scores
At day 7, kidneys were harvested, fixed in 4% buffered formalin and embedded in paraffin wax. Coronal sections (4 µm) were dehydrated using ethanol and were stained with haematoxylin–eosin and Masson's trichrome. Degree of renal injury after ischaemia was evaluated by two observers blind towards the group categories, using a semiquantitative method. Twenty randomly selected microscope fields (×400) per kidney were evaluated and scored using a scale ranging from 0 to 4. The following items were recorded: presence of focal necrosis, inflammatory cell infiltration, dilated glomerular chamber, casts, dilated tubules and abnormality of the glomerular tuft. A score of 0 signified absence of lesions, and scores of 1, 2, 3 and 4 corresponded to 1–10%, 11–25%, 26–50% and 51–100% involvement of the microscope field, respectively, for all items.
Cell proliferation and apoptosis
Kidney sections were deparaffinized and were investigated for cell proliferation, from 7‐µm sections. Monoclonal mouse anti‐proliferating cell nuclear antigen (PCNA) (clone PC10, Dako) was used at dilution 1 : 150 and developed using Dako Envision/HRP kit. Positive cells were identified, counted and analysed under the light microscope. Apoptosis was assessed using a terminal deoxynucleotidyl transferase‐mediated digoxigenin‐deoxyuridine nick‐end labelling (TUNEL) assay (In Situ Cell Death Detection Kit, POD, Roche Diagnostics). TUNEL staining was performed on 7‐µm paraffin wax sections to detect nuclear DNA fragmentation at the cellular level. Positive cells were identified, counted and analysed using a fluorescence microscope. Scoring for PCNA‐ and TUNEL‐positive cells was carried out by counting numbers of positive nuclei per high power field in 20 randomly chosen sections of kidney parenchyma.
Real‐time polymerase chain reaction
At harvesting, kidneys were dissected and four different samples were obtained: outer cortex, inner cortex, outer medulla and inner medulla. Fifty milligrams of each sample were immediately immersed into TRIzol reagent (Invitrogen) and frozen in liquid nitrogen to be stored at –80 °C until further use. Total RNA from each sample was isolated and quantified by optical density spectrophotometry. cDNA was synthesized by reverse transcription of 1–2 µg of target RNA, using Superscript II (Invitrogen) and random primers (Roche Diagnostics) to minimize potential contamination with genomic DNA. Controls with no reverse transcription were also included, and results showed that there was no polymerase chain reaction (PCR) product produced for these samples. Quantitative real‐time PCR was carried out using the Light Cycler 480 system (Roche Diagnostics) according to the manufacturer's instructions. Expression level of each mRNA was normalized using GAPDH housekeeping gene product as endogenous reference.
Sequences of primers used to amplify genes are as following: GAPDH: 5′‐ATC ACT GCC ACC CAG AAG ACT‐3′ forward, 5′‐CAT GCC AGT GAG CTT CCC GTT‐3′ reverse; VEGF‐A: 5′‐TGT AAT GAC GAA AGT CTG GAG‐3′ forward, 5′‐TCA CCG CCT CGG CTT GTC ACA‐3′ reverse; TNF‐α: 5′‐AGA ACC CCC TGG AGA TAA CC‐3′ forward, 5′‐AAG TGC AGC AGG CAG AAG AG‐3′ reverse; caspase‐3: 5′CCA ATG GAC CCG TCG ATC T‐3′ forward, 5′‐GTC TGC CTC AAC TGG TAT TTT CTG A‐3′ reverse; Bcl‐2: 5′‐GTG GCC TTC TTT GAG TTC G‐3′ forward, 5′‐CAT CCC AGC CTC CGT TGT CCT‐3′ reverse.
Samples were run in duplicate, and average crossing point value was used for calculations. The crossing point was obtained to quantify initial starting number of copies. Relative quantity of mRNA expression was calculated with the previously published crossing point method formula (21, 22) described hereunder in which E is the real‐time PCR efficiency and CP is the crossing point:
| Ratio = (Etarget)CPtarget(control‐sample)/(Eref)CPref(control‐sample) |
In order to avoid unspecific DNA amplification, we included DNase digestion procedure in the RNA extraction protocol and performed reactions with appropriate negative (template‐free) controls. In addition, analysis of melting curves of amplified products confirmed uniform amplification of the products and gel electrophoresis showed correct size of amplification products and absence of unspecific bands.
Statistical analysis
Data are expressed as mean ± standard error. Differences between the groups were evaluated using one‐way analysis of variance. P‐values less than 0.05 were considered statistically significant.
Results
MSCs showed typical properties of multipotent mesenchymal stromal cells in vitro
The sheep MSCs fulfilled the criteria defining multipotent MSCs (23): (i) they exhibited spindle‐shaped morphology (Fig. 1a) and were plastic‐adherent when maintained in standard culture conditions; (ii) functional characterization in culture confirmed their potential to differentiate into osteogenic (Fig. 1b), chondrogenic (Fig. 1c) and adipogenic cells (Fig. 1d) in vitro; and (iii) in addition, flow cytometric analysis showed that these cells were positive for CD44 and CD58 surface markers; in contrast, they were negative for CD45 and CD31 as previously demonstrated in sheep (24, 25, 26) (Fig. 1e).
Figure 1.

Mesenchymal stem cells (MSC) in culture, in vitro differentiation and cytofluorometry analysis. MSCs were characterized by their property to adhere to cell culture plastic dish and exhibit spindle shape morphology in culture (a). They were further identified by their canonical ability to differentiate into osteocytes (b), chondrocytes (c) and adipocytes (d) in vitro. Cell surface markers of MSCs were studied using flow cytometry analysis (e). Sheep MSCs were positive for CD44 and CD58 whereas being negative for CD31 and CD45.Original magnification: ×200 in (a); ×100 in (b) and (d); ×400 in (c).
MSCs localized in the kidney 7 days after intra‐renal arterial injection but did not differentiate into renal cells
At day 7 after MSC transplantation, all kidneys were evaluated to assess MSC localization. cm‐DiI‐specific fluorescence was found only in the renal cortex and not in the medulla. In the cortex, MSCs were found in glomeruli (Fig. 2a), with 85 ± 5.1% of glomeruli containing them (Fig. 2d). MSCs were rarely identified in the renal microvasculature (data not shown) and not in tubules. The cortex was further analysed. External cortex showed an average of 87 ± 9.2% of glomeruli containing MSCs and thet internal cortex 81% ± 7.1% (Fig. 2d). There was no significant difference in the number of positive glomeruli nor in number of MSCs per positive glomeruli in the different regions of the cortex. Positive glomeruli in the whole cortex contained an average of 3.8 ± 1.2 MSCs/glomeruli (Fig. 1e).
Figure 2.
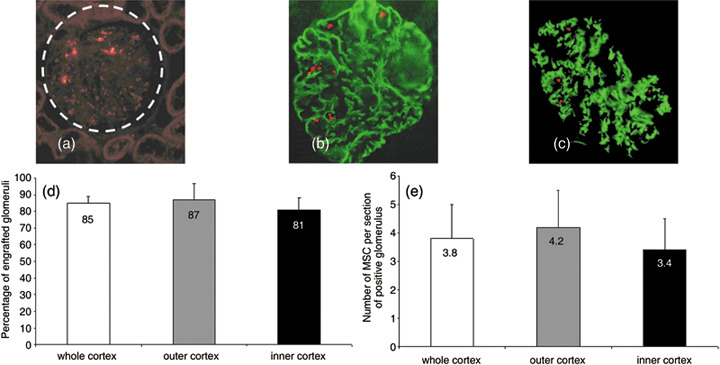
Mesenchymal stem cell (MSC) localization and counting in the kidney and phenotypical analysis. MSCs were tracked using cm‐DiI fluorescence (a). MSCs were present in glomeruli (white circle) whereas no MSCs could be found in tubules (a). MSC differentiation into glomerular cell type was investigated (b, c). Glomeruli were stained with an antibody against podocin (b) and von Willebrand factor (c) to assess podocyte or endothelial cell phenotype, respectively. No MSCs expressed either marker. MSCs did not differentiate into glomerular cells, and they remained located inside the capillary lumen. Counting MSCs showed that they were mainly present in the cortex localizing in glomeruli (d). Up to 85 ± 5.1% of the glomeruli presented MSCs. There was no significant difference when comparing outer (87 ± 9.2%) and inner cortex (81 ± 7.1%). There were approximately 4 MSCs per glomerulus section (e). No significant difference was observed in the number of MSCs per glomerulus in the outer or inner cortex. Original magnification: ×200 in (a), (b) and (c).
To determine whether MSCs had acquired a glomerular cell phenotype, kidney sections were serially costained with antibodies against markers of specific glomerular cells. The results of costaining demonstrated that MSCs localized in glomeruli were not positive for podocin (Fig. 2b) or von Willbrand factor (Fig. 2c) demonstrating that MSCs did not acquire epithelial podocyte or endothelial phenotype characteristics. In summary, in the present study the MSCs did not differentiate into renal cells.
Intra‐arterial transplantation of MSCs did not improve renal function
BUN and serum creatinine concentrations Before induction of renal ischaemia‐reperfusion injury and MSC treatment, groups A, B and C had identical values. In sham‐operated group A, parameters remained normal. In group B, when ischaemia was induced, BUN and serum creatinine concentrations demonstrated a 2‐fold increase at day 1, then slowly decreased towards baseline levels by day 7. In group C, BUN and serum creatinine followed the same trend as in group B (Fig. 3a,b).
Figure 3.

Blood urea nitrogen (BUN), plasma creatinine concentration and plasma renin activity (PRA). BUN and plasma creatinine values did not change in sham‐operated group. IRI medium‐treated group showed significant increase in BUN and plasma creatinine at days 1 and 2, with values returning to baseline by day 7. In IRI MSC‐treated group, values followed the same trend as in IRI medium‐treated. White bars = group A (sham‐operated group); grey bars = group B (IRI medium‐treated group); black bars = group C (IRI MSC‐treated group). §P < 0.05 vs. group A at corresponding time points. PRA values did not change in sham‐operated group. IRI medium‐treated group and IRI MSC‐treated group showed significant increase in PRA at 10 min and 30 min, with values returning to almost baseline at day 7. There was no significant difference observed after MSCs infusion in IRI sheep. White bars = group A (sham‐operated group); grey bars = group B (IRI medium‐treated group); black bars = group C (IRI MSC‐treated group). §P < 0.05 vs. group A at corresponding time points.
Plasma renin activity In sham‐operated group A, PRA values remained unchanged all through experimentation. In group B, PRA was significantly increased at 10 min and 30 min after injury, then decreased to reach almost baseline values. In group C, a significant elevation was also observed that followed the same trend as in group B (Fig. 3c).
Urine analysis In sham‐operated group A, urinary parameters showed no modification. In groups B and C, Na/creatinine and Ca/creatinine ratios had a significant increase at 10 min and 30 min then decreased towards baseline values (Fig. 4a,b).
Figure 4.

Urine Na/creatinine and Ca/creatinine ratios. Effect of mesenchymal stem cell (MSC) transplantation on urinary Na/creatinine (a) and Ca/creatinine (b) ratios. Ratios’ values did not change in the sham‐operated group. IRI medium‐treated group showed significant increase in either ratios at 10 min and 30 min, with values returning to almost baseline at day 7. In IRI MSC‐treated group values followed the same trend as in IRI medium‐treated group. White bars = group A (sham‐operated group); grey bars = group B (IRI medium‐treated group); black bars = group C (IRI MSC‐treated group). §P < 0.05 vs. group A at corresponding time points.
No significant difference was observed in BUN, serum creatinine, PRA and urinary parameters in MSC‐treated animals when compared to medium‐treated sheep. Altogether these functional results do not show any beneficial effect of MSC infusion in the course of renal IRI.
MSC treatment did not improve ischaemic renal lesions
All kidneys submitted to balloon occlusion showed morphological features of IRI. In IRI groups, renal lesions consisted mainly of focal acute tubular necrosis and cortical necrosis. Acute tubular necrosis ranged from dilated tubules with regenerating flattened tubular epithelium, blebbing, shedding of tubular epithelial cells, cast formation and slight necrosis with minimal accompanying interstitial infiltrate to more important lesions, such as degeneration and frank necrosis of tubular segments. Both acute tubular necrosis and cortical necrosis were found in most sections with variable extent (Fig. 5a–c). The evaluation showed that renal lesion score of MSC‐treated group C (1.12 ± 0.36) was not significantly different from the score of the medium‐treated group B (score of 1.17 ± 0.42) (Fig. 5d). In IRI MSC‐treated group C, infused MSCs did not trigger vasculature thrombosis nor enhance glomerular lesions when compared to IRI medium‐treated group B. These results show that MSC injection neither improved nor worsened ischaemia reperfusion‐induced renal lesions in our large‐animal model.
Figure 5.

Weight of kidneys, renal injury scores and morphological analysis. In the sham‐operated group, no renal damage could be observed (a). In IRI medium‐treated group, typical ischaemia reperfusion‐induced damage was present with tubular dilatation (black arrows) and necrosis, epithelial cell shedding (white arrows), inflammatory cell infiltration (dashed black arrows), cast formation and cortical necrosis (b). In IRI MSC‐treated group, the type and extent of lesions were the same as in IRI medium‐treated group (c). Injury scores confirmed that there was no damage in sham‐operated group, and when comparing IRI medium‐treated group and IRI MSC‐treated group, no improvement of renal lesions could be observed after MSCs treatment (d). §P < 0.05 vs. group A. Original magnification: ×200 in (a), (b) and (c).
MSCs did not modulate cell proliferation nor apoptosis
Failure to assess the beneficial effect of injected MSCs on kidney function and morphology led us to investigate apoptotic and proliferative processes at the cellular level. Indeed, it has been described that the primary effect of infused stem cells may be on endogenous tubular cell function (27). For examination of this question, kidneys were investigated with TUNEL and PCNA methods. In the sham‐operated group, TUNEL‐positive and PCNA‐positive cells were rarely observed (respectively, 0.2 ± 0.02 and 0.5 ± 0.07) (Fig. 6a,e). Results showed that there was a significant increase in cell proliferation and apoptosis in our large animals in groups submitted to IRI when compared to the sham‐operated group. There were approximately 9–10 TUNEL‐positive cells (9.2 ± 3.4) per high‐power field in medium‐treated IRI group B, and 10–11 positive cells (11 ± 4.4) in the MSC‐treated IRI group (Fig. 6b,c). There were approximately 55 PCNA‐positive cells (55.3 ± 6.2) in the medium‐treated IRI group and 60 positive cells (60.4 ± 16.3) in the MSC‐treated IRI group (Fig. 6f,g). There was no statistically significant difference observed in apoptosis or cell proliferation in IRI animals treated with MSCs compared to IRI animals receiving vehicle medium (Fig. 6d,h).
Figure 6.

Renal cell apoptosis and proliferation. Renal cell apoptosis and proliferation was assed in all groups. Terminal deoxynucleotidyl transferase‐mediated digoxigenin‐deoxyuridine nick‐end labelling (TUNEL) staining was performed on kidneys at day 7 in sham‐operated group (a), medium‐treated IRI group (b) and MSC‐treated IRI group (c). Quantification of TUNEL‐positive cells/high‐power field was carried out (d). Proliferating cell nuclear antigen (PCNA) staining was performed on sections of kidneys at day 7 in sham‐operated group (e), medium‐treated IRI group (f) and MSC‐treated IRI group (g). Quantification of PCNA was performed (h). There was a significant increase in TUNEL and PCNA expression in IRI groups when compared to sham group, but no statistically significant difference was observed when MSCs were injected compared to medium treatment in IRI groups. §P < 0.05 vs. sham‐operated group. Original magnification: ×400 in (a–g).
MSCs did not influence gene expression
Because it has been reported that MSCs may be of benefit to the post‐ischaemic kidney through paracrine mechanisms (28, 29), and because MSCs are known to secrete different types of cytokines (30, 31), we screened kidneys for changes in expression of apoptosis‐, inflammatory‐ and growth factor‐related genes. Quantitative real time PCR, with relative quantification of target gene copy numbers in relation to GAPDH transcripts, was carried out. GAPDH was equally expressed in groups A, B and C.
Seven days after ischaemia, renal TNF‐α expression was up‐regulated mainly in the outer medulla of kindneys; Bcl‐2 and caspase expressions were increased in all compartments except in the outer medulla. There was no clear change in VEGF‐α expression in any of the areas. Gene expression was not influenced by MSC transplantation (Fig. 7); results of the molecular evaluation confirmed those observed at the functional, histological and cellular level.
Figure 7.

Gene expression analysis. Comparative gene expression ratios in all groups. Four different regions of the kidney have been individually investigated: outer cortex, inner cortex, outer medulla and inner medulla. GAPDH was used as internal control. Black bars on all graphs illustrate gene expression ration of 1 (i.e. a value obtained when gene expression is identical in compared groups). Values lower or higher than 1 indicate gene expression is decreased or increased in one group when compared to the other. No significant difference in genes expression was observed after MSC treatment in IRI groups. *P < 0.05.
Discussion
To the best of our knowledge, this is the first time that the effect of MSC transplantation has been evaluated in a large‐animal model of bilateral renal ischaemia‐reperfusion disease. In our experimental setting, autologous MSCs localized to the kidney, but did not provide any beneficial effect on the course of the ischaemia‐reperfusion injury. Our results contradict several reports in rodent models that demonstrated that stem cells, including MSCs, can ameliorate the outcome of toxic‐ or ischaemic‐induced nephropathies (3, 27, 29, 32, 33).
In our sheep model, as expected, IRI was followed by significant increase in plasma renin activity, creatinine/BUN concentrations, and urinary sodium excretion, consequences of renal vasoconstriction, reduced glomerular filtration rate and tubular injury secondary to renal ischaemia. No significant difference in these parameters was observed after MSCs infusion during the 7‐day period following our IRI. In contrast, in rodents, an extensively investigated, albeit imperfect model of clinical ARF (34), most studies have shown beneficial effect of MSC infusion on renal function in the early period after IRI (3, 28, 29). However, the mechanism of such a glomerular filtration rate (GFR) improvement has not been defined; an effect on total renal outer cortical blood flow or renovascular resistance was not found. Some studies (35, 36, 37), however, have failed to demonstrate such a beneficial effect; in the study performed by Lin et al., administration of bone marrow cells (BMC) did not improve renal function in mice during the first week after ischaemic injury (37).
At the tissue level, 60‐min bilateral ischaemia induced mild renal injury with focal acute tubular necrosis; this pattern of lesions is closer to that observed in humans than that reported in mice or rats; in the latter species, lesions are reported to be more severe (34). In accordance with our functional results, MSC infusion neither improved nor worsened renal scores observed 7 days after IRI. In published studies on the effect of stem cells on renal injury, renal morphological scores have rarely been performed and no dramatic improvement of renal lesions was reported. In the study of Tögel el al. (29), improvement in renal injury was demonstrated but inflammation was persistent, as leucocyte infiltration was still present; evaluation was performed early (24 h) after MSC transplantation, not predicting what evolution of tissue damage would be. In the study of Kale et al. (3), performed 7 days after IRI, animals receiving stem cells showed substantial resolution of necrotic injury in large areas but tubular injury was still evident in significant regions.
At the cellular level, when tracking injected cells in our ovine model, MSCs were found in the kidney, in glomeruli, but did not differentiate into renal cells. Initial studies in different models of acute renal failure have suggested that MSCs engrafted in the damaged kidney and differentiated into tubular epithelial cells (3, 33, 37, 38). Since then, other groups have found that MSCs do not engraft into tubules, labelled MSCs being detected mostly in glomeruli, still ameliorating renal function (28, 29, 39). However, these results, as in the majority of studies that failed to find MSC in tubules, correspond to early examination of kidneys after MCS injection. In a previous study (19) in which kidneys were evaluated later, 3 weeks after MSC injection, we observed presence of MSCs both in glomeruli and in tubules; moreover, the cells had acquired markers of renal cells in glomeruli as well as in tubules, suggesting they underwent trans‐differentiation. In a recent study, Herrera et al. (40) found that after 24 h injected MSCs were mainly detectable within peritubular capillaries and interstitium, whereas after 8 days MSCs were found mainly in tubules. Broekema et al. (41) showed that bone marrow derived cells (BMDC) engraftment in tubules was optimal on day 14, decreasing by day 28 after the ischaemic insult. We hypothesize that injected MSCs are present in glomerular capillaries and renal vasculature during the first week after injection; some of them stay in glomeruli but others migrate to tubules; after some time (2–3 weeks) some of these cells may trans‐differentiate. At any rate, integration of stem cells into the kidney may not be the key to improvement, and the precise mechanism involved in beneficial effect of MSCs remains unknown. As it has been observed in the presence or absence of cell engraftment, and at a time point too early for tubular replacement via ‘trans‐differentiation’ of stem cells, it cannot be directly related to reparation; even in the cases in which transplanted cells were observed in tubules, the low level of engraftment cannot explain the renal reparation observed. Lately, the study of Bi et al. (27) confirmed that infusion of bone marrow stem cells (BMSC) reduced the severity of cisplatin‐induced acute renal failure in adult rodent model without stem cell integration into kidney. This effect was also seen when BMSC were given by intraperitoneal injection. Furthermore, intraperitoneal administration of the conditioned medium from BMSC culture diminished tubular cell apoptosis, increased animals’ survival and limited renal injury. Bi et al. (2007) concluded that BMSC protect the kidney by secreting factors that limit apoptosis and enhance proliferation of the endogenous tubular cells, suggesting that transplantation of the cells themselves is not necessary.
Thus, a growing body of evidence suggests that the paracrine role of stem cells is critical. Therefore, we further investigated our large‐animal model at the molecular level. In the course of IRI, modification in expression of different molecules involved in inflammatory (TNF‐α, IL‐10), apoptotic (Bcl‐2, caspase) and cell growth/proliferation (VEGF‐α) processes have not been well established (42). Different studies have shown that MSC infusion was associated with modification of expression of different molecules (28, 29, 32, 43), some of them showing anti‐inflammatory or vasculotropic properties. In the study of Tögel et al. (29), expression of pro‐inflammatory cytokines IL‐1β and TNF‐α was significantly reduced and that of anti‐inflammatory IL‐10 and Bcl‐2 was highly up‐regulated in MSC‐treated kidneys, suggesting that the beneficial effect of MSCs results from complex paracrine mechanisms acting on cellular function. In our ovine model, as expected, TNF‐α and both Bcl‐2 and caspase expressions increased in response to IRI, but no significant difference in their expression was observed after MSC infusion. In the recent report of Imberti et al. (32), it was shown that MSCs exert beneficial effect on tubular cell repair by producing mitogenic factor IGF‐1 that enhances cell proliferation and repair. In our study, when investigating cell proliferation and apoptosis, we observed a significant increase in both phenomena after IRI, but again, no modulation in cell proliferation or death was found after MSC transplantation. These results differ from studies in rodents that showed both a decrease in apoptosis and increase in cell proliferation after stem cell grafts. In summary, the investigation at the functional, morphological, cellular and molecular levels lead to consensual results showing that MSC infusion in our large‐animal experimental setting did not benefit the post‐ischaemic kidney.
When comparing experimental studies, an outcome may be deeply influenced by the methodology and material used. We thereafter analyse important issues concerning the experimental strategy that provide insight into our results. The design of our study, apparently, is comparable to the one adopted by other teams that demonstrated encouraging potential of stem cells in kidney restoration.
We performed autologous transplantation that we believe to be a valuable strategy when evaluating stem‐cell based therapeutics as it rules out any problems related to cell rejection. In most rodent experiments, animals had to be submitted to irradiation to enable cell engraftment, which may modify a recipient's biology and thus the results obtained. However, the beneficial effect of MSCs has also been observed in non‐irradiated syngeneic animals (28, 32), a model that approaches the autologous method we used.
Timing of stem cell transplantation and assessment may affect the results (41). In most studies, stem cells have been injected immediately or 24 h after creation of injury, and renal consequences have been examined during the early course of IRI, from 48 h to 7 days. As we adopted the same strategy, we believe timing can not explain the discrepancies in results.
The route of stem cell injection differs among experimental studies: intravenous (32, 33, 40), intra‐arterial (28, 29, 39, 44) and intraperitoneal (27) methods of infusion have been reported. We have chosen the intra‐arterial route that has been successfully used by others (28, 39, 44) in order to deliver stem cells to the targeted organ more efficiently. Potential embolism of injected stem cells using the intra‐arterial route may be a concern, but no such observation was reported here. In a first study, Kunter et al. (44) reported that injection of MSCs into the renal artery provided protection in an experimental model of glomerulonephritis and that no clinical or histological signs of embolism were noted. In a second study, Kunter et al. (45) again demonstrated beneficial effect of intra‐arterially injected MSCs in his experimental model, but maldifferentiation into glomerular adipocytes was reported. In our study, we did not observe such ‘unorthodox’ cells within the glomeruli of our large‐animal model. Additionally, we checked vascular permeability after MSC infusion and did not note any evidence of venous or capillary obstruction. Moreover, in a previous study (19), we demonstrated that intra‐arterial renal injection of MSCs in our sheep did not lead to any renal lesions in healthy kidneys.
The type, the number and preparation of stem cells may also influence the results. Most studies have used MSCs that proved efficient in experimental ARF models (19, 28, 29, 32, 33, 40, 43). In a comparative study, Morigi et al. (33) clearly demonstrated the superiority of MSCs on haematopoietic stem cells (HSCs) in a cisplatin‐induced model of renal damage. The amount of stem cells to be injected may be critical; we injected approximately 1.5 × 106 MSCs/kg of body weight intra‐arterially. This amount matches the ones used by authors reporting successful use of stem cell transplantation via intra‐arterial route (28, 29, 39, 44). Therefore, an insufficiency in the number of cells injected cannot explain the absence of effect. Preparation of cells may play a role as in the study of Duffield et al. (35), infusion of MSCs did not reduce severity of ARF, unless the cells were grown on matrigel. In our study, we did not culture MSCs on matrigel; this difference in the preparation of the cells may affect their potential. Independently from stem cell specificity, unknown factors may influence their paracrine or endocrine capacities. Although we followed carefully the usual protocol for isolation and culture that proved efficient in other studies, ovine MSCs may respond differently.
In summary, we adopted the same experimental strategy as the one used in rodent experiments that showed beneficial effect of MSCs in kidney acute failure, but MSCs in our large‐animal model failed to demonstrate regenerative potential. Besides, it is still unclear how the potential of MSCs can be safely and reasonably exploited for therapeutic use. Indeed, MSC may present drawbacks. In vitro and in vivo studies have reported a malignant potential of MSC. Wang et al. (46) identified a subpopulation of cells in human MSC culture that upon transplantation into NOD/SCID mice, formed multiple macroscopic solid tumours in multiple organs or tissues. In a clinical study, Ning et al. (47) reported that co‐transplantation of MSCs and HSCs may prevent graft‐versus‐host disease in haematologic malignancy patients, but the relapse rate is obviously higher when MSC are added.
Finally, the experimental model itself, a large‐animal species, may be incriminated. Models of renal injury differ among species (34) and stem cell capabilities of restoration in nephropathies may therefore vary. The pattern of lesions observed in our ovine model seems to better mimic human ischaemic nephropathy. Additionally, differences in stem cell behaviour and their regenerative potential among species may account for discrepancies observed between results obtained from experiments in rodents and those from our large‐animal model. It has been shown that stem cell turnover is remarkably higher in small animals than in larger ones (15). Such a difference in this fundamental ‘stemness biology’ property might account for absence of effect of MSCs in our study. Even in mice, Peister et al. (48) demonstrated that, using MSCs from five different strains, they differed in their media requirements for optimal growth, rates of propagation, and presence of the surface epitopes CD34, stem cell antigen‐1, and vascular cell adhesion molecule 1. Peister et al. conclude that differences among MSCs from different strains may explain some of the conflicting data recently published on engraftment of mouse MSCs or other bone marrow cells, into non‐haematopoietic tissues. We believe that large‐animal models must be used in order to confirm results obtained in murine experimental studies. Indeed, large‐animal models have been invaluable tools for evaluating combined stem cell and gene transfer therapy protocols before they were used in clinical studies. Progress in this area has benefited greatly from the use of large‐animal models where efficacy has often not correlated with results in murine models (49). Successful gene transduction of HSCs in large outbred animals has been difficult to achieve (50, 51), in contrast to efficient gene transfer into mouse HSCs. This discrepancy was in part explained by fundamental differences in species at the genetic level (52) and offered new insight into HSC biology that helped translating the technique to human.
In summary, the use of autologous stem cell transplantation techniques in a relevant large‐animal model of kidney IRI provides a reliable setting for the evaluation of the effect of MSCs. The extensive investigation at the functional, morphological, cellular and molecular level lead to consensual results showing that infusion of MSCs in our large‐animal experimental setting did not modulate ischaemia reperfusion‐induced kidney injury. We believe that there may be fundamental differences in species, which make the translation to human of results obtained in rodents hazardous. The extent to which we will be able to achieve efficient cell therapy will depend on assimilating a rapidly developing base of scientific data with the practical considerations of design, delivery and, ultimately, host response specificity. To date, no successful therapies that alter the outcome of ARF in patients have been found. Individual growth factors have shown promise in mouse models but have not proved effective in human trials (53). Therefore, we advocate that the development of alternative large‐animal models would benefit to the evaluation of stem‐cell therapeutic potential in the kidney, in order to reproduce results obtained in rodents and fine‐tune strategies before their ultimate use in humans.
Acknowledgements
This work was performed at IMM Research Laboratory, Paris, France, and was supported by a grant from La Fondation de l’Avenir and l’Union du crédit du Bâtiment (UCB), Paris, France (grant N° UCB). The authors wish to thank La Fondation de l’Avenir and UCB for their support and IMM Research's team for expert technical assistance.
Institutions where the work has been carried out: 1. All in vivo experiments (together with the functional evaluation) have been carried out at IMM‐RECHERCHE, Institut Mutualiste Montsouris, 75014 Paris. 2. Histological analysis has been performed at INSERM U574, Hospital Necker‐Enfant‐Malades, University Paris Descartes F‐75006, France. 3. Real‐time polymerase chain reaction analysis has been carried out at INSERM U872, Centre de Recherche des Cordeliers, Paris, F‐75006 France; Université Pierre et Marie Curie, Paris 6, UMR S872, Paris, F‐75006 France; and Université Paris Descartes, UMR S 872, Paris, F‐75006 France.
References
- 1. Abdel Aziz MT, Atta HM, Mahfouz S, Fouad HH, Roshdy NK, Ahmed HH et al (2007) Therapeutic potential of bone marrow‐derived mesenchymal stem cells on experimental liver fibrosis. Clin. Biochem. 40, 893–899. [DOI] [PubMed] [Google Scholar]
- 2. Airey JA, Almeida‐Porada G, Colletti EJ, Porada CD, Chamberlain J, Movsesian M et al (2004) Human mesenchymal stem cells form Purkinje fibers in fetal sheep heart. Circulation 109, 1401–1407. [DOI] [PubMed] [Google Scholar]
- 3. Kale S, Karihaloo A, Clark PR, Kashgarian M, Krause DS, Cantley LG (2003) Bone marrow stem cells contribute to repair of the ischemically injured renal tubule. J. Clin. Invest. 112, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz‐Gonzalez XR et al (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49. [DOI] [PubMed] [Google Scholar]
- 5. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147. [DOI] [PubMed] [Google Scholar]
- 6. Aggarwal S, Pittenger MF (2005) Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105, 1815–1822. [DOI] [PubMed] [Google Scholar]
- 7. Kh Haider H, Ashraf M (2005) Bone marrow stem cells in the infarcted heart. Coron. Artery. Dis. 16, 99–103. [DOI] [PubMed] [Google Scholar]
- 8. Vassilopoulos G, Wang PR, Russell DW (2003) Transplanted bone marrow regenerates liver by cell fusion. Nature 422, 901–904. [DOI] [PubMed] [Google Scholar]
- 9. Menasche P (2002) Cell therapy of heart failure. C. R. Biol. 325, 731–738. [DOI] [PubMed] [Google Scholar]
- 10. Slynarski K (2000) The osteogenetic potential of bone‐marrow mesenchymal stem cells. Ortop. Traumatol. Rehabil. 2, 31–32. [PubMed] [Google Scholar]
- 11. Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD et al (2006) Bone marrow‐derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells 24, 2448–2455. [DOI] [PubMed] [Google Scholar]
- 12. Guo JK, Schedl A, Krause DS (2006) Bone marrow transplantation can attenuate the progression of mesangial sclerosis. Stem Cells 24, 406–415. [DOI] [PubMed] [Google Scholar]
- 13. Steindler DA (2007) Stem cells, regenerative medicine, and animal models of disease. ILAR J. 48, 323–338. [DOI] [PubMed] [Google Scholar]
- 14. Wakeman DR, Crain AM, Snyder EY (2006) Large animal models are critical for rationally advancing regenerative therapies. Regen Med. 1, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagner JL, Storb R (1996) Preclinical large animal models for hematopoietic stem cell transplantation. Curr. Opin. Hematol. 3, 410–415. [DOI] [PubMed] [Google Scholar]
- 16. Vulliet PR, Greeley M, Halloran SM, MacDonald KA, Kittleson MD (2004) Intra‐coronary arterial injection of mesenchymal stromal cells and microinfarction in dogs. Lancet 363, 783–784. [DOI] [PubMed] [Google Scholar]
- 17. Menasche P, Alfieri O, Janssens S, McKenna W, Reichenspurner H, Trinquart L et al (2008) The Myoblast Autologous Grafting in Ischemic Cardiomyopathy (MAGIC) Trial: first randomized placebo‐controlled study of myoblast transplantation. Circulation 117, 1189–1200. [DOI] [PubMed] [Google Scholar]
- 18. Stamm C, Liebold A, Steinhoff G, Strunk D (2006) Stem cell therapy for ischemic heart disease: beginning or end of the road? Cell Transplant. 15(Suppl. 1), S47–S56. [DOI] [PubMed] [Google Scholar]
- 19. Behr L, Hekmati M, Fromont G, Borenstein N, Noel LH, Lelievre‐Pegorier M et al (2007) Intra renal arterial injection of autologous mesenchymal stem cells in an ovine model in the postischemic kidney. Nephron. Physiol. 107, 65–76. [DOI] [PubMed] [Google Scholar]
- 20. Mirshahi P, Toprak SK, Faussat AM, Dubrulle S, Marie JP, Soria C et al (2006) Malignant hematopoietic cells induce an increased expression of VEGFR‐1 and VEGFR‐3 on bone marrow endothelial cells via AKT and mTOR signalling pathways. Biochem. Biophys. Res. Commun. 349, 1003–1010. [DOI] [PubMed] [Google Scholar]
- 21. Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group‐wise comparison and statistical analysis of relative expression results in real‐time PCR. Nucleic Acids Res. 30, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pfaffl MW (2001) A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dominici M, Le Blanc K, Mueller I, Slaper‐Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317. [DOI] [PubMed] [Google Scholar]
- 24. Hoerstrup SP, Cummings Mrcs I, Lachat M, Schoen FJ, Jenni R, Leschka S et al (2006) Functional growth in tissue‐engineered living, vascular grafts: follow‐up at 100 weeks in a large animal model. Circulation 114, I159–I166. [DOI] [PubMed] [Google Scholar]
- 25. Schoeberlein A, Holzgreve W, Dudler L, Hahn S, Surbek DV (2005) Tissue‐specific engraftment after in utero transplantation of allogeneic mesenchymal stem cells into sheep fetuses. Am. J. Obstet. Gynecol. 192, 1044–1052. [DOI] [PubMed] [Google Scholar]
- 26. Sutherland FW, Perry TE, Yu Y, Sherwood MC, Rabkin E, Masuda Y et al (2005) From stem cells to viable autologous semilunar heart valve. Circulation 111, 2783–2791. [DOI] [PubMed] [Google Scholar]
- 27. Bi B, Schmitt R, Israilova M, Nishio H, Cantley LG (2007) Stromal cells protect against acute tubular injury via an endocrine effect. J. Am. Soc. Nephrol. 18, 2486–2496. [DOI] [PubMed] [Google Scholar]
- 28. Tögel F, Weiss K, Yang Y, Hu Z, Zhang P, Westenfelder C (2007) Vasculotropic, paracrine actions of infused mesenchymal stem cells are important to the recovery from acute kidney injury. Am. J. Physiol. Renal. Physiol. 292, F1626–F1635. [DOI] [PubMed] [Google Scholar]
- 29. Tögel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C (2005) Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation‐independent mechanisms. Am. J. Physiol. Renal. Physiol. 289, F31–F42. [DOI] [PubMed] [Google Scholar]
- 30. Bai L, Caplan A, Lennon D, Miller RH (2007) Human mesenchymal stem cells signals regulate neural stem cell fate. Neurochem. Res. 32, 353–362. [DOI] [PubMed] [Google Scholar]
- 31. Caplan AI, Dennis JE (2006) Mesenchymal stem cells as trophic mediators. J. Cell. Biochem. 98, 1076–1084. [DOI] [PubMed] [Google Scholar]
- 32. Imberti B, Morigi M, Tomasoni S, Rota C, Corna D, Longaretti L et al (2007) Insulin‐like growth factor‐1 sustains stem cell mediated renal repair. J. Am. Soc. Nephrol. 18, 2921–2928. [DOI] [PubMed] [Google Scholar]
- 33. Morigi M, Imberti B, Zoja C, Corna D, Tomasoni S, Abbate M et al (2004) Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J. Am. Soc. Nephrol. 15, 1794–1804. [DOI] [PubMed] [Google Scholar]
- 34. Rosen S, Heyman SN (2001) Difficulties in understanding human ‘acute tubular necrosis’: limited data and flawed animal models. Kidney Int. 60, 1220–1224. [DOI] [PubMed] [Google Scholar]
- 35. Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T et al (2005) Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow‐derived stem cells. J. Clin. Invest. 115, 1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin F, Moran A, Igarashi P (2005) Intrarenal cells, not bone marrow‐derived cells, are the major source for regeneration in postischemic kidney. J. Clin. Invest. 115, 1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin F, Cordes K, Li L, Hood L, Couser WG, Shankland SJ et al (2003) Hematopoietic stem cells contribute to the regeneration of renal tubules after renal ischemia‐reperfusion injury in mice. J. Am. Soc. Nephrol. 14, 1188–1199. [DOI] [PubMed] [Google Scholar]
- 38. Imasawa T, Utsunomiya Y, Kawamura T, Zhong Y, Nagasawa R, Okabe M et al (2001) The potential of bone marrow‐derived cells to differentiate to glomerular mesangial cells. J. Am. Soc. Nephrol. 12, 1401–1409. [DOI] [PubMed] [Google Scholar]
- 39. Lange C, Togel F, Ittrich H, Clayton F, Nolte‐Ernsting C, Zander AR et al (2005) Administered mesenchymal stem cells enhance recovery from ischemia/reperfusion‐induced acute renal failure in rats. Kidney Int. 68, 1613–1617. [DOI] [PubMed] [Google Scholar]
- 40. Herrera MB, Bussolati B, Bruno S, Morando L, Mauriello‐Romanazzi G, Sanavio F et al (2007) Exogenous mesenchymal stem cells localize to the kidney by means of CD44 following acute tubular injury. Kidney Int. 72, 430–441. [DOI] [PubMed] [Google Scholar]
- 41. Broekema M, Harmsen MC, Koerts JA, Petersen AH, Van Luyn MJ, Navis G et al (2005) Determinants of tubular bone marrow‐derived cell engraftment after renal ischemia/reperfusion in rats. Kidney Int. 68, 2572–2581. [DOI] [PubMed] [Google Scholar]
- 42. Shi Y, Melnikov VY, Schrier RW, Edelstein CL (2000) Downregulation of the calpain inhibitor protein calpastatin by caspases during renal ischemia‐reperfusion. Am. J. Physiol. Renal. Physiol. 279, F509–F517. [DOI] [PubMed] [Google Scholar]
- 43. Semedo P, Wang PM, Andreucci TH, Cenedeze MA, Teixeira VP, Reis MA et al (2007) Mesenchymal stem cells ameliorate tissue damages triggered by renal ischemia and reperfusion injury. Transplant. Proc. 39, 421–423. [DOI] [PubMed] [Google Scholar]
- 44. Kunter U, Rong S, Djuric Z, Boor P, Muller‐Newen G, Yu D et al (2006) Transplanted mesenchymal stem cells accelerate glomerular healing in experimental glomerulonephritis. J. Am. Soc. Nephrol. 17, 2202–2212. [DOI] [PubMed] [Google Scholar]
- 45. Kunter U, Rong S, Boor P, Eitner F, Müller‐Newen G, Djuric Z et al (2007) Mesenchymal stem cells prevent progressive experimental renal failure but maldifferentiate into glomerular adipocytes. J. Am. Soc. Nephrol. 18, 1754–1764. [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J et al (2005) Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy 7, 509–519. [DOI] [PubMed] [Google Scholar]
- 47. Ning H, Yang F, Jiang M, Hu L, Feng K, Zhang J et al (2008) The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia 22, 593–599. [DOI] [PubMed] [Google Scholar]
- 48. Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ (2004) Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 103, 1662–1668. [DOI] [PubMed] [Google Scholar]
- 49. Trobridge G, Beard BC, Kiem HP (2005) Hematopoietic stem cell transduction and amplification in large animal models. Hum. Gene Ther. 16, 1355–1366. [DOI] [PubMed] [Google Scholar]
- 50. Kiem HP, Leisenring W, Raff R, Deeg HJ, Schuening FG, Appelbaum FR et al (1996) Failure of recombinant stem cell factor to enhance engraftment of 1‐leucyl‐L‐leucine methyl ester treated canine marrow after irradiation. Blood 88, 1896–1897. [PubMed] [Google Scholar]
- 51. Bodine DM, Moritz T, Donahue RE, Luskey BD, Kessler SW, Martin DI et al (1993) Long‐term in vivo expression of a murine adenosine deaminase gene in rhesus monkey hematopoietic cells of multiple lineages after retroviral mediated gene transfer into CD34+ bone marrow cells. Blood 82, 1975–1980. [PubMed] [Google Scholar]
- 52. Orlic D, Girard LJ, Jordan CT, Anderson SM, Cline AP, Bodine DM (1996) The level of mRNA encoding the amphotropic retrovirus receptor in mouse and human hematopoietic stem cells is low and correlates with the efficiency of retrovirus transduction. Proc. Natl. Acad. Sci. USA 93, 11097–11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirschberg R, Kopple J, Lipsett P, Benjamin E, Minei J, Albertson T et al (1999) Multicenter clinical trial of recombinant human insulin‐like growth factor I in patients with acute renal failure. Kidney Int. 55, 2423–2432. [DOI] [PubMed] [Google Scholar]